

In Brief
Stanger, de Beer, Dranow et al. describe the novel BID domain fold, revealing a compact four-helix bundle. Their analyses suggest that the conserved shape of BID domains is critical to function as a secretion signal, while low surface conservation facilitates the evolution of secondary functions.
SUMMARY
The BID (Bep intracellular delivery) domain functions as secretion signal in a subfamily of protein substrates of bacterial type IV secretion (T4S) systems. It mediates transfer of (1) relaxases and the attached DNA during bacterial conjugation, and (2) numerous Bartonella effector proteins (Beps) during protein transfer into host cells infected by pathogenic Bartonella species. Furthermore, BID domains of Beps have often evolved secondary effector functions within host cells. Here, we provide crystal structures for three representative BID domains and describe a novel conserved fold characterized by a compact, antiparallel four-helix bundle topped with a hook. The conserved hydrophobic core provides a rigid scaffold to a surface that, despite a few conserved exposed residues and similarities in charge distribution, displays significant variability. We propose that the genuine function of BID domains as T4S signal may primarily depend on their rigid structure, while the plasticity of their surface may facilitate adaptation to secondary effector functions.
Graphical Abstract
INTRODUCTION
Bacterial type IV secretion (T4S) systems are supramolecular protein assemblies that mediate contact-dependent (1) interbacterial transfer of relaxases and the covalently attached DNA into recipient cells during bacterial conjugation and (2) interkingdom transfer of host cell-targeted effector proteins of pathogenic bacteria such as Helicobacter pylori, Legionella pneumophila, Brucella spp., and Bartonella spp. (Christie et al., 2014). T4S substrates harbor a C-terminal non-cleavable T4S signal that is considered to interact with the T4S coupling protein (T4CP), a T4S system-associated ATPase (Christie et al., 2014). T4S signals are typically only a few tens of amino acids long and consist of clusters of positively charged or hydrophobic residues (Christie et al., 2014). However, a subfamily of T4S systems prominently found in the a-proteobacteria display T4S signals with a more complex bipartite structure composed of the approximately 140 amino-acid long BID (Bep intracellular delivery) domain and a short positively charged C-terminal segment that is similar to the genuine T4S signal of other T4S systems (Schulein et al., 2005). T4CPs that are associated with BID domain-containing effectors form a monophyletic cluster within the phylogenetic tree of T4CPs (Schulein et al., 2005), indicating that this sublineage evolved and maintained specific adaptations to facilitate interaction with the BID domain.
BID domains are found in relaxases and in the Beps (Bartonella effector proteins) representing numerous host cell-targeted effectors of pathogenic Bartonella spp. Beps are translocated by the VirB T4S system and its associated T4CP VirD4 (Saenz et al., 2007). Beps have evolved by gene duplication, diversification, and reshuffling from a single ancestor containing an N-terminal FIC (filamentation induced by cAMP) domain, a C-terminal BID domain, and a connecting central OB (oligonucleotide/oligosaccharide binding) fold, resulting in diverse derived modular architectures (Engel et al., 2011; Saenz et al., 2007). Providing a striking example of parallel evolution, this process occurred independently in two distinct lineages of Bartonella, giving rise to Bep1 to Bep10 in lineage 3 (L3) and BepA to BepJ in lineage 4 (L4) (Engel et al., 2011). In the N-terminal part, Beps harbor either a FIC-OB fold, tandem-repeat tyrosine-phosphorylation motifs, or additional BID domains (Engel et al., 2011), which mediate diverse effector functions within host cells (Harms et al., 2016; Selbach et al., 2009; Siamer and Dehio, 2015). The original function of the C-terminal BID domain present in each Bep is to facilitate protein transfer via the VirB/VirD4 T4S system. However, several studies in the model pathogen Bartonella henselae and other L4 species showed that individual BID domains, including those in multi-BID domain architectures, have secondarily evolved discrete effector functions within host cells (Siamer and Dehio, 2015) that are considered to be mediated by specific protein-protein interactions with host proteins. The single BID Structure 25, 203–211, January 3, 2017 domain of BepA binds to host adenylyl cyclase and potentiates GαS-dependent cyclic AMP (cAMP) production, ultimately resulting in inhibition of apoptosis (Pulliainen et al., 2012). The BID domains of BepE are required for normal migration of host cells during infection in vitro and in vivo (Okujava et al., 2014), and those of BepF or BepG trigger actin-dependent uptake of bacterial aggregates into a unique cellular structure known as invasome (Rhomberg et al., 2009; Truttmann et al., 2011).
To pave the way for addressing structure/function-related questions concerning the BID domain, we determined crystal structures of BID domains from three different Bartonella effector proteins, describe the new fold, and analyze site-specific determinants for structure and potential function by sequence comparison.
RESULTS
Structure Determination
Crystals of the BID domains from Bartonella rochalimae Bep6 (BroBep6_tBID1), Bartonella clarridgeiae Bep9 (BclBep9_tBID1), and B. henselae BepE (BheBepE_BID1) were obtained and complete datasets collected to a resolution of 2.2 Å or higher (Table 1). Since there were no homologs in the PDB with significant sequence identity (>25%), the crystal structure of BroBep6_tBID1 was determined by SeMet-SAD phasing. The resulting model was then used for the structure determination of BclBep9_tBID1 and BheBepE_BID1 by molecular replacement. Refinement yielded BID domain models with Rwork/Rfree (%) values of 17.7/20.3, 17.1/21.4, and 18.3/22.5, respectively. Data collection and final refinement statistics are given in Table 1.
Table 1.
Crystallographic Data and Refinement Statistics
| BroBep6_tBID1 | Bc/Bep9_tBID1 | BheBepE_BID1 | |
|---|---|---|---|
| PDB ID | 4YK1 | 4YK2 | 4YK3 |
| Data Collection Statistics | |||
| X-ray source | CLS 08ID-1 | APS 21-ID-G | APS 21-ID-G |
| X-ray detector | Mar300 CCD | Mar300 CCD | Mar300 CCD |
| Wavelength (Å) | 0.9796 | 0.9786 | 0.9786 |
| Space group | P 41 2 2 | P 21 21 2 | P 1 21 1 |
| Cell dimensions a, b, c (Å), β (°) | 79.25, 79.25, 85.97, 90.00 | 76.68, 62.31, 71.04, 90.00 | 58.07, 77.42, 67.51, 93.66 |
| Resolution limits (Å) | 50.0–2.10 (2.15–2.10) | 50.0–2.05 (2.10–2.05) | 50.0–2.20 (2.26–2.20) |
| Rmergea (%) | 7.0 (58.2) | 6.1 (54.8) | 4.4 (52.9) |
| Rmeasb (%) | 7.2 (60.2) | 6.8 (61.1) | 5.0 (59.6) |
| CC1/2 (%) | 100 (94.3) | 99.9 (89.0) | 99.9 (84.2) |
| <I/s(I)> | 29.99 (5.32) | 19.33 (3.18) | 23.49 (2.91) |
| Wilson B factor | 25.9 | 19.7 | 33.7 |
| Total reflections | 473,920 (34,650) | 106,806 (7,902) | 140,015 (10,368) |
| Unique reflections | 30,572 (2,234) | 21,863 (1,597) | 30,337 (2,230) |
| Multiplicity | 15.5 (13.7) | 4.9 (4.9) | 4.6 (4.6) |
| Completeness (%) | 100.0 (100.0) | 99.2 (99.3) | 99.8 (99.9) |
| Mosaicity (°) | 0.190 | 0.161 | 0.273 |
| Refinement Statistics | |||
| Rworkc (%) | 17.7 (20.0) | 17.1 (21.2) | 18.3 (23.2) |
| Rfreed (%) | 20.3 (22.4) | 21.4 (24.0) | 22.5 (25.5) |
| No. of non-H atoms | 1,174 | 1,943 | 3,340 |
| Macromolecules | 1,075 | 1,733 | 3,204 |
| Ligands | 0 | 39 | 12 |
| Solvent | 99 | 171 | 124 |
| Protein residues | 137 | 213 | 434 |
| Rmsd from ideal | |||
| Bond lengths (Å) | 0.008 | 0.007 | 0.008 |
| Bond angles (°) | 0.93 | 0.98 | 0.97 |
| Ramachandran favorede (%) | 99 | 100 | 99 |
| Ramachandran allowede (%) | 0.72 | 0.45 | 1.2 |
| Ramachandran outlierse (%) | 0 | 0 | 0 |
| Clashscoree | 2.77 | 3.11 | 2.91 |
| Average B values (Å2) | 42.51 | 37.04 | 54.01 |
| Macromolecules | 42.17 | 34.95 | 54.19 |
| Ligands | 110.70 | 61.34 | |
| Solvent | 46.23 | 41.36 | 48.63 |
| TLS groups | 4 | 9 | 18 |
Numbers in parentheses refer to the outermost shell.
Rmerge = ∑hkl∑i|li(hkl) − <I(hkl)>|/∑hkl∑iIi(hkl), where Ii(hkl) is the observed intensity for a reflection and <I(hkl)> is the average intensity obtained from multiple observations of symmetry-related reflections. b
Rmeas = ∑hkl [N/(N−1)]1/2 ∑i|li(hkl) − <I(hkl)>|/∑hkl∑iIi(hkl) where Ii(hkl) is the observed intensity for a reflection, <I(hkl)> is the average intensity obtained from multiple observations of symmetry-related reflections and N is the number of observations of intensity I(hkl).
Rwork = ∑hkl||Fobs| − |Fcalc||/∑hkl|Fobs|.
Rfree is the R value calculated for 5% of the dataset that was not included in the refinement.
MolProbity.
BID Domain Structures and Sequence Conservation
All three BID domains are folded to an antiparallel four-helix bundle and adopt an elongated shape with a length of 70 Å and a diameter of 25 Å (Figures 1B–1D). Superposition of residues 319–413 of BroBep6_tBID1 with the corresponding residues of BclBep9_tBID1 and BheBepE_BID1 yielded a root-mean-square deviation (rmsd) of 1.15 Å and 1.76 Å for 95 Cα atoms (sequence identity of 36% and 21%, respectively). The three structures are virtually identical in their core but display significant conformational variability at the extremities of the polypeptide chain (Figures 1B–1D and S1). Noteworthy, the first and last helices (a1 and a4, respectively) can adopt either a straight or a kinked conformation. In BheBepE_BID1 the kinks in helices α1 and α4 coincide with proline residues P154 and P253, respectively (Figure S1C). Except when stated otherwise, in the following we take the BID structure of BroBep6 as reference along with its corresponding residue numbering.
Figure 1. Crystal Structures of the BID Domain of Bartonella Effector Proteins Reveal a Novel Fold.

(A) Domain architectures of Bep6 from Bartonella rochalimae (BroBep6_tBID1), Bep9 from Bartonella clarridgeiae (BclBep9_tBID1), and BepE from Bartonella henselae (BheBepE_BID1) (from left to right). Vertical bars represent tandem-repeat tyrosine-phosphorylation motifs.
(B–D) Side views of the crystal structures of (B) BroBep6_tBID1, (C) BclBep9_tBID1, and (D) BheBepE_BID1.
(E) Superposition of the three structures shown in (B), (C), and (D), highlighting the similarity and compact nature of the BID fold. A stereo view is provided in Figure S1D.
(F) Detailed structure of the α3-α4 linker, referred to as the hook, of BroBep6_tBID1 as representative for the three BID domain structures. The Ca trace is shown as cartoon and H bonds are shown as black dashed lines. The position of glycine residues is highlighted by spheres at their respective Ca positions. Residues discussed in the text are shown in stick representation. Secondary structure elements are labeled in light pink.
(G) Sequence logo of three structures shown in (B) to (D) with secondary structure elements of the reference structure indicated. Residues of structural importance are marked with black triangles and residues of potential functional relevance are marked with red triangles. In addition, green arches indicate kinks in helices a1 and a4. The core of the BID domain is highlighted in beige.
(H) Structure of BroBep6_tBID1 with residues conserved among the three structures shown as sticks.
See also Figures S1, S2, and S5.
Strikingly, 3D structure comparison using the DaliLite server (Holm and Park, 2000) and several other servers (see Supplemental Experimental Procedures) revealed no significant structural homology with any other known structures. Some structural similarity between the BID domain structures and various other a-helix bundles or coiled-coil structures were found (best Z scores between 6.6 and 6.8), but with superposition of only some of the a helices at best. For example, the best hit for our reference structure is a four-helix bundle of a histidine kinase sensor domain (PDB: 3I9Y) (Moore and Hendrickson, 2009), of which only one helix superposed with helix a4 of the BID domain. Thus, we can safely describe the BID domain as a novel fold.
The N and C termini of the BID domain are located at the same pole with the a3-a4 linker located at the opposite pole (Figures 1 and S1). This helix linker (residues 368–384 of BroBep6_tBID1) adopts a well-defined, apparently conserved structure (Figure 1E) and comprises a short 310 helix (h1) and a b hairpin (b1-b2). Because of its shape, we named this structure the hook. A detailed view of the hook of our reference structure, BroBep6_tBID1, is shown in Figure 1F. The h1 helix is initiated by a conserved proline (P368) and ends with a conserved serine (S370). S370 interacts with the main-chain carbonyl of residue 367 (immediately preceding the 310 helix), which explains the conservation of this serine. The ƞ1 helix is followed by a short loop that leads to the β1-β2 b hairpin located at the top of the structure that further connects to helix α4. The hydrophobic side chain of residue L374 stabilizes the loop by burying itself in the BID core. Strikingly, residue G376 at the C-terminal end of this short loop is strictly conserved in BID domains, which allows the subsequent b hairpin to closely pack with the N-terminal end of helix α4. The β1-β2 hairpin itself contains a regular reverse turn (β turn) stabilized by an H bond between carbonyl 379 (i) and amide 382 (i + 3) as shown in Figure 1F. The β turn is of type II’ in BroBep6_tBID1 and of type I’ in BclBep9_tBID1 and BheBepE_BID1, with a glycine at position i + 1 and i + 2, respectively (Sibanda and Thornton, 1985). In addition to the main-chain/main-chain interactions between the β1 and β2 strands, the b hairpin forms an H bond between the side chain of S384 (i + 5) and the main-chain amide of residue 377 (i 2) of our reference structure (Figure 1F). Interestingly, a serine (S384 of BroBep6_tBID1), asparagine (N150 of BclBep9_tBID1), or threonine (T218 of BheBepE_BID1) can occupy this position and allows the formation of the aforementioned interaction. A few residues at the surface of the hook appear partially conserved, potentially for functional reasons. This includes two positively charged residues (K383 and K388), a negatively charged residue (E391), and two hydrophobic residues (I379 and I382). Noteworthy, the two aforementioned isoleucines are variable, and any medium to large hydrophobic side chain seems to be accommodated at these positions. We anticipate that the hook may constitute (part of) the interface for the contact with the T4CP. The sequence alignment of the three BID domains reveals only a few more conserved residues (Figure 1G). These are mostly hydrophobic, located at the center of the a-helical bundle and probably crucial for the integrity of the four-helix bundle (Figures 1G and 1H).
The BID Fold: Conserved but Specialized
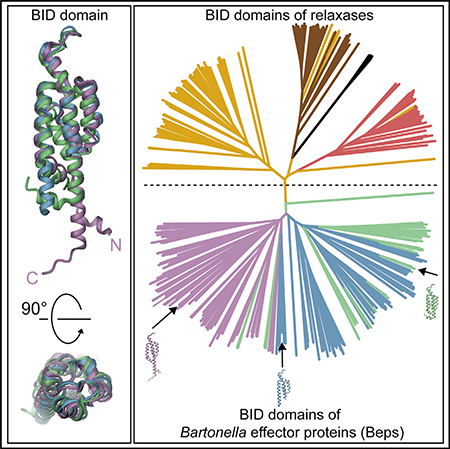
To gain insight into the conservation of the BID domain, we performed a BLAST search and retrieved 351 sequences (with less than 90% redundancy), all Beps or relaxases. The neighbor joining distance-based tree of the BID domain sequences is clearly divided into two classes, representing relaxases and Beps (Figure 2B; a high-resolution image with individual species names and sequence references is shown in Figure S3). Based on the Bep and relaxase multi-domain architectures (Figure 2A) as well as the clusters seen in the neighbor-joining tree, we devised a systematic nomenclature to classify the BID domains (Figure 2C). The BIDs are divided into terminal (tBIDx) and non-terminal BIDx, with “x” indicating the order of the BID domain from the N terminus. For the Beps, the tBIDx class is subdivided into tBIDx domains found in either the “ancestral” FIC-OB-tBIDx architecture or the diverse “derived” domain arrangements. In relaxases, which have either one or two BID domains, the tBIDx class is subdivided into tBID1 and tBID2, while the BIDx class consists exclusively of BID1 domains. In Beps, the ancestral tBID1 subclass forms a distinct cluster, which, in comparison with other subclasses, is well conserved at both the N and C termini as revealed by the respective sequence logos in Figure 3A. It is tempting to speculate that these conserved residues may interact with the adjacent OB fold as deduced by the short length (five residues) of the connecting segment. The “derived” tBIDx subclass, covering all remaining C-terminal BID domains of Beps, forms a separate cluster. Compared with ancestral tBID1, derived tBIDx have fewer conserved domain borders, which, due to the lack of an adjacent OB fold, may reflect the lack of conserved inter-domain interactions. Given that only the most C-terminal BID domain constitutes the T4S signal (Schulein et al., 2005), the additional BID domains present in multi-BID domain Beps (BIDx class) are likely released from selection pressure and may thus more easily have adapted to novel functions. Consistent with this notion, the BIDx class does not form a uniform cluster (Figure 2B). The BID domains of relaxases cluster into three discrete subclasses tBID1, BID1, and tBID2 (Figure 2B). A few tBID1 domains derived from homologs of the VbhT toxin (Engel et al., 2012; Harms et al., 2015). Due to the low number of sequences, this group was not investigated further in the current study.
Figure 2. BID Domain Arrangements and Subclasses in Beps and Relaxases.

(A) The BID domain architecture as seen in two relaxases (TraA from Agrobacterium fabrum and Riorf112 from Agrobacterium rhizogenes), a VbhT homolog (VbhT from Bartonella schoenbuchensis), and representative B. henselae Beps, with vertical black lines indicating Tyr phosphorylation motifs and +++ the positively charged C terminus. MobA, mobilization proteins of the MobA/MobL family; AAA_30, ATPases associated with diverse cellular activities (AAA) domain; FIC, filamentation induced by cAMP domain; OB, oligonucleotide/oligosaccharide binding fold.
(B) Simplified neighbor-joining distance-based tree representation of the multiple sequence alignment of the BID domains from Beps and relaxases. The branches corresponding to Bep_tBIDx (ancestral) are colored in pink, Bep_tBIDx (derived) in blue, and Bep_BIDx in green (Engel et al., 2011). The relaxase tBID1 domains are colored in yellow, tBID2 in brown, and the BID1 in red. See Figure S3 for the full high-resolution tree shown here with species names and UniProt IDs.
(C) Classification scheme of BID domains developed in this study.
See also Figures S3 and S4.
Figure 3. Conservation Analysis of BID Domains.

(A) Comparison of residue conservation in BID subclasses defined in Figure 2C. For each subclass, the sequence logo is shown and the background is colored using the ConSurf conservation color scale ranging from cyan to magenta for low to high conservation. An overall sequence logo is shown above the subclasses, indicating only a low amount of globally conserved residues.
(B and C) Overall conservation score mapped to the BroBep6_tBID1 structure. (B) Surface representation colored by overall conservation. The L374[A/R/K]G376 motif appears at the start of the hook. (C) Ribbon representation with conserved residues (ConSurf score ⩾8) shown as sticks.
(D) Surface representation of BroBep6_tBID1 colored by conservation of the BID domain in an ancestral tBID1 arrangement (last row of the alignment in A). The N and C termini appear well conserved when compared with the overall conservation shown in (B).
The overall conservation as well as the conservation within the six defined subclasses was mapped to the BID domain sequence and structure (Figure 3). Upon mapping the ConSurf conservation scores to the structure, it appears that the overall conservation at the surface (Figure 3B) is rather low compared with the conservation of the buried residues (median ConSurf score for surface residues is 5.0 versus 8.0 for buried residues in BroBep6_tBID1, Figure 3C). However, a single hotspot appears highly conserved in all BID domains: the P368xxxxxL374[A/R/K]G376 motif located directly at the N terminus of the short b hairpin (β1-β2) at the tip of the BID domain (Figures 1F and 3B). This region, as already mentioned in the previous paragraph, may interact with the T4CP. In addition, several prolines are very well conserved and appear critical for the proper folding of BID domains, e.g., P354 in BroBep6_tBID1 (structurally homologous to P120 in BclBep9_tBID1 and P188 in BheBepE_BID1 in Beps) and P368 at the start of h1 in both Beps and relaxases.
When looking at the six BID domain subclasses individually, some specific conservation patterns appear. In the Beps, only the ancestral tBID1 subclass has a highly conserved L298IPxE302 motif directly at the N terminus that could potentially interact with the preceding OB fold. There is an additional conserved R425xxxx[V/I]xxP433 motif located ~140 residues further downstream. In Beps, an additional R387[K/R]xAE391 motif occurs immediately after the hook. In contrast, the relaxases are more conserved across the subclasses and share a very prominent [V/I]429P[A/G]LS433 motif at the C terminus. The relaxase BID1 class has a motif, L296[I/L]PP299, similar to the ancestral tBID1 domain, whereas the relaxase tBID1 and tBID2 domains have an M296[V/L]A[G/A]299 motif. As in the Beps, there is a conserved motif after the hook, although in relaxases this motif is R387xxA390. For relaxases, residue numbers correspond to the numbering in the alignment of Figure 3A.
The sequence alignment combined with the three BID domain structures provide a solid basis to refine the boundaries of the BID domain described initially by Schulein et al. (2005). As a general domain definition for the BID fold (based on the structural superposition), we propose to use the highly conserved proline located at the N terminus of helix h1 as an “anchor” (P368 in BroBep6_tBID1, P134 in BclBep9_tBID1, and P202 in BheBepE_BID1) and define the domain boundaries as 50 amino acids in the N-terminal direction and 50 amino acids in the C-terminal direction, resulting in a BID domain of ~100 amino acids (Figure 1G, highlighted in beige). This domain definition is based on the best-conserved superimposable part of the BID domain and thus excludes the variable N-terminal part of helix a1 and the variable C-terminal part of a4. Noteworthy, the neighbor-joining distance-based tree of BID domain trimmed to the new boundaries (Figure S4) closely resembles the tree shown in Figure 2B (compare Figures S3 and S4).
The sequence and structure analyses have shown low sequence conservation over the entire BID domain (on average 14% between Beps and relaxases). To assess whether at least the surface charge distribution is conserved, we performed electrostatic surface calculations using APBS and PDB2PQR. Figures 4A–4C shows that in all three BID domain structures, the electrostatic potential on the surface appears to be rather consistent. It consists of two highly positively charged areas that are separated by a small patch of negative charges, mostly generated by E310, E317, and E344. The hook region in all three structures is highly positively charged, suggesting that it may interact with a negatively charged partner. This analysis was expanded to homology models of BID domains of other Beps, i.e., BheBepA_tBID1 (ancestral), BheBepE_tBID2 (derived), and BroBep9_BID1, revealing similar surface properties (Figures 4D–4F), suggesting that this feature was acquired early during evolution of the BID domain. Due to low sequence similarity between the BID domains of Beps and relaxases (on average ~14%) and uncertain placement of indels (Figure 3A), no accurate relaxase homology models could be built for surface charge conservation comparisons.
Figure 4. Electrostatic Potential of Experimentally Determined and Modeled BID Domain Structures.

The potentials have been mapped onto the respective protein surfaces with color code as defined in the inset. Protein backbones are shown in cartoon representation. (A) BroBep6_tBID1 (ancestral) (PDB: 4YK1), (B) BclBep9_tBID1 (derived) (PDB: 4YK2), (C) BheBepE_BID1 (PDB: 4YK3), and homology models of (D) BheBepA_tBID1 (ancestral), (E) BheBepE_tBID2 (derived), and (F) BroBep9_BID1.
DISCUSSION
The C-terminal BID domain and adjacent positively charged tail sequence function as an evolutionary conserved bipartite signal for T4S in both Beps and a subset of relaxases (Schulein et al., 2005), likely by mediating protein-protein interaction with the T4CP as initial step of the T4S process (Schroder et al., 2011). The determination of three BID domain structures reveals a well-conserved novel fold formed by a four-helix bundle (Figure 1) lacking significant structural homology to known protein structures. The core of the domain is formed by highly conserved apolar residues that likely provide rigidity to the domain (Figure 3). Despite the constraint to maintain a functional T4S signal, residue conservation at the surface of the protein appears to be generally low, even though charge distribution is preserved (Figures 3 and 4). In particular a conserved structural feature of the BID domain, which due to its shape we named the hook, is invariantly positively charged. Further to the positively charged tail sequence adjacent to the BID domain, we thus envisage that the hook may serve as anchoring point for conserved interactions of the BID domain with the T4CP. Future structure/function-related studies should characterize the interaction interface of the hook and possibly other surfaces of the BID domain with the T4CP and address possible cooperative binding of the adjacent tail sequence.
The discrete clustering of defined subclasses of BID domains based on their origin (from relaxases or Beps) and position in multi-domain architectures (terminal or non-terminal) probably reflects more their evolutionary history than their function (Figure 2). The exception may be the less conserved non-terminal BID domains (BIDx) of Beps derived from domain duplication that—likely as they are relieved from selection pressure to maintain interaction with the T4CP—diversified faster, facilitating the evolution of novel effector functions within host cells (Siamer and Dehio, 2015). However, some of the more conserved terminal BID domains (tBIDx) also have evolved secondary effector functions. As an example, the interaction of BhBepA_tBIDx with the C2 subunit of human adenylyl cyclase has been demonstrated (Pulliainen et al., 2012), but the interaction surface remains unknown.
The sole other study revealing the 3D structure of a secretion signal is of the TSA domain of the conjugative relaxase TraI of the R1 plasmid (Redzej et al., 2013), revealing a globular structure that contrasts with the elongated BID domain. TSA forms, similarly to the BID domain, a domain with both termini in proximity. The BID domain (Figure S5A) shares some structural features with the unrelated oligomeric proteins IpaD (Figure S5B) and prefoldin (Figures S5C and S5D). IpaD consists of an a-helical bundle with a small b sheet at its apex (Figure S5B) that is located at the tip of the type III secretion system’s needle of Shigella flexneri. Upon IpaD oligomerization, IpaB and IpaC are recruited and translocated in a contact-dependent manner into host cell membranes to form a pore (Cheung et al., 2015). Interestingly, the N-terminal domain of IpaD acts as an intramolecular chaperone that prevents premature oligomerization (Johnson et al., 2007). Could the BID domain also represent an intramolecular chaperone? Secreted substrates can potentially cross the inner membrane through the VirB4/TrwK hexamer (Low et al., 2014). The inner diameter of the TrwK hexamer has been measured to 42 A˚ (Pen˜ a et al., 2012). Therefore, the dimensions of the BID domains, measured to an apparent width of 25 Å, appear compatible with translocation via a T4S system in a fully folded state, while globular protein domains should require at least partial unfolding during translocation (Christie et al., 2014). BID domains may thus act as folding seeds to refold Beps or relaxases from their C terminus, thereby acting as intramolecular chaperones upon transfer into target cells. Prefoldin found in archaea or eukaryotes is formed by two long coiled coils topped by one or two b sheets that are reminiscent of the hook of BID domains (Siegert et al., 2000) (Figure S5C). Prefoldin oligomerizes to form hexameric rings containing a large cavity that captures unfolded protein or folding intermediates (Siegert et al., 2000). Thus, it is tempting to speculate that multi-BID domain effectors, e.g., BepF or BepG (Figure 2A), may form BID oligomers in a similar manner as prefoldin forms its cavity (Figure S5D).
Our structural work sets the stage for follow-up structure-function studies that will, among others, aim at investigating the interactions of the BID domain with (1) T4CP and other T4S system components during protein translocation, and upon translocation with (2) other domains encoded in the same polypeptide chain, e.g., FIC domain, OB fold, tandem-repeat tyrosine-phosphorylation motifs, and additional BID domains, as well as (3) in cases of the evolution of secondary effector functions in individual BID domains, with their specific interaction partners.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
The BID domains from B. rochalimae Bep6 (BroBep6_tBID1), B. clarridgeiae Bep9 (BclBep9_tBID1), and B. henselae BepE (BheBepE_BID1) were cloned, expressed, and purified as described in detail in Supplemental Experimental Procedures.
Protein Crystallization, X-Ray Data Collection, and Structure Determination
BroBep6_tBID1, BclBep9_tBID1, and BheBepE_BID1 were crystallized, X-ray data collected, and structures determined as described in detail in Supplemental Experimental Procedures. All data reduction and refinement statistics are reported in Table 1.
Sequence Analysis
The sequence dataset used in this analysis was generated by BLAST against the UniProtKB database. All the sequences of our working dataset were then aligned using ClustalX 2.0. Neighbor-joining distance-based trees were constructed and visualized with iTOL. Sequence logos were generated with Weblogos and alignments visualized with Aline. Conservation scores were generated using ConSurf. Electrostatic potentials were calculated with the APBS-Tools and PDB2PQR plugins for PyMOL using the default settings. Details on the matrices and cutoff used and references are given in Supplemental Experimental Procedures.
ACCESSION NUMBERS
The coordinates and structure factors of BroBep6_tBID1, BclBep9_tBID1, and BheBepE_BID1 have been deposited in the PDB with accession numbers PDB: 4YK1, 4YK2, and 4YK3, respectively. Sequence abbreviations are given in Supplemental Experimental Procedures.
Supplementary Material
Highlights.
BID domains share a novel fold with a compact four-helix bundle and a look
The rigid fold is determined by a conserved core of hydrophobic amino acids
Charged surface areas and few exposed residues are preserved
Low surface conservation facilitates the evolution of secondary functions
ACKNOWLEDGMENTS
We acknowledge Peter J. Myler and the SSGCID, Seattle, USA for support of the project. This work was supported by the NIH/NIAID contracts nos. HHSN272200700057C and HHSN272201200025C, the ERC AdG FICModFun 340330 (to C.D.), and SNF grants 310030B-149886 (to C.D.), and 31003A_138414 (to T.S.). We thank the staff of beamlines 08ID-1 of the Canadian Light Source (Saskatchewan, Canada) and 21-ID-G of the Advanced Photon Source (Argonne National Laboratory, USA) for their excellent support.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, five figures, and three 3D molecular models and can be found with this article online at http://dx.doi.org/10.1016/j.str.2016.10.010.
REFERENCES
- Cheung M, Shen DK, Makino F, Kato T, Roehrich AD, Martinez Argudo I, Walker ML, Murillo I, Liu X, Pain M, et al. (2015). Three-dimensional electron microscopy reconstruction and cysteine-mediated crosslinking provide a model of the type III secretion system needle tip complex. Mol. Microbiol 95, 31–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie PJ, Whitaker N, and Gonzalez-Rivera C (2014). Mechanism and structure of the bacterial type IV secretion systems. Biochim. Biophys. Acta 1843, 1578–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel P, Salzburger W, Liesch M, Chang CC, Maruyama S, Lanz C, Calteau A, Lajus A, Medigue C, Schuster SC, et al. (2011). Parallel evolution of a type IV secretion system in radiating lineages of the host-restricted bacterial pathogen Bartonella. PLoS Genet. 7, e1001296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel P, Goepfert A, Stanger FV, Harms A, Schmidt A, Schirmer T, and Dehio C (2012). Adenylylation control by intra- or intermolecular active-site obstruction in Fic proteins. Nature 482, 107–110. [DOI] [PubMed] [Google Scholar]
- Harms A, Stanger FV, Scheu PD, de Jong IG, Goepfert A, Glatter T, Gerdes K, Schirmer T, and Dehio C (2015). Adenylylation of gyrase and topo IV by FicT toxins disrupts bacterial DNA topology. Cell Rep. 12, 1497–1507. [DOI] [PubMed] [Google Scholar]
- Harms A, Stanger FV, and Dehio C (2016). Biological diversity and molecular plasticity of FIC domain proteins. Annu. Rev. Microbiol 70, 341–360. [DOI] [PubMed] [Google Scholar]
- Holm L, and Park J (2000). DaliLite workbench for protein structure comparison. Bioinformatics 16, 566–567. [DOI] [PubMed] [Google Scholar]
- Johnson S, Roversi P, Espina M, Olive A, Deane JE, Birket S, Field T, Picking WD, Blocker AJ, Galyov EE, et al. (2007). Self-chaperoning of the type III secretion system needle tip proteins IpaD and BipD. J. Biol. Chem 282, 4035–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low HH, Gubellini F, Rivera-Calzada A, Braun N, Connery S, Dujeancourt A, Lu F, Redzej A, Fronzes R, Orlova EV, et al. (2014).Structure of a type IV secretion system. Nature 508, 550–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JO, and Hendrickson WA (2009). Structural analysis of sensor domains from the TMAO-responsive histidine kinase receptor TorS. Structure 17, 1195–1204. [DOI] [PubMed] [Google Scholar]
- Okujava R, Guye P, Lu YY, Mistl C, Polus F, Vayssier-Taussat M, Halin C, Rolink AG, and Dehio C (2014). A translocated effector required for Bartonella dissemination from derma to blood safeguards migratory host cells from damage by co-translocated effectors. PLoS Pathog. 10, e1004187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena A, Matilla I, Martín-Benito J, Valpuesta JM, Carrascosa JL, la Cruz De F, Cabezon E, and Arechaga I (2012). The hexameric structure of a conjugative VirB4 protein ATPase provides new insights for a functional and phylogenetic relationship with DNA translocases. J. Biol. Chem 287, 39925–39932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulliainen AT, Pieles K, Brand CS, Hauert B, Bohm A, Quebatte M, Wepf A, Gstaiger M, Aebersold R, Dessauer CW, et al. (2012). Bacterial effector binds host cell adenylyl cyclase to potentiate Galphas-dependent cAMP production. Proc. Natl. Acad. Sci. USA 109, 9581–9586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redzej A, Ilangovan A, Lang S, Gruber CJ, Topf M, Zangger K, Zechner EL, and Waksman G (2013). Structure of a translocation signal domain mediating conjugative transfer by type IV secretion systems. Mol.Microbiol 89, 324–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhomberg TA, Truttmann MC, Guye P, Ellner Y, and Dehio C (2009). A translocated protein of Bartonella henselae interferes with endocytic uptake of individual bacteria and triggers uptake of large bacterial aggregates via the invasome. Cell Microbiol. 11, 927–945. [DOI] [PubMed] [Google Scholar]
- Saenz HL, Engel P, Stoeckli MC, Lanz C, Raddatz G, Vayssier-Taussat M, Birtles R, Schuster SC, and Dehio C (2007). Genomic analysis of Bartonella identifies type IV secretion systems as host adaptability factors.Nat. Genet 39, 1469–1476. [DOI] [PubMed] [Google Scholar]
- Schroder G, Schuelein R, Quebatte M, and Dehio C (2011). Conjugative DNA transfer into human cells by the VirB/VirD4 type IV secretion system of the bacterial pathogen Bartonella henselae. Proc. Natl. Acad. Sci. USA 108, 14643–14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulein R, Guye P, Rhomberg TA, Schmid MC, Schroder G, Vergunst AC, Carena I, and Dehio C (2005). A bipartite signal mediates the transfer of type IV secretion substrates of Bartonella henselae into human cells. Proc. Natl. Acad. Sci. USA 102, 856–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selbach M, Paul FE, Brandt S, Guye P, Daumke O, Backert S, Dehio C, and Mann M (2009). Host cell interactome of tyrosine-phosphorylated bacterial proteins. Cell Host Microbe 5, 397–403. [DOI] [PubMed] [Google Scholar]
- Siamer S, and Dehio C (2015). New insights into the role of Bartonella effector proteins in pathogenesis. Curr. Opin. Microbiol 23, 80–85. [DOI] [PubMed] [Google Scholar]
- Sibanda BL, and Thornton JM (1985). Beta-hairpin families in globular proteins. Nature 316, 170–174. [DOI] [PubMed] [Google Scholar]
- Siegert R, Leroux MR, Scheufler C, Hartl FU, and Moarefi I (2000). Structure of the molecular chaperone prefoldin: unique interaction of multiple coiled coil tentacles with unfolded proteins. Cell 103, 621–632. [DOI] [PubMed] [Google Scholar]
- Truttmann MC, Guye P, and Dehio C (2011). BID-F1 and BID-F2 domains of Bartonella henselae effector protein BepF trigger together with BepC the formation of invasome structures. PLoS One 6, e25106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.