

Summary
Objective:
Microglial abnormalities have been reported in pathological specimens from patients with tuberous sclerosis complex, a genetic disorder characterized by epilepsy, intellectual disability, and autism. However, the pathogenic role of microglia in epilepsy in TSC is poorly understood, particularly whether microglia defects may be a primary contributor to epileptogenesis or are secondary to seizures or simply epiphenomona. In this study, we tested the hypothesis that Tsc1 gene inactivation in microglia is sufficient to cause epilepsy in mouse models of TSC.
Methods:
Using a chemokine receptor, Cx3cr1, to target microglia, conventional Tsc1Cx3cr1-CreCKO mice and postnatal-inducible Tsc1Cx3cr1-CreERCKO mice were generated and assessed for molecular and histopathological evidence of microglial abnormalities, mTORC1 pathway activation, and epilepsy.
Results:
Tsc1Cx3cr1-CreCKO mice exhibited a high efficiency of microglia Tsc1 inactivation, mTORC1 activation, increased microglial size and number, and robust epilepsy, which were rapamycin-dependent. However, Cre reporter studies demonstrated that constitutive Cx3cr1 expression affected not only microglia, but also a large percentage of cortical neurons, confounding the role of microglia in epileptogenesis in Tsc1 Cx3cr1-CreCKO mice. In contrast, postnatal inactivation of Tsc1 utilizing a tamoxifen-inducible Cx3cr1-CreER resulted in a more selective microglia Tsc1 inactivation with high efficiency, mTORC1 activation, and increased microglial size and number, but no documented epilepsy.
Significance:
Microglia abnormalities may contribute to epileptogenesis in the context of neuronal involvement in TSC mouse models, but selective Tsc1 gene inactivation in microglia alone may not be sufficient to cause epilepsy, suggesting that microglia have more supportive roles in the pathogenesis of seizures in TSC.
Keywords: epilepsy, seizure, microglia, tuberous sclerosis, rapamycin
Introduction
Tuberous sclerosis complex (TSC) is an autosomal dominant genetic disorder, typically involving severe neurological comorbidities, including drug-resistant epilepsy, intellectual disability, and autism spectrum disorder.1–3 TSC occurs in about 1 in 6000 people and is caused by germline mutations in either the TSC1 or TSC2 genes. As TSC1 and TSC2 normally function to suppress the mechanistic target of rapamycin 1 (mTORC1) pathway, abnormal mTORC1 hyperactivation in TSC promotes increased cellular growth and proliferation, leading to tumor or hamartoma formation in multiple organs such as the brain, eyes, skin, heart, lungs, and kidneys.4–6 In the brain, focal lesions, including cortical tubers and subependymal giant cell astrocytomas, are characteristic and may contribute to neurological symptoms in TSC patients. In particular, cortical tubers, which represent focal malformations of cortical development, are often thought to be the foci for epileptic seizures.
Despite the potential role of tubers as gross structural lesions in causing neurological manifestations of TSC, the cellular pathophysiology of epilepsy and cognitive dysfunction remains unclear. Furthermore, independent of tubers, there is increasing evidence that more remote, non-lesional (i.e., structurally normal on MRI) brain regions may also contribute to or cause the neurological phenotypes of TSC.7 Not surprisingly, on the cellular level, abnormalities in neurons have been strongly implicated in TSC pathophysiology, including physiological defects as well as morphological abnormalities, such as in the form of the classic dysmorphic neurons seen in TSC brains.8 In addition, increasing evidence from both animal models and human TSC tissue suggest that physiological and pathological abnormalities in astrocytes also play a major role in contributing to neurological phenotypes of TSC.9; 10
Microglia are another cell type that has been increasingly implicated in neurological disorders, including TSC. Microglia act as resident macrophages and are involved in innate immunity and inflammatory responses in the brain.11; 12 In addition, microglia participate in other basic processes related to brain development and function, such as synaptogenesis and synaptic pruning.13 However, microglia can also assume a pathological state of activation, involving increased cell size and number, that may contribute to the pathophysiology of some neurological disorders. In TSC patients, pathological abnormalities in microglia have been demonstrated in cortical tuber specimens,14; 15 but whether microglia are primary contributors to epilepsy and other neurological dysfunction in TSC or are part of a secondary, reactive process has not been definitively established. A recent study indicates that microglia abnormalities could be a primary cause of epilepsy in TSC.16 In the present study, we similarly aim to determine whether Tsc1 gene inactivation in microglia is sufficient to cause epilepsy using mouse models of TSC.
Materials and Methods
Animals and drug treatment
Care and use of all mice were conducted according to an animal protocol approved by the Washington University Animal Studies Committee, and consistent with National Institutes of Health (NIH) guidelines on the Care and Use of Laboratory Animals. In addition, NIH guidelines on Rigor and Reproducibility in Preclinical Research were followed, including use of randomization, blinding, both sexes, and statistical/power analyses.
Tsc1flox/flox-Cx3cr1-Cre knock-out (Tsc1Cx3cr1-CreCKO) mice with conditional inactivation of the Tsc1 gene targeting microglia were generated by crossing Cx3cr1-Cre mice (B6J. B6N(Cg)-Cx3cr1tm1.1(cre)Jung, obtained from Jackson Laboratory)17 with Tsc1flox/flox mice9. Cx3cr1-CreER mice (B6.129P2(Cg)-Cx3cr1tm2.1(cre/ERT2)Litt/Wgan, Jackson Laboratory)18; 19, were crossed with Tsc1flox/flox mice to generate tamoxifen inducible Tsc1 knockout (Tsc1Cx3cr1-CreERCKO) mice. Tsc1flox/+-Cx3cr1-Cre, Tsc1flox/+-Cx3cr1-CreER, and Tsc1flox/flox littermate mice were used as controls. Mice were on a mixed C57/BL6 and SV129 background.
In separate Cre reporter studies, CgGt(Rosa)26Sortm6(CAG-ZsGreen1)Hze/J (Rosa-Green; Jackson Laboratory) reporter mice were crossed with Cx3cr1-Cre or Cx3cr1-CreER mice to confirm the cellular localization of Cre-mediated recombination with or without tamoxifen treatment.
Genotyping was performed for Cx3cr1-Cre according to the protocol from the Jackson Laboratory website and for Tsc1-flox as previously described9. Briefly, toes were cut and lysed with protease K ( Sigma, St. Louis, MO), DNA was extracted, and PCR Amplification was used to genotype mice by detection of sequences not normally present in the genome.
In some experiments, three-week-old Tsc1Cx3cr1-CreCKO mice were randomized to treatment with rapamycin (3mg/kg/day) or vehicle for one week, with brain tissue then harvested for western blot or immunohistochemistry analysis (8 mice per group), or for 10 weeks with video-EEG monitoring. Rapamycin (LC Labs, Woburn, MA) was initially dissolved in 100% ethanol, stored at −20°C, and diluted in a vehicle solution containing 5% Tween 80, 5% PEG 400 (Sigma, St. Louis, MO), and 4% ethanol. In other experiments, two-week old Tsc1Cx3cr1-CreERCKO mice were randomized into tamoxifen or vehicle-treatment groups. Tamoxifen (Sigma, St. Louis, MO) was dissolved in corn oil (Sigma, St. Louis, MO) at a concentration of 20 mg/ml, and was administered (9 mg/40 g body weight; i.p.) to mice every other day for five times starting at 2 weeks of age.
Western blotting
Western blot analysis was used to measure protein levels of P-S6 in the brains of Tsc1Cx3cr1-CreCKO, Tsc1Cx3cr1-CreERCKO and control mice as a measure of mTORC1 activity, using standard methods as described previously.20 Briefly, the entire neocortex and hippocampus were dissected and homogenized separately. Equal amounts of total protein extract were separated by gel electrophoresis and transferred to nitrocellulose membranes. After incubating with primary antibodies to P-S6 (Ser240/244) or S6 (#2215, #2217, Cell Signaling Technology), the membranes were reacted with a peroxidase-conjugated secondary antibody (#7074,Cell Signaling Technology). Signals were detected by enzyme chemiluminescence (GE Healthcare Life Science) and quantitatively analyzed with ImageJ software (NIH, Bethesda, MD).
Immunohistochemistry
Histological analysis was performed in a blinded fashion to assess microglia cell size and number by standard methods, as previously described.20 In brief, brains were perfusion-fixed with 4% paraformaldehyde and cut into 45 μm sections with a cryotome. Immunohistochemistry was performed for Iba1 to assess microglial cells, by labeling with primary antibody, anti-Iba1 (#019–19741 Wako), followed by labeling with secondary antibody Alexa-488 conjugated goat anti-rabbit IgG (#A11034, Life Technologies. In addition, sections were counterstained with TO-PRO-3 Iodide (Life Technologies) for the nonspecific nuclear staining of all cells. In other experiments, after Iba1 staining, double labeling for P-S6 was performed with primary anti P-S6 antibody (#2217, Cell Signaling Technology) followed by Cy3-conjugated goat anti-rabbit IgG (#A10520, Life Technologies).
Images were acquired with a Zeiss LSM PASCAL confocal microscope (Zeiss Thornwood, NY). In images from coronal sections at approximately 2 mm posterior to bregma and approximately 1 mm from midline, regions of interest were marked in neocortex by a 200 μm-wide box spanning from the neocortical surface to the bottom of layer VI, and in hippocampus by a 200×200 μm2 box within the striatum radiatum of CA1 and dentate gyrus. Iba1 positive cells and TO-PRO-3 Iodide stained cell numbers were counted in single standardized regions of interest from two sections per mouse from a total of five to six mice per group, and Iba1 positive cell numbers were normalized to TO-PRO-3 Iodide stained cell number, and were presented as Iba1 positive cells/100 cells. The size of Iba1 positive cells in each group was measured by outlining the cell body and calculating area using ImageJ software (NIH, Bethesda, MD).
In other experiments, sections of Cx3cr1-Cre or Cx3cr1-CreER mice crossed with Rosa-Green Cre reporter mice were double labeled with primary antibodies to Iba1 and GFAP (#019–19741, Wako; #3670, Cell Signaling Technology), or Iba1 and NeuN (#019–19741, Wako; MAB377, Millipore), then followed by labeling with secondary antibodies of Cy5-conjugated goat anti-rabbit IgG (#ab97077, Abcam) and Cy3-conjugated goat anti-mouse IgG (#M30010, Life Technologies). Similar methods for identification for quantification of brain sections were performed as described above. The efficiency of Cre recombination was calculated by the number of double-labeled cells (Rosa-Green & Iba1, Rosa-Green & NeuN, and Rosa-Green & GFAP) divided by total number of Iba1, NeuN and GFAP positive cells, respectively. Cellular specificity was calculated by the number of double-labeled cells (Rosa-Green & Iba1, Rosa-Green & NeuN, and Rosa-Green & GFAP) divided by total number of Rosa-Green positive cells.
Video-electroencephalography monitoring and seizure threshold testing
Vehicle- and rapamycin-treated Tsc1Cx3cr1-CreCKO mice or tamoxifen-treated Tsc1Cx3cr1-CreERCKO mice underwent video-EEG monitoring starting at 3 weeks of age (48 hrs at 3 weeks, and then continuous 24/7 starting at 4 weeks), using established methods for implanting epidural electrodes and performing continuous video-EEG recordings, as described previously.21; 22 Briefly, mice were anesthetized with isoflurane and placed in a stereotaxic frame. Epidural screw electrodes were surgically implanted and secured using dental cement for long term EEG recordings. Four electrodes were placed on the skull: one right and one left central electrodes (1 mm lateral to midline, 2 mm posterior to bregma), one frontal electrode (0.5 mm anterior and 0.5 mm to the right or left of bregma) and one occipital electrode (0.5 mm posterior and 0.5 mm to the right or left lambda). The typical recording montage involved two EEG channels with the right and left central “active” electrodes being compared to either the frontal or occipital “reference” electrode. Video and EEG data were acquired simultaneously with an AD Instruments PowerLab video-EEG system. Continuous 24/7 video-EEG data were obtained every week from Tsc1Cx3cr1-CreCKO mice, for the life of the animal. For the tamoxifen-treated Tsc1Cx3cr1-CreERCKO mice, video-EEG monitoring was terminated after 2.5 – 6 months after tamoxifen treatment. The entire EEG record was analyzed for electrographic seizures by a blinded reviewer, with video confirmation to rule out artifacts and confirm clinical seizure activity. Electrographic seizures were identified by their characteristic pattern of discrete periods of rhythmic spike discharges that evolved in frequency and amplitude lasting at least 10 s, typically ending with repetitive burst discharges and voltage suppression. On video analysis, the behavioral correlate to these seizures typically involved head bobbing, rearing with forelimb clonus, and occasional generalized convulsive activity. Seizure frequency (number of seizures per week period, based on analysis of the entire EEG record) was calculated from each week epoch.
An additional cohort of tamoxifen- and vehicle-treated Tsc1Cx3cr1-CreERCKO mice underwent pentylenetetrazole seizure threshold testing (PTZ, 75 mg/kg, i.p.). The latency to first tonic-clonic seizure was measured as an indicator of seizure threshold.
Statistics
All statistical analysis was performed using GraphPad Prism (GraphPad Software). Quantitative differences between groups were analyzed by student’s t-test, or one-way ANOVA with Tukey’s multiple comparisons post hoc tests. Chi-Square test was used for survival analysis. Quantitative data are expressed as mean ± SEM. Statistical significance was defined as p < 0.05.
Results
Microglial and mTORC1 activation in Tsc1Cx3cr1-CreCKO mice.
To test the potential role of microglia in the neurological phenotype of TSC, we first created a conventional conditional knockout mouse targeting Tsc1 gene inactivation with a Cx3cr1-Cre driver (Tsc1Cx3cr1-CreCKO mice), as Cx3cr1 represents a chemokine receptor that has been reported to be selectively expressed by microglia during embryonic brain development.23; 24 Tsc1Cx3cr1-CreCKO mice were viable and initially appeared grossly normal. Tsc1Cx3cr1-CreCKO mice demonstrated evidence of microglial activation, as seen by increased cell size and number of microglia as assayed by Iba1 immunohistochemistry (Fig. 1). The increased microglial size and number was evident in four-week-old Tsc1Cx3cr1-CreCKO mice in both neocortex and hippocampus and was at least partially reversible by the mTORC1 inhibitor rapamycin, indicating dependence on mTORC1. As additional evidence for the functional effect of Tsc1 inactivation in Tsc1Cx3cr1-CreCKO mice, mTORC1 activity was found to be increased in both neocortex and hippocampus of 4-week-old Tsc1Cx3cr1-CreCKO mice as assayed by western blotting of downstream P-S6 expression, which was also blocked by rapamycin (Fig. 2).
Figure 1. Microglial activation in Tsc1Cx3cr1-CreCKO mice.
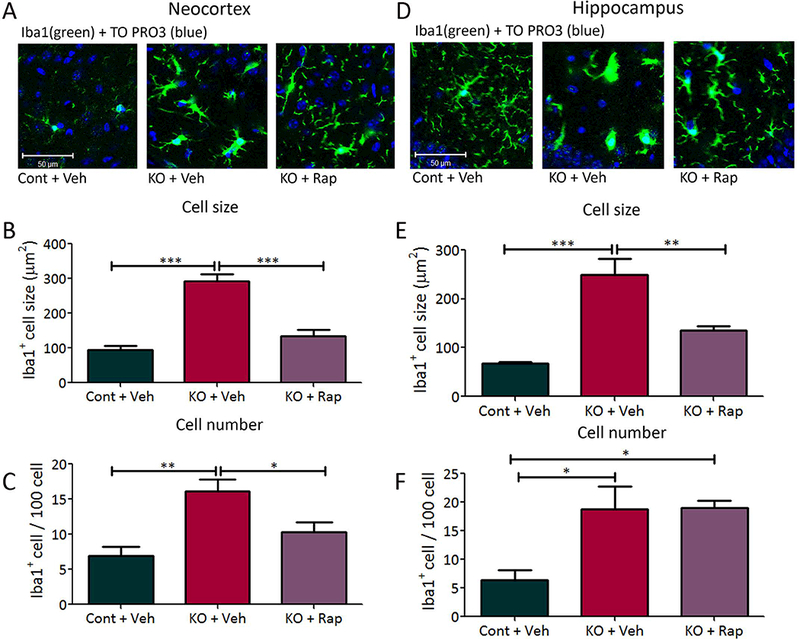
Tsc1Cx3cr1-CreCKO mice exhibit increased microglial cell size and number. Microglial activation was assessed by Iba1 (green) immunohistochemistry, followed by counterstaining with TO-PRO-3 Iodide (blue) for the nonspecific nuclear staining of all cells, in neocortex (A) and hippocampus (D). Four-week-old Tsc1Cx3cr1-CreCKO mice (KO+Veh) had significantly increased Iba1 positive cell size and number in neocortex (B,C) and hippocampus (E, F), compared with control mice (Cont+Veh). Rapamycin treatment (3 mg/kg/day i.p. for one week starting at 3 weeks of age) of Tsc1Cx3cr1-CreCKO mice (KO+Rap) significantly reduced the Iba1 positive cell size and number, except for that of cell number in hippocampus (F). * p<0.05, ** p<0.01, *** p<0.001, by one-way ANOVA (n = 5–6 mice/group). Cont = control mice, KO = Tsc1Cx3cr1-CreCKO mice, Veh = vehicle, Rap = rapamycin. Scale bar = 50 μm.
Figure 2. mTORC1 activation in Tsc1Cx3cr1-CreCKO mice.
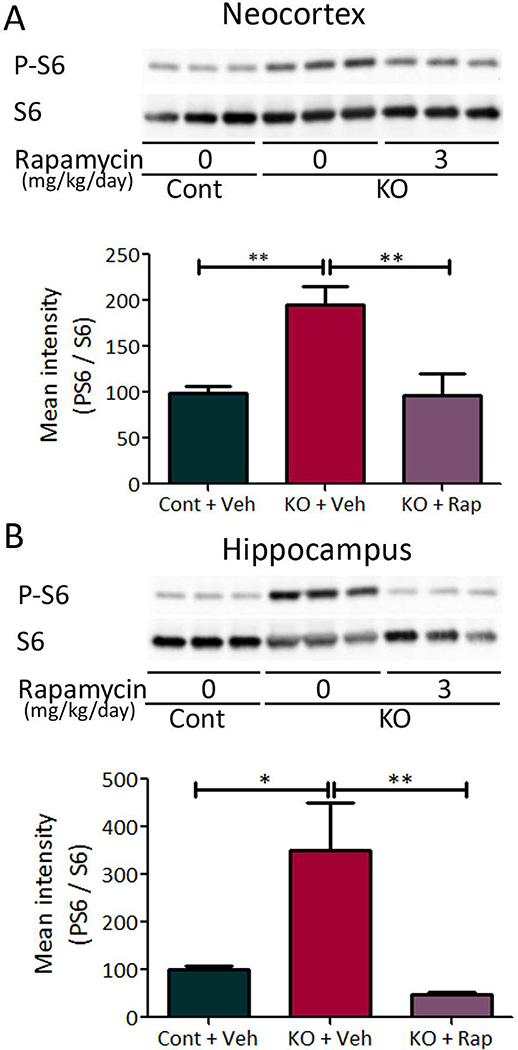
mTORC1 activation was assessed by western blotting of protein expression of P-S6 and S6 in the neocortex (A) and hippocampus (B) of Tsc1Cx3cr1CKO mice, and the effect of rapamycin was tested. Four week old Tsc1Cx3cr1-CreCKO mice (KO+Veh) had significantly increased P-S6 levels compared with control mice (Cont+Veh). Rapamycin treatment (3 mg/kg/day, i.p. for seven days) significantly decreased P-S6 levels in Tsc1Cx3cr1-CreCKO mice (KO+Rap). The ratio of P-S6/S6 was normalized to the vehicle treated control group. * p<0.05, ** p<0.01, by one-way ANOVA (n = 8 mice/group). Cont = control mice, KO = Tsc1Cx3cr1-CreCKO mice, Veh = vehicle, Rap = rapamycin.
Furthermore, co-immunostaining of P-S6 and Iba1 demonstrated abundant double-labeled cells, which were increased in size and number in four-week-old Tsc1Cx3cr1-CreCKO mice (KO) compared with control mice (Cont), indicating that mTORC1 activity was increased in KO microglia (Suppl Fig. S1).
mTORC1-dependent megalencephaly, epilepsy and decreased survival in Tsc1Cx3cr1-CreCKO mice
Tsc1Cx3cr1-CreCKO mice exhibited diffuse megalencephaly of the brain, as grossly visible and also reflected by increased brain weight at four weeks of age (Fig. 3A), which was prevented by rapamycin treatment. Vehicle-treated Tsc1Cx3cr1-CreCKO mice had significantly decreased body weight compared with vehicle treated control mice at four weeks of age, but not 3 weeks of age, while rapamycin treatment did not prevent the decreased weight (Fig. 3B). Tsc1Cx3cr1-CreCKO mice were monitored for seizures with continuous video-EEG monitoring starting at 3 weeks of age. 100% (12 of 12 mice) had documented clinical-electrographic seizures, as well as interictal bursts of spikes (Fig. 4A). Behaviorally the seizures were characterized primarily by rearing up and repetitive forelimb clonus. Electrographically, seizures demonstrated stereotypic development of repetitive spikes which evolved in frequency and amplitude into periodic bursts, lasting at least 10 seconds and followed by voltage suppression. Overall, average seizure frequency was relatively low (~one seizure/week), but individual mice tended to develop acute clusters of seizures between 4–9 weeks of age correlating with a rapid onset of premature death (Fig. 4B). About 50% of Tsc1Cx3cr1-CreCKO mice had died by about 5 weeks of age and all by 13 weeks (Fig. 4C). Most mice were documented on video-EEG to die in the context of or immediately after a seizure.
Figure 3. Megalencephaly and decreased body weight in Tsc1Cx3cr1-CreCKO mice.
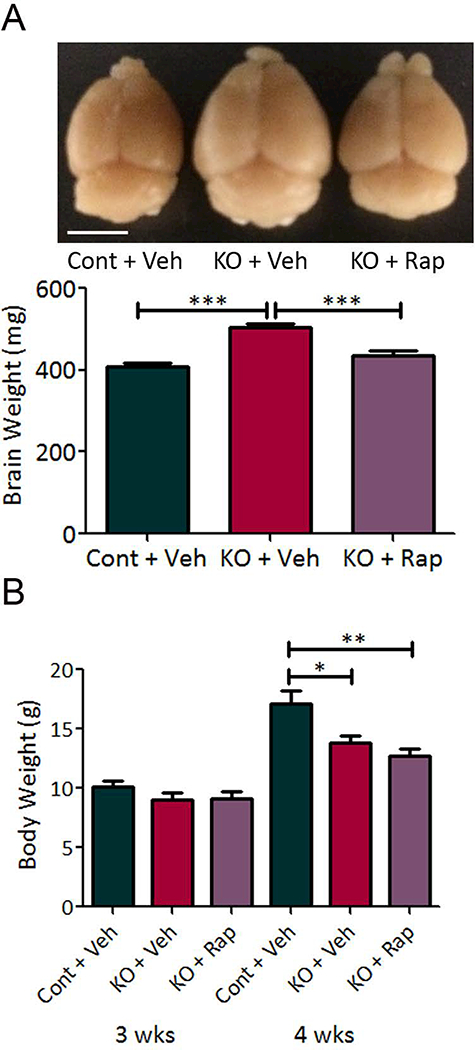
(A) Tsc1Cx3cr1-CreCKO mice (KO+Veh) exhibited diffuse megalencephaly of the brain, as grossly visible and also reflected by increased brain weight at four weeks of age, compared with control mice (Cont+Veh). Rapamycin treatment (3 mg/kg/day, i.p. for seven days) significantly decreased brain weight in Tsc1Cx3cr1-CreCKO mice (KO+Rap). Scale bar = 5 mm. *** p<0.001 by one-way ANOVA (n = 9–10 mice/group). (B) Body weight of mice of each group at three and four weeks of age. There was no significant difference in body weight between groups at three weeks of age. At four weeks, Tsc1Cx3cr1-CreCKO mice (KO + Veh) had significantly decreased body weight compared with control mice (Cont + Veh). Rapamycin treatment did not reverse this decreased body weight, * p<0.05, ** p<0.01, by one-way ANOVA (n = 8 mice/group). Cont = control mice, KO = Tsc1Cx3cr1-CreCKO mice, Veh = vehicle, Rap = rapamycin.
Figure 4. mTORC1-dependent epilepsy and decreased survival in Tsc1Cx3cr1-CreCKO mice.
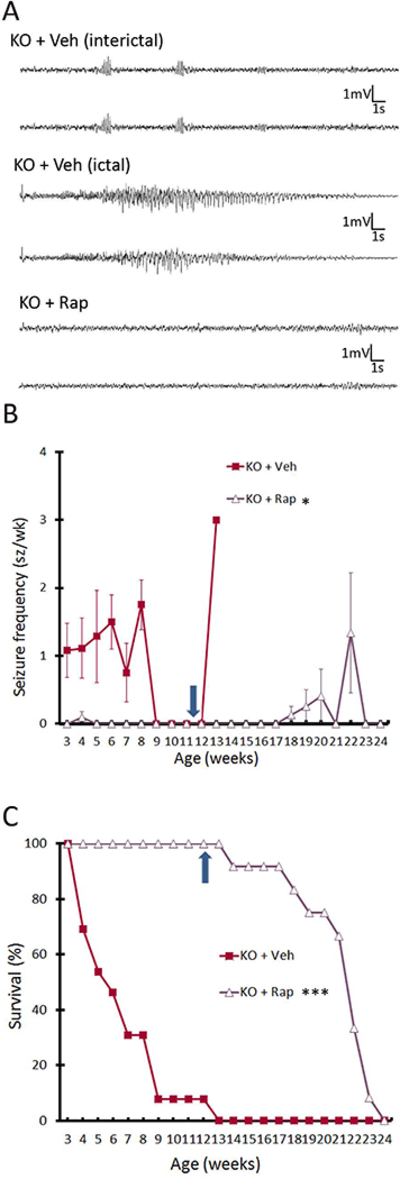
(A) Representative bilateral EEG recordings of Tsc1Cx3cr1-CreCKO mice treated with vehicle (KO+Veh) and rapamycin (KO+Rap). Tsc1Cx3cr1-CreCKO mice exhibit bursts of interictal spikes and clinical-electrographic seizures, which were both suppressed by rapamycin. (B) Seizures start to develop in vehicle treated Tsc1Cx3cr1-CreCKO mice (KO+Veh) around 3 weeks of age, and 100% (12 of 12) of mice had documented clinical-electrographic seizures. Rapamycin treatment (3 mg/kg/day, i.p.) almost completely suppressed the development of seizures in Tsc1Cx3cr1CKO mice. *p<0.05 compared with vehicle-treated Tsc1Cx3cr1-CreCKO mice by one-way ANOVA (n = 12 mice/group). (C) Survival analysis showed that vehicle-treated Tsc1Cx3cr1-CreCKO mice die prematurely with 50% mortality at 5–6 weeks of age and 100% mortality by 13 weeks, while all rapamycin treated mice survived during rapamycin treatment. ***p<0.001 by Chi-Square test, comparing the two groups (n = 12 mice/group). However, once rapamycin was stopped at 12 weeks of age (arrows in B and C), seizures subsequently occurred in some mice and all mice died by 24 weeks of age. Cont = control mice, KO = Tsc1Cx3cr1-CreCKO mice, Veh = vehicle, Rap = rapamycin.
Rapamycin treatment initiated at 3 weeks of age almost completely suppressed the development of seizures and prolonged survival, with all rapamycin-treated Tsc1Cx3cr1-CreCKO mice surviving to at least 12 weeks of age (Fig. 4B,C). However, once rapamycin was stopped at 12 weeks of age, seizures subsequently occurred in some mice and all mice died by 24 weeks of age (Fig. 4B,C).
Specificity of constitutive Cx3cr1-Cre versus inducible Cx3Cr1-CreER for microglia
The documentation of robust seizures in 100% of Tsc1Cx3cr1-CreCKO mice provides initial evidence that Tsc1 inactivation in microglia is sufficient to cause epilepsy, but this assumes that the Cx3cr1-Cre driver is specific for microglia. To test for cellular specificity, we crossed Rosa-Green Cre reporter mice with our Cx3cr1-Cre mice to confirm the cellular localization of Cre mediated recombination. In a quantitative analysis, the expression of the GFP reporter gene was detected in almost 90% of Iba1 positive cells in hippocampus and cortex, indicating a high efficiency of Cre-recombination within microglia (Suppl. Fig. S2). However, when double-labeling for other cellular markers, a large percentage of other Rosa-Green positive cells were identified that were negative for Iba1, but positive for NeuN or to a lesser degree GFAP, indicating that neurons and some astrocytes were also affected by Cre-recombination. Hence, the Cx3cr1-Cre does not appear to be specific for microglia, at least under conditions of constitutive expression, which includes the prenatal embryonic period. Thus, while microglia may contribute to an epilepsy phenotype, it is difficult to conclude that Tsc1 inactivation in microglia in itself is solely responsible for causing seizures in conventional Tsc1Cx3cr1-CreCKO mice.
While the above studies indicate that constitutive Cx3cr1-driven inactivation may not be microglia-specific, it’s possible that temporally targeted, postnatal Cx3cr1 expression may be more specific for microglia, such as in using an inducible Cx3cr1-CreER that can be activated by tamoxifen injection postnatally. To test this possibility, Rosa-Green Cre reporter mice were crossed with Cx3cr1-CreER mice and injected with tamoxifen starting at two weeks of age. Similar to Cx3cr1-Cre mice without tamoxifen, the efficiency of Cre-expression was around 90% in cortex and hippocampus in Cx3cr1-CreER mice following tamoxifen injection (Suppl. Fig. S3). Moreover, in contrast to constitutive Cx3cr1-Cre mice, inducible Cx3cr1-CreER appeared to be more specific to microglia as seen in Iba1 double-labeling compared with NeuN and GFAP, although about 5% of neurons in cortex still showed evidence of Cre-recombination. Thus, compared with Tsc1Cx3cr1-CreCKO mice, postnatally-activated Tsc1Cx3cr1-CreERCKO mice are more appropriate for testing the role of microglia in neurological phenotypes of TSC.
Tsc1Cx3cr1-CreERCKO mice demonstrate microglia and mTORC1 activation, but do not have epilepsy.
Tsc1Cx3cr1CreERCKO mice injected with tamoxifen starting at two weeks of age were first analyzed for evidence of microglial activation. Similar to conventional Tsc1Cx3cr1-CreCKO mice, microglia in cortex and hippocampus had both increased cell size and number following tamoxifen injection in Tsc1Cx3cr1-CreERCKO mice (Fig. 5). Correspondingly, mTORC1 activation was increased in cortex and hippocampus of Tsc1Cx3cr1-CreERCKO mice, as reflected by downstream P-S6 expression (Fig. 6). However, in contrast to Tsc1Cx3cr1-CreCKO mice, continuous video-EEG monitoring for up to 6 months following tamoxifen injection did not detect any seizures in Tsc1Cx3cr1-CreERCKO mice or any definite interictal epileptiform abnormalities in all mice (Suppl. Fig. S4 A, n=9 mice recorded continuously for at least 10 weeks after tamoxifen, range 10–26 weeks, mean 16.0±2.5 weeks; an additional 2 mice recorded for 3–4 weeks after tamoxifen). Furthermore, there was no significant difference between 8 week old tamoxifen- and vehicle-injected Tsc1Cx3cr1-CreERCKO mice in the PTZ-induced seizure threshold (Suppl. Fig. S4 B, latency to first generalized tonic-clonic seizure = 106.2±19.4 s in tamoxifen-injected mice versus 94.8±21.6 s in controls; n=12 mice per group). In addition, there was no evidence of megalencephaly or significantly increased brain weight in tamoxifen-injected Tsc1Cx3cr1-CreERCKO mice (Suppl. Fig. S4 C, brain weight =458.2±13.0 in tamoxifen-injected mice vs. 434.5±7.0 in vehicle-injected mice). Thus, in contrast to the Tsc1Cx3cr1-CreCKO mice which lacked microglia specificity, the more microglia-selective Tsc1Cx3cr1-CreERCKO mouse did not provide evidence that Tsc1 inactivation in microglia by itself is sufficient to cause epilepsy.
Figure 5. Postnatal, tamoxifen-induced microglial activation in Tsc1Cx3cr1-CreERCKO mice.
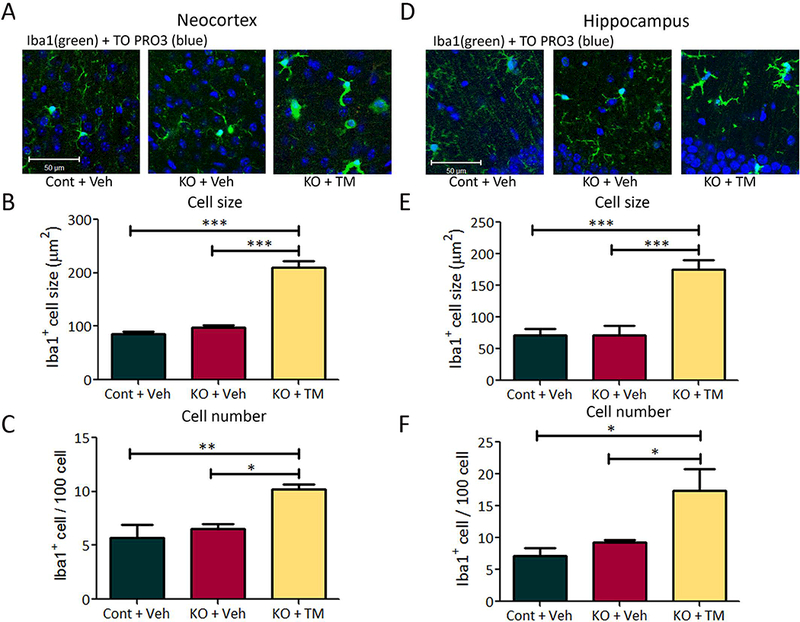
Tsc1Cx3cr1-CreERCKO mice treated with tamoxifen at 2 weeks of age exhibit increased microglial cell size and number. Microglial activation was assessed by Iba1 (green) immunohistochemistry, followed by counterstaining with TO-PRO-3 Iodide (blue) for the nonspecific nuclear staining of all cells in neocortex (A) and hippocampus (D). Eight week old tamoxifen treated Tsc1Cx3cr1-CreERCKO mice (KO+TM) had significantly increased Iba1-positive cell size and number in neocortex (B,C) and hippocampus (E, F), compared with vehicle-treated control and Tsc1Cx3cr1-CreERCKO mice (Cont+Veh, KO+Veh). * p<0.05, ** p<0.01, *** p<0.001, by one-way ANOVA (n = 5 mice/group). Cont = control mice, KO = Tsc1Cx3cr1-CreERCKO mice, Veh = vehicle, TM = tamoxifen. Scale bar = 50 μm.
Figure 6. Tamoxifen induced mTORC1 activation in Tsc1Cx3cr1-CreERCKO mice.
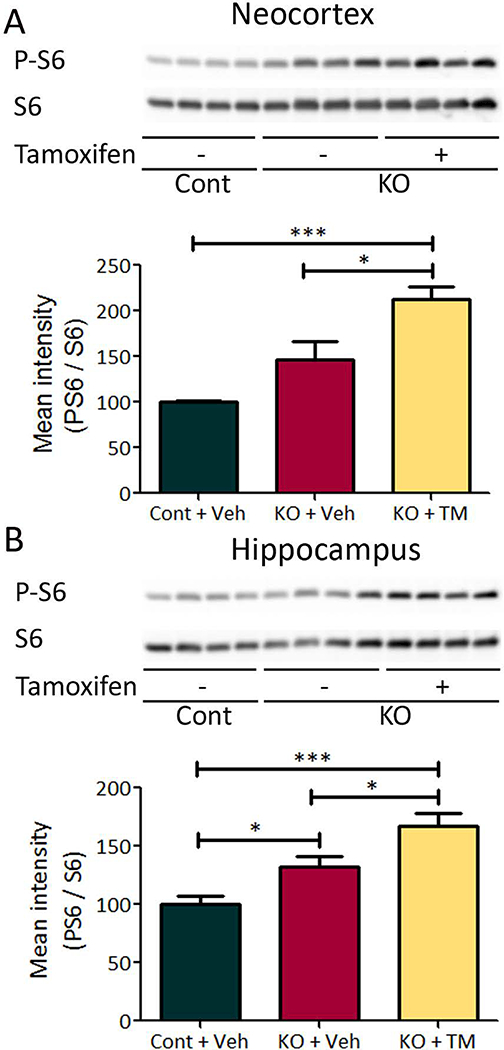
mTORC1 activation was assessed by western blotting of protein expressions of P-S6 and S6 in the neocortex (A) and hippocampus (B) of tamoxifen treated Tsc1Cx3cr1-CreERCKO mice and control mice. Starting at 2 weeks of age, tamoxifen was administered (9 mg/40 g body weight; i.p) to mice every other day for five times. Six weeks after tamoxifen treatment, Tsc1Cx3cr1-CreERCKO mice had significantly increased P-S6 levels compared with vehicle treated Tsc1Cx3cr1-CreERCKO mice and control mice in both neocortex (A), and hippocampus (B). The ratio of P-S6/S6 was normalized to the vehicle treated control group. * p<0.05, *** p<0.001, by one-way ANOVA (n = 8 mice/group). Cont = control mice, KO = Tsc1Cx3cr1-CreERCKO mice, Veh = vehicle, TM = tamoxifen.
Discussion
The present study directly tests the hypothesis that Tsc1 gene inactivation and corresponding mTORC1 activation specifically in microglia are sufficient to cause an epilepsy phenotype in TSC mouse models. Using a constitutive Cx3cr1-Cre approach, Tsc1Cx3cr1-CreCKO mice demonstrate increased microglia size and number, mTORC1 hyperactivation, and robust epilepsy; however, the Cx3cr1 expression did not appear to be specific for microglia, but also directly involved a large percentage of cortical neurons, making it difficult to isolate the role of microglia in epileptogenesis. In contrast, utilizing an inducible Cx3cr1-Cre ER approach that was more selective for microglia, postnatal induction of Tsc1 inactivation led to similar microglial abnormalities and mTORC1 activation in cortex and hippocampus, but no detectable epilepsy. Comparing the two models, it appears that Tsc1 inactivation in microglia alone, in the absence of neuronal involvement, may not be sufficient to cause epilepsy.
There has been accumulating evidence for a primary role of microglia in epileptogenesis, but this remains controversial and likely depends on the type or cause of epilepsy. Without doubt, microglial abnormalities have been reported in pathological specimens from epilepsy patients and in multiple animal models of epilepsy.25; 26 However, most of these findings are correlative and do not prove a primary role of microglia in contributing to epileptogenesis, as opposed to being a secondary response to seizures or an epiphenomenon. The most direct evidence for a primary role of microglia in causing epilepsy derives from animal models in which microglial activation can be reversed, typically by pharmacological means, such as through the use of minocycline, an inhibitor of microglial activation.27 Minocycline decreases epileptogenesis in rodent models of temporal lobe epilepsy.28; 29 In TSC patients, abnormalities in microglia have been reported in cortical tuber specimens,14; 15 but again whether these microglial abnormalities are primary or secondary related to epilepsy is unknown. In a previous study, a mouse model of TSC, involving inactivation of the Tsc1 gene in glia and neurons driven by a GFAP promoter (Tsc1GFAPCKO mice), leads to increases in microglial cell size and number, which is reversible by minocycline.20 However, the epilepsy in Tsc1GFAPCKO mice is not reversed by minocycline, indicating that the microglial reaction in this model is a secondary response.
In the present study, we adopted a genetic approach to directly test the primary role of microglia in epileptogenesis in TSC mouse models, using Cx3cr1-Cre to induce Tsc1 inactivation under the assumption that Cx3cr1 is specifically expressed in the brain in microglia.23; 24 While conventional Tsc1Cx3cr1-CreCKO did show a very high efficiency in affecting microglia, somewhat surprisingly Cre-reporter mice studies did not confirm specificity for microglia but also indicated substantial direct neuronal involvement, confounding interpretation of the cellular origin of epilepsy in the Tsc1Cx3cr1-CreCKO mice. While the Cx3cr1 promoter has been reported to be an appropriate driver for targeting microglia, the issue of microglial specificity of Cx3cr1 in the brain is not definitive, as recent reports have demonstrated Cx3cr1 expression in neurons,30–32 suggesting that use of a Cx3cr1 driver is not necessarily specific for microglia in the brain. As many gene promoters are developmentally regulated and more active in certain cell types in early development before becoming silent, it is possible that specificity of Cxc3cr1 expression in microglia increases with age. In fact, utilizing an inducible Cxc3cr1-CreER with postnatal activation did appear to significantly increase microglia specificity in Tsc1Cx3cr1ERCKO mice, but in our hands, this was not sufficient to cause epilepsy.
By comparison, a recently published study also generated a similar Tsc1Cx3cr1-CreCKO mouse with comparable microglia abnormalities and robust seizures, leading the authors to conclude that specific microglia abnormalities were sufficient to cause epilepsy.16 However, while the Cre-driver in that study was stated to be specific for microglia, quantitative analysis and, in particular, double-labeling for neurons in their Cre reporter experiments were not reported, so it is possible that their model also had neuronal, as well as microglial, knock-out of Tsc1, as we found in our quantitative Cre reporter studies with double labeling for microglia, neurons, and astrocytes. On the other hand, the previous study also tested effect of postnatal induced, Cx3Cr1-CreER driven Tsc1 inactivation and, in contrast to our study, still found very robust seizures.16 However, the previous study did not include any Cre reporter data for postnatal Cre induction. By extrapolation, our quantitative reporter studies with postnatal Cre-induction in Cx3cr1-Cre ER mice showed substantially increased microglia specificity compared to the conventional Tsc1Cx3cr1-CreCKO mouse, suggesting that postnatal microglial Tsc1 inactivation may have been sufficient to cause epilepsy in the previous study.16 However, there was still a small percentage of cortical and hippocampal neurons demonstrating Cre-recombination in our postnatal reporter studies, which in itself might be sufficient to cause epilepsy. For example, postnatal mTOR activation in less than 10% of dentate granule neurons has been previously shown to be sufficient to cause epilepsy.33 Another difference between our studies was the source of the Cx3cr1-Cre mice and the genetic background, which could influence seizure susceptibility. However, if it is accepted that Cx3cr1 is expressed in neurons30–32, then these additional factors should not negate the underlying biological principle that use of Cx3cr1 as a Cre-driver is not specific for microglia and the potential contribution of neurons cannot be ignored in these models. Finally, there could have been a difference in efficiency in microglia (or neuronal) Cre-recombination in our respective inducible-Cre mice between the two studies, but our detailed reporter studies document a very high efficiency of Cre-recombination in microglia. Thus, based on the available data, it may still not be definitive whether any epilepsy phenotype reported with either pre- or postnatal Cx3Cr1-driven Tsc1 inactivation in the previous16 or current study was specifically due to microglia abnormalities.
In summary, with rigorous reporter data correlating microglia specificity and efficiency of Tsc1Cx3cr1-CreCKO mice and Tsc1Cx3cr1-CreERCKO mice with the presence and absence of epilepsy, respectively, the most parsimonious conclusion with the currently-available data is that Tsc1 inactivation in microglia alone may not be sufficient to cause epilepsy, but primary neuronal involvement may also be required. However, this does not exclude the possibility that microglia abnormalities contribute to or are necessary for epileptogenesis in the context of neuronal abnormalities. As a future direction, a “two-hit” model combining an acquired insult, such as a pro-inflammatory stimulus, in the Tsc1Cx3cr1-CreERCKO mice, may be sufficient to cause epilepsy. Furthermore, while in our hands, postnatal inactivation of Tsc1 alone in Tsc1Cx3cr1-CreERCKO mice was not sufficient to cause epilepsy, it is possible that prenatal microglia-specific Tsc1 inactivation may be sufficient to cause epilepsy, but unfortunately the constitutive Tsc1Cx3cr1-CreCKO mice with presumed embryonic induction could not test this possibility due to clear lack of specificity for microglia. Future studies and approaches that improve selectivity for targeting microglia embryonically may test this possibility more directly.
Supplementary Material
Key Points.
Tsc1Cx3cr1-CreCKO mice have a high efficiency of microglia Tsc1 inactivation, increased microglial size and number, and robust epilepsy.
However, Tsc1Cx3cr1-CreCKO mice also demonstrate direct neuronal Tsc1 inactivation, confounding the role of microglia in epilepsy.
Postnatal inactivation of Tsc1 in inducible Tsc1Cx3cr1-CreERCKO mice causes more selective microglia Tsc1 inactivation, but no epilepsy.
Microglia abnormalities in themselves may contribute to, but may not be sufficient for, epileptogenesis in TSC mouse models.
Acknowledgements
This work was supported by the National Institutes of Health (R01 NS056872 to MW and S10 RR027552 to Washington University), the McDonnell Center for Systems Neuroscience, the Alafi Neuroimaging Lab at Washington University, the Intellectual and Developmental Disabilities Research Center (U54 HD087011) at Washington University, and Missouri State Tuberous Sclerosis Fund.
Footnotes
Ethical Publication Statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Disclosures
None of the authors has any conflict of interest to disclose.
References
- 1.Orlova KA, Crino PB. The tuberous sclerosis complex. Ann N Y Acad Sci 2010;1184:87–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DiMario FJ Jr., Sahin M, Ebrahimi-Fakhari D. Tuberous Sclerosis Complex. Pediatr Clin North Am 2015;62:633–648. [DOI] [PubMed] [Google Scholar]
- 3.Chu-Shore CJ, Major P, Camposano S, et al. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia 2010;51:1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lipton JO, Sahin M. The neurology of mTOR. Neuron 2014;84:275–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012;149:274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017;168:960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong M Mechanisms of epileptogenesis in tuberous sclerosis complex and related malformations of cortical development with abnormal glioneuronal proliferation. Epilepsia 2008;49:8–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meikle L, Talos DM, Onda H, et al. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci 2007;27:5546–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uhlmann EJ, Wong M, Baldwin RL, et al. Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol 2002;52:285–296. [DOI] [PubMed] [Google Scholar]
- 10.Sosunov AA, Wu X, Weiner HL, et al. Tuberous sclerosis: a primary pathology of astrocytes? Epilepsia 2008;49 Suppl 2: 53–62. [DOI] [PubMed] [Google Scholar]
- 11.Town T, Nikolic V, Tan J. The microglial “activation” continuum: from innate to adaptive responses. J Neuroinflammation 2005;2:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devinsky O, Vezzani A, Najjar S, et al. Glia and epilepsy: excitability and inflammation. Trends Neurosci 2013;36:174–184. [DOI] [PubMed] [Google Scholar]
- 13.Paolicelli RC, Bolasco G, Pagani F, et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011;333:1456–1458. [DOI] [PubMed] [Google Scholar]
- 14.Boer K, Jansen F, Nellist M, et al. Inflammatory processes in cortical tubers and subependymal giant cell tumors of tuberous sclerosis complex. Epilepsy Res 2008;78:7–21. [DOI] [PubMed] [Google Scholar]
- 15.Boer K, Troost D, Jansen F, et al. Clinicopathological and immunohistochemical findings in an autopsy case of tuberous sclerosis complex. Neuropathology 2008;28:577–590. [DOI] [PubMed] [Google Scholar]
- 16.Zhao X, Liao Y, Morgan S, et al. Noninflammatory Changes of Microglia Are Sufficient to Cause Epilepsy. Cell Rep 2018;22:2080–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jung S, Aliberti J, Graemmel P, et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 2000;20:4106–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parkhurst CN, Yang G, Ninan I, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013;155:1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao L, Zabel MK, Wang X, et al. Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol Med 2015;7:1179–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang B, Zou J, Han L, et al. Microglial activation during epileptogenesis in a mouse model of tuberous sclerosis complex. Epilepsia 2016;57:1317–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeng LH, Xu L, Gutmann DH, et al. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol 2008;63:444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erbayat-Altay E, Zeng LH, Xu L, et al. The natural history and treatment of epilepsy in a murine model of tuberous sclerosis. Epilepsia 2007;48:1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wieghofer P, Knobeloch KP, Prinz M. Genetic targeting of microglia. Glia 2015;63:1–22. [DOI] [PubMed] [Google Scholar]
- 24.Wolf Y, Yona S, Kim KW, et al. Microglia, seen from the CX3CR1 angle. Front Cell Neurosci 2013;7:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Najjar S, Pearlman D, Miller DC, et al. Refractory epilepsy associated with microglial activation. Neurologist 2011;17:249–254. [DOI] [PubMed] [Google Scholar]
- 26.Shapiro LA, Wang L, Ribak CE. Rapid astrocyte and microglial activation following pilocarpine-induced seizures in rats. Epilepsia 2008;49 Suppl 2:33–41. [DOI] [PubMed] [Google Scholar]
- 27.Yrjanheikki J, Keinanen R, Pellikka M, et al. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci U S A 1998;95:15769–15774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang N, Mi X, Gao B, et al. Minocycline inhibits brain inflammation and attenuates spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neuroscience 2015;287:144–156. [DOI] [PubMed] [Google Scholar]
- 29.Abraham J, Fox PD, Condello C, et al. Minocycline attenuates microglia activation and blocks the long-term epileptogenic effects of early-life seizures. Neurobiol Dis 2012;46:425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meucci O, Fatatis A, Simen AA, et al. Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc Natl Acad Sci U S A 2000;97:8075–8080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J, Gan Y, Han P, et al. Ischemia-induced Neuronal Cell Death Is Mediated by Chemokine Receptor CX3CR1. Sci Rep 2018;8:556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yeo SI, Kim JE, Ryu HJ, et al. The roles of fractalkine/CX3CR1 system in neuronal death following pilocarpine-induced status epilepticus. J Neuroimmunol 2011;234:93–102. [DOI] [PubMed] [Google Scholar]
- 33.Pun RY, Rolle IJ, Lasarge CL, et al. Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron 2012;75:1022–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.