

Harmful algal blooms (HABs) significantly influence biogeochemical cycles driven by bacteria. The shifts in microbial communities during HABs have been studied intensively, but the assembly mechanisms of microbial communities during HABs are poorly understood, with limited investigation of the balance of deterministic and stochastic processes in shaping microbial communities in HABs. In this study, the dynamics and assembly of microbial communities in an early-spring diatom bloom process were investigated. Our data both confirm previously observed general microbial successional patterns and show new detailed mechanisms for microbial assembly in HABs. These results will facilitate deeper understanding of the ecological processes shaping microbial communities in HABs. In addition, predictions of metabolic potential in this study will facilitate understanding of the influence of HABs on nitrogen metabolism in marine environments.
KEYWORDS: microbial communities, assembly mechanism, diatom bloom, metagenomic prediction
ABSTRACT
Harmful algal blooms (HABs) are serious ecological disasters in coastal areas, significantly influencing biogeochemical cycles driven by bacteria. The shifts in microbial communities during HABs have been widely investigated, but the assembly mechanisms of microbial communities during HABs are poorly understood. Here, using 16S rRNA gene amplicon sequencing, we analyzed the microbial communities during an early-spring diatom bloom, in order to investigate the dynamics of microbial assembly processes. Rhodobacteraceae, Flavobacteriaceae, and Microbacteriaceae were the main bacterial families during the bloom. The 30 most abundant operational taxonomic units (OTUs) segregated into 4 clusters according to specific bloom stages, exhibiting clear successional patterns during the bloom process. The succession of microbial communities correlated with changes in the dynamics of algal species. Based on the β-nearest taxon distance, we constructed a simulation model, which demonstrated that the assembly of microbial communities shifted from strong heterogenous selection in the early stage of the bloom to stochasticity in the middle stage and then to strong homogeneous selection in the late and after-bloom stages. These successions were driven mainly by chlorophyll a contents, which were affected mainly by Skeletonema costatum. Moreover, functional prediction of microbial communities showed that microbial metabolic functions were significantly related to nitrogen metabolism. In summary, our results clearly suggested a dominant role of determinacy in microbial community assembly in HABs and will facilitate deeper understanding of the ecological processes shaping microbial communities during the algal bloom process.
IMPORTANCE Harmful algal blooms (HABs) significantly influence biogeochemical cycles driven by bacteria. The shifts in microbial communities during HABs have been studied intensively, but the assembly mechanisms of microbial communities during HABs are poorly understood, with limited investigation of the balance of deterministic and stochastic processes in shaping microbial communities in HABs. In this study, the dynamics and assembly of microbial communities in an early-spring diatom bloom process were investigated. Our data both confirm previously observed general microbial successional patterns and show new detailed mechanisms for microbial assembly in HABs. These results will facilitate deeper understanding of the ecological processes shaping microbial communities in HABs. In addition, predictions of metabolic potential in this study will facilitate understanding of the influence of HABs on nitrogen metabolism in marine environments.
INTRODUCTION
Phytoplankton blooms (e.g., diatoms, haptophytes, and autotrophic dinoflagellates) account for a significant fraction of marine primary productivity. Diatoms alone are estimated to contribute 20% to 40% of global carbon fixation (1). Harmful algal blooms (HABs) induced by diatoms in the open ocean or in coastal areas can be a serious problem for aquaculture and fisheries (2). Extensive efforts have been made to reveal the factors that trigger HABs, as it is crucial to understand their dynamics and thus control them. The occurrence and development of HABs have been explained by a complex combination of environmental conditions and microbial factors (1). Because of their roles in biogeochemical cycling during phytoplankton blooms, prokaryotes are now considered significant factors influencing the development of HABs (3).
Bacterial communities are considered a fundamental component of marine ecosystems, and they are a major driver of nutrient cycles in coastal waters. Moreover, bacteria have extensive interactions with phytoplankton (4, 5), and these interactions may influence microbial community compositions (MCCs). Bacterial clades, responding sensitively to phytoplankton blooms, belong to Gammaproteobacteria, Flavobacteriia (phylum Bacteroidetes), and the Roseobacter clade (6). This response is not so specific and includes distinct clades of taxa that bloom one after another. Study of the MCCs in mosaic phytoplankton blooms showed that Alphaproteobacteria, Gammaproteobacteria, and Flavobacteria represented the majority of the bacterial communities (7). SAR11 and the Roseobacter clade, within the class Alphaproteobacteria, are of particular importance for the turnover of dissolved organic matter (DOM) in the bloom (7). This trophic connection leads to synchronized blooms of bacteria during phytoplankton blooms (8, 9). Studies of the variation of MCCs during the HAB process have been widely reported, but few studies have revealed MCC assembly mechanisms during the bloom process.
Elucidating the processes regulating MCCs would make a significant contribution toward a comprehensive understanding of bacterial dynamics. It is well recognized that determinacy and stochasticity are two major types of assembly processes influencing MCCs (10, 11). The deterministic process involves selection from the abiotic environment and species interactions (12). In contrast, the stochastic process is induced by unpredictable disturbances, probabilistic dispersal of microbes, and random birth and death events (13). Although deterministic and stochastic processes have been found in animal and plant communities, studies of these two processes in microbial communities have been reported only in recent years (12, 14). Based on current findings, the deterministic process may either sort species adapted to a given environment or exclude species that may otherwise enhance ecosystem function (15). The balance of the stochastic process has a significant role in defining the assembly of microbial communities (16). Deterministic and stochastic processes have been widely discussed in relation to various ecological processes, but little is known about their roles in the assembly of microbial communities during natural HAB processes.
Xiangshan Bay, located in Zhejiang Province of China, is one of the most important aquaculture bases with poor water exchange ability. Thousands of fish cages, combined with the input of residual feed and fish excretion, lead to severe eutrophication in this semienclosed harbor (17). Consequently, HABs occur frequently, resulting in MCC disturbances (9). An algal bloom is a strong environmental filtering process that affects MCCs, but the assembly mechanisms of microbial communities during the bloom process need to be elucidated. In the present study, we investigated microbial communities during a diatom bloom process in Xiangshan Bay. We sought to elucidate (i) the composition and succession patterns of microbial communities during the HAB process and (ii) changes in the relative influences of stochastic and deterministic processes in the assembly of microbial communities during distinct bloom stages. We also evaluated the predicted metabolic functions of microbial communities during the HAB process, which few studies have reported previously. We think that the results of this study will facilitate understanding of the importance of bacterial roles in the fate of HABs and the mechanisms for the microbial assembly process during HABs.
RESULTS
Bloom characteristics.
The sudden early-spring bloom in Xiangshan Bay was induced by Skeletonema costatum and Guinardia delicatula. Chlorophyll a (Chl a) contents reached 40 μg/liter in the early stage (ES) (Fig. 1B). Cell densities of S. costatum and G. delicatula ranged from 5.7 × 106 to 8.1 × 106 cells/liter in the ES; the dominant algal species changed from S. costatum to G. delicatula during this stage. After the ES, the abundance of S. costatum decreased dramatically, but the G. delicatula cell density reached its maximum in the middle stage (MS), contributing to the large contents of Chl a. In the late stage (LS), the abundance of S. costatum was 2.1 × 105 cells/liter, which was 30 times lower than that in the ES. The contents of Chl a were mainly accompanied by the occurrence of S. costatum. In the after-bloom (AB) stage, the concentration of Chl a decreased to 0.98 μg/liter and we could not precisely quantify S. costatum and G. delicatula; therefore, the abundance data are missing.
FIG 1.

Map of Xiangshan Bay, indicating the sampling sites (A), and the abundance of algal species (B). ES1 and ES2 indicate samples from 10 February 2017 and 15 February, respectively, and MS1 and MS2 indicate samples from 19 February and 23 February. LS indicates samples from 27 February. AB indicates samples from 8 March. ES1 and ES2 were integrated into the ES of the bloom according to the Chl a contents, and MS1 and MS2 were integrated into the MS of the bloom. The map was created using Surfer 13 (Golden Software).
Microbial α-diversity.
We observed clear differences in richness and phylogenetic diversity at distinct bloom stages, gradually decreasing from the ES to the AB stage (see Fig. S1 in the supplemental material). There were no remarkable changes in the Shannon index and evenness during the whole bloom process. Richness and phylogenetic diversity showed the strongest positive correlations with Chl a contents (r = 0.426 [P < 0.05] and r = 0.494 [P < 0.01], respectively), followed by dissolved oxygen (DO) (r = 0.386 [P < 0.05] and r = 0.430 [P < 0.05], respectively). Richness and phylogenetic diversity showed the strongest negative correlations with salinity (r = −0.491 [P < 0.01] and r = −0.531 [P < 0.01], respectively), ammonium (r = −0.467 [P < 0.05] and r = −0.475 [P < 0.01], respectively), and water temperature (r = −0.423 [P < 0.05] and r = −0.465 [P < 0.05], respectively) (Table S1). The environmental variables were not correlated with the Shannon index and evenness. These results indicated that richness and phylogenetic diversity were susceptible indices during algal bloom processes.
MCCs during the bloom process.
The dominant bacterial classes were Alphaproteobacteria (relative abundance, 32.9%), Flavobacteriia (28.8%), Gammaproteobacteria (18.6%), Actinobacteria (8.2%), Betaproteobacteria (3.7%), and Verrucomicrobiae (1.9%), which constituted 94.1% of the total number of sequences (Fig. 2A). Furthermore, Rhodobacteraceae (23.8%), Flavobacteriaceae (20.4%), Microbacteriaceae (8.1%), Cryomorphaceae (8.1%), Surface1 (SAR11) (6.6%), Oceanospirillaceae (5.7%), SAR86 (3.9%), Verrucomicrobiaceae (1.8%), and Puniceicoccaceae (1.2%) were the main bacterial families, accounting for 79.6% of the total number of sequences (Fig. 2B).
FIG 2.

Relative abundances of microbial lineages at the class (relative abundance of >0.5%) (A) and family (relative abundance of >5%) (B) levels during the diatom bloom process.
MCCs changed greatly during the bloom process (Fig. 2). Rhodobacteraceae was the most abundant taxon (average relative abundance, 37.1%) in the ES but decreased significantly in the AB stage (15.2% abundance). Compared with the bloom stages (ES, MS, and LS), the proportions of SAR11 (from 2.9% to 17.9%), Comamonadaceae (from 0.11% to 6.4%), SAR86 (from 1.7% to 7.3%), and Vibrionaceae (from 0.03% to 1.6%) increased significantly at the AB stage. Oceanospirillaceae and Verrucomicrobiaceae reached maximum abundance in the MS. Flavobacteriaceae had relatively stable abundance throughout the bloom process. In addition, the 30 most abundant operational taxonomic units (OTUs) were affiliated mainly with Rhodobacteraceae, Flavobacteriaceae, Oceanospirillaceae, Verrucomicrobiaceae, and SAR11 (Fig. 3). With only these OTUs involved in the random forest analysis, the overall out-of-bag (OOB) error was only 3.45% (Table S2), with 100% classification accuracy in the MS, the LS, and the AB stage, suggesting that the 30 most abundant OTUs could indicate the distinct algal bloom stages precisely. The cluster of columns in the heatmap (Fig. 3) revealed approximately 4 clusters according to the bloom stages, exhibiting clear successional patterns during the bloom process.
FIG 3.

Abundance of the 30 most abundant OTUs in 4 bloom stages. Samples from the same stage clustered together. Microbial abundance was scaled with log transformation in the heatmap.
We also investigated the correlation of the 30 most abundant OTUs with the abundance of S. costatum. Fourteen of the 30 most abundant OTUs were strongly correlated with S. costatum (Fig. S2). Five OTUs showed significant (P < 0.001) positive correlations with S. costatum, i.e., OTU12736 (R2 = 0.827), OTU7254 (R2 = 0.696), OTU5946 (R2 = 0.678), OTU10900 (R2 = 0.646), and OTU12316 (R2 = 0.450). Negative linear correlations with S. costatum were observed for OTU2635 (R2 = 0.598 [P < 0.001]), OTU4140 (R2 = 0.418 [P < 0.001]), OTU3566 (R2 = 0.373 [P < 0.01]), OTU10614 (R2 = 0.306 [P < 0.01]), OTU8289 (R2 = 0.305 [P < 0.01]), and OTU6302 (R2 = 0.240 [P < 0.05]). Three OTUs (OTU1301, OTU5613, and OTU10568) showed negative nonlinear correlations with S. costatum. In summary, 9 of the 30 most abundant OTUs were closely associated with the decline of S. costatum, which indicated that those OTUs might inhibit the growth of S. costatum or promote its death during the algal bloom process.
Successional patterns of microbial communities during the bloom process.
The nonmetric multidimensional scaling (NMDS) plot showed that samples clustered together according to distinct bloom stages (Fig. 4A), suggesting that there were variations in community structure over the bloom stages. Permutational multivariate analysis of variance (PERMANOVA) revealed that the structures of the microbial communities varied significantly (R2 = 0.527 [P < 0.0001]) among distinct bloom stages (Table 1), which indicated that the succession of microbial communities was induced mainly by the diatom bloom. Moreover, there were significant correlations between microbial communities and all environmental variables (Table 1; also see Fig. S3 in the supplemental material), as evidenced by Mantel tests and PERMANOVA (Table 1). In addition, the succession of microbial communities occurred mainly along the NMDS1 axis, which was significantly correlated with the Chl a content (R2 = 0.478 [P < 0.01]) (Fig. 4B). Therefore, even if this sudden algal bloom occurred during dramatic environmental changes, the dynamics of algal species might be a key factor driving the succession of microbial communities.
FIG 4.

NMDS ordinations of microbial community dissimilarity in the bloom process (A), and correlation between Chl a contents and NMDS1 (B). Colors correspond to different sampling stages, indicating the succession of microbial communities together with NMDS1.
TABLE 1.
PERMANOVA (Adonis) analysis based on Bray-Curtis dissimilarity with 999 permutations and simple Mantel tests for correlations between environmental variables (Euclidean distance) and β-diversity of microbial communities (Bray-Curtis dissimilarity) with 999 permutations
| Variablea | ρ (Mantel test) | R2 (Adonis) |
|---|---|---|
| Bloom stage | 0.527 | |
| DO | 0.593 | 0.261 |
| pH | 0.591 | 0.275 |
| Phosphate level | 0.454 | 0.234 |
| Salinity | 0.450 | 0.210 |
| Nitrate level | 0.430 | 0.203 |
| Nitrite level | 0.382 | 0.233 |
| Chl a content | 0.367 | 0.202 |
| Ammonium level | 0.366 | 0.183 |
| Temperature | 0.364 | 0.237 |
| COD | 0.353 | 0.205 |
| Silicate level | 0.279 | 0.178 |
DO, dissolved oxygen; COD, chemical oxygen demand. All P values were <0.01.
Community assembly processes in successive stages.
The β-nearest taxon index (βNTI) was used to provide deep insight into the potential roles of deterministic or stochastic factors in phylogenetic community succession of microbial communities. Based on the analysis of phylogenetic turnover, βNTI values were greater than 2 in the ES (Fig. 5A). After that, βNTI values decreased significantly throughout the bloom process. In the MS, most βNTI values ranged from −2 to 2, suggesting a role of stochasticity. In the LS and the AB stage, almost all of the βNTI values were lower than −2, indicating a deterministic process. These results revealed that βNTI distributions progressively shifted along the bloom stages, and the mechanisms driving the succession of microbial communities might have been different.
FIG 5.

Patterns of distribution of βNTI values across bloom stages (A), and effect of algal biomass (indicated by Chl a contents) on the distribution of βNTI values (B). In panel A, horizontal dashed lines indicate lower and upper significance thresholds at −2 and 2, respectively; the asterisks denote the βNTI average in each group.
Based on the pattern of βNTI values and on environmental heterogeneity in different bloom stages, we assembled microbial communities at 4 bloom stages in a simulation model (Fig. 6). Environmental heterogeneity was the highest in the ES, indicating that selection by environmental variables ruled the deterministic process. In the MS, there was a transition from selection of heterogenous variables to stochasticity, as environmental heterogeneity decreased significantly. In the LS and the AB stage, environmental heterogeneity continuously decreased, and the deterministic assembly process of the microbial communities was induced mainly by homogeneous selection. The distribution of βNTI values changed in the simulation, corresponding closely to the shifts in Chl a contents (Mantel ρ = 0.264 [P = 0.004]) (Table 2 and Fig. 5B) and silicate levels (Mantel ρ = 0.202 [P = 0.003]). The simulation showed that assembly of microbial communities shifted from strong heterogenous selection (βNTI of >2, in the ES) to stochasticity (∣βNTI∣ of <2, in the MS) and then to strong homogeneous selection (βNTI of less than −2, in the LS and the AB stage), accompanied mainly by the dynamics of algal species.
FIG 6.
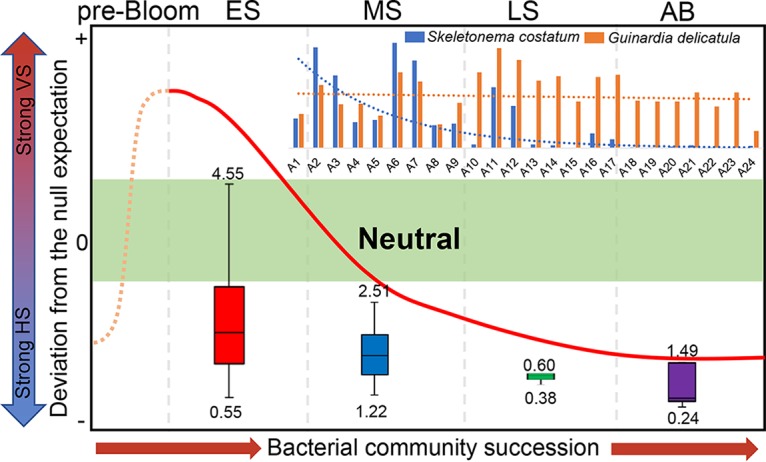
Simulation model of the successional pattern of microbial communities based on βNTI distributions. Ecological selection is weak from −2 to 2 (green area), while selection is strong toward positive and negative values on the y axis, resulting from strong variable selection (VS) and homogeneous selection (HS), respectively. The boxplot shows the environmental heterogeneity in each bloom stage. The bar plot shows the abundance of 2 harmful algal species. The prebloom stage, without bloom disturbance, may have ecological selection similar to that of the AB stage.
TABLE 2.
Simple Mantel tests for correlations between environmental variables (Euclidean distance) and βNTI distance matrix for microbial communities with 999 permutations
| Variablea | ρ (Mantel test) | P |
|---|---|---|
| Chl a content | 0.264 | 0.004 |
| Silicate level | 0.202 | 0.003 |
| Salinity | 0.179 | 0.038 |
| DO | 0.137 | 0.068 |
| pH | 0.129 | 0.096 |
| COD | 0.104 | 0.125 |
| Nitrate level | 0.044 | 0.251 |
| Temperature | −0.006 | 0.462 |
| Ammonium level | −0.016 | 0.509 |
| Phosphate level | −0.045 | 0.604 |
| Nitrite level | −0.137 | 0.894 |
DO, dissolved oxygen; COD, chemical oxygen demand.
Predicted metabolic potential during the bloom process.
The nearest sequenced taxon index (NSTI) values ranged from 0.056 to 0.113; the mean value was 0.090 ± 0.013 for all samples (see Data Set S2 in the supplemental material). The Bray-Curtis distance-based redundancy analysis (db-RDA) showed a clear shift in the composition of functional gene families throughout the 4 bloom stages (Fig. S4). The most frequent predicted functions were amino acid metabolism (12.2%), membrane transport (11.4%), carbohydrate metabolism (10.4%), replication and repair (7.1%), and energy metabolism (5.8%) (Fig. S5).
Nutrient levels, including nitrate, nitrite, ammonium, phosphate, and silicate levels, were positively correlated with predicted metabolic potential in the MS and LS, indicating greater relative potential of nitrogen metabolic dynamics than in the other stages (Fig. S4 and Table S3). Therefore, gene families of nitrogen metabolism were investigated in detail (Fig. 7). Seventeen genes segregated into 4 clusters according to their predicted abundance (Fig. 7). Cluster I included mainly NO/N2O-reducing-related genes (norB, norC, and nosZ). Cluster II included mainly nitrogen-fixation-related genes (nifK, nifH, and nifD), which were particularly frequent in most samples from the ES. Cluster III genes, including nitrite-reducing-related genes (nirB and nirD), had greater relative abundance than genes in other clusters. Genes in cluster IV (napB, narG_narZ_nxrA, narl_narV, and narH_narY_nxrB) were related to nitrate reduction.
FIG 7.

Relative abundances of nitrogen metabolism-related genes during the bloom process, as predicted by PICRUSt. The predicted genes could be segregated into 4 clusters in a row according to their functions.
Regarding the contributions of bacterial families to the selected genes (Fig. S6), genes in cluster I were predicted mainly to occur in Flavobacteriaceae (>43.9% of contributions), which contributed only to this cluster. The high prevalence of cluster II genes in the ES was attributed mainly to Puniceicoccaceae (Verrucomicrobia; >49.1% of contributions). The predicted source of cluster III genes was primarily the Rhodobacteraceae family (>54.4% of contributions). The predicted sources of cluster IV genes were Comamonadaceae (>49.8% of contributions) and Halomonadaceae (47.8% of contributions in napB). The taxonomic metagenomic contributions identified here revealed that each of the gene clusters was contributed mainly by a single bacterial family.
DISCUSSION
Microbial α-diversity variations during the bloom process.
Microbial α-diversity decreased significantly during the diatom bloom process (see Fig. S1 in the supplemental material), as observed previously in Xiangshan Bay (9). Factors correlated with bacterial richness and growth would be expected to determine the microbial succession. Chl a is one of those factors, which can be used to estimate phytoplankton biomass directly (18). The spatial and temporal distributions of Chl a are closely related to marine bacterial abundance (19, 20). Moreover, a microcosm study found that the presence of phytoplankton could affect bacterial richness and production significantly (21). In line with these studies, we found that microbial α-diversity was strongly influenced by diatom biomass (Table S1). Here we also showed that the dominant algal species shifted during the bloom process (Fig. 1B), which may also affect microbial α-diversity, because distinct algal species could release DOM, facilitating differential bacterial growth (22).
We should note that environmental variables, including salinity and water temperature, were related to microbial α-diversity. During this early-spring bloom, the water temperature increased significantly from the ES to the AB stage, which would evidently affect bacterial abundance (19). Salinity is generally associated with dramatic changes in bacterial abundance and community composition (23, 24). Consequently, microbial α-diversity was influenced mainly by bloom disturbances, together with water temperature and salinity.
MCCs associated with the diatom bloom.
Members of the Alphaproteobacteria/Gammaproteobacteria and Flavobacteria constituted the major parts of MCCs in this early-spring bloom (Fig. 2A). Flavobacteriaceae and Rhodobacteraceae were the most dominant bacteria during the bloom process (Fig. 2B). The relative abundances of these taxa were similar to those in previously reported HABs in the Xiangshan Bay (9) and in other locations (21, 25). Flavobacteriaceae, which can utilize a broad range of biopolymers (particularly polysaccharides and proteins) (7), are known for their ability to thrive under bloom conditions in temperate waters, as well as in polar systems (7, 25). The relative abundance of Flavobacteria in the present study was stable, possibly as a result of eutrophication and input of organic matter from aquaculture in Xiangshan. Marine Rhodobacteraceae taxa are major vitamin suppliers for B12-auxotrophic prokaryotes and for eukaryotic primary producers (e.g., diatoms, dinoflagellates, and brown algae) (26, 27). Interestingly, about 90% of Rhodobacterales species possess the complete vitamin B12 synthesis pathways (28), indicating that Rhodobacterales might directly support the diatom bloom by supplying vitamins. Hence, a decrease in Rhodobacteraceae abundance during the bloom process might promote the decline in diatoms.
The bacterial taxa SAR11 and SAR86 are oligotrophic bacterial clades whose abundance may increase with diatom biomass and with Chl a contents (29, 30). Increases in Vibrio abundance have been associated with algal blooms and, in some cases, with specific species of algae (31). High Vibrio abundance may occur in diatom-dominated phytoplankton assemblages (32). Moreover, Vibrio abundance was related to water temperature (31). Therefore, diatom blooms and water temperature may contribute to the progression of Vibrio. Oceanospirillaceae was tightly linked to DOM (33), so that DOM derived from algae would significantly influence their growth. Verrucomicrobiaceae were also enriched in this bloom, because members of this taxon could use diverse glucoside hydrolases to degrade polysaccharides during the bloom process (34).
According to an analysis of the 30 most abundant OTUs, the abundance of Rhodobacteraceae was closely linked to that of S. costatum, or the decrease in Rhodobacteraceae was unfavorable for the growth of S. costatum. Nine OTUs with negative correlations with S. costatum belong to Oceanospirillaceae and Flavobacteriaceae. Members of Oceanospirillaceae and Flavobacteriaceae have long been reported to have algicidal activity (35, 36), which may inhibit the growth of S. costatum. Therefore, our results indicate that some dominant bacterial members may change the course of diatom blooms, according to the interactions between bacteria and diatoms.
Linking of microbial community variations with diatom blooms.
Algal blooms could drive the variations in MCCs significantly, because DOM, allelochemical matter, and other growth factors derived from algal species could strongly influence bacteria growing in alga-related circumstances. Our results revealed that samples could cluster into the corresponding bloom stages (Fig. 3 and 4A; also see Fig. S3), and the diatom bloom could explain the largest MCC variations (Table 1), indicating that MCC variations were closely associated with bloom dynamics. Among the environmental variables analyzed, DO and pH were major abiotic factors governing the dynamics of microbial community structures (Table 1; also see Fig. S3). DO and pH are closely linked to algal growth, due to photosynthesis and the respiration of algal cells. Previous studies proved that DO and pH are major factors affecting microbial community structures in various ecosystems (37, 38). Taken together, our results demonstrated that diatom bloom dynamics would significantly determine the MCCs in Xiangshan Bay.
MCCs varied dramatically over the whole process, which supported the idea of strong temporal shifts in microbial assemblages, and clustered according to bloom stages, which suggested a potential repeatable bloom succession. The cluster of the 30 most abundant OTUs and random forest analysis revealed that MCCs shifted precisely with bloom dynamics (Fig. 3; also see Table S2). Moreover, random forest classification achieved a high level of prediction accuracy with the 30 most abundant OTUs (Table S2), suggesting the great bloom-indicating ability of these OTUs. This result has evident ecological significance for improving the accuracy of the forecast model for HABs. There are many heuristic models capable of simulating various aspects of HABs (39), but most of them have a relatively low level of forecast accuracy. The input parameters in these models are mainly physiochemical variables and not biological factors (e.g., bacteria or algae) that are inherent factors in HABs (2). Therefore, bacteria with bloom-indicating abilities should be integrated into forecast models to improve their accuracy in the future.
Relative roles of deterministic versus stochastic processes.
Variations in the importance of deterministic and stochastic assembly processes are prevalent in natural ecosystems (12, 14, 40). To the best of our knowledge, the present study is somewhat novel in estimating the relative strengths of deterministic and stochastic processes in defining microbial communities across whole HABs. Our results revealed that diatom biomass was the most important factor driving microbial assembly throughout the HAB (Table 2 and Fig. 5B), but there might have been other factors driving microbial assembly in specific bloom stages.
According to βNTI values (Fig. 5A) and the simulation model (Fig. 6), we observed a greater influence of determinacy in the initial successional stages. This result is consistent with previous studies that reported microbial primary successions in various ecosystems (14, 16, 40). The high level of determinacy in the ES may be the result of eutrophication. The nutrient loads from catchments and excessive aquaculture activities exacerbate the degree of eutrophication in Xiangshan Bay (41). Consequently, strong selective forces imposed by high nutrient inputs may drive succession of the microbial communities in the initial or prebloom stages. In the MS, stochasticity mainly drove bacterial assembly. We inferred that physiochemical conditions are not as extreme as in the ES, because environmental heterogeneity decreased significantly. In contrast to conditions in other bloom stages, there was a strong wind on the MS sampling day, which would promote the immigration of bacteria by strong waves. Thus, random dispersal could have been an important ecological factor in the MS. In the LS and the AB stage, the deterministic process again governed bacterial assembly, but it was induced mainly by environmental homogeneity (Fig. 6). In the LS, many of the S. costatum organisms were lysed, and DOM derived from this algal species was released into the seawater, leading to strong homogeneous selection (5). In the AB stage, we observed an evident increase in temperature, which might also be an important factor driving MCCs, as reported previously (42). We inferred that the influence of the rapidly rising temperatures might override the effects of other environmental variables, inducing homogeneous selection at the AB stage.
Although our analyses explained the MCC successional patterns in the whole HAB, the repeatability in Xiangshan Bay and other algal bloom processes still needs to be tested. Nonetheless, the present data helped to elucidate the relative magnitude of forces driving MCCs, which are crucial components of both natural and agricultural ecosystems. In the future, more samples will be integrated into this model to test its repeatability and to improve its accuracy.
Predicted metabolic potential in the diatom bloom.
Evidence for variation in MCCs in HABs has been widely reported, but few studies focused on the metabolic potential of the microbial communities. To gain more insights into the metabolic potential of bacteria in the HAB process, PICRUSt was used to predict the metagenome of each bloom stage. The NSTI values (see Data Set S2 in the supplemental material) were either similar to or lower than the mean NSTI value reported for bacterial communities in samples collected from various environments (43–45). Therefore, we inferred that our predicted metabolic potential is supported by the microbial genomic reference database.
The present study revealed that the metabolic functions of microbial communities were significantly related to nitrogen (Table S3 and Fig. S4), and distinct bacterial families contributed to the different metabolic potentials (Fig. 7; also see Fig. S6), which may be induced by the nitrogen nutrient levels. Cluster I consisted mainly of genes encoding nitric oxide and nitrous oxide reductases. This metabolic potential might be due to the family Flavobacteriaceae (46). The metabolic potential in cluster II was related to nitrogen fixation, with Puniceicoccaceae (phylum Verrucomicrobia) being the main contributor. A novel bacterium of the family Puniceicoccaceae, with nitrogen-fixing ability, was found last year (47). In addition, nitrogen fixation by heterotrophic bacterial communities could be significantly stimulated by high nutrient loads (48); therefore, there may be a relatively greater nitrogen fixation potential in the ES, in comparison to other stages (Fig. 7). Rhodobacteraceae, the main clade in cluster III, is an important denitrifying family (49) related to nitrite reduction. Members of Comamonadaceae and Halomonadaceae in cluster IV have been observed to catalyze the reduction of nitrate (50, 51). These results indicated that nitrogen metabolism is mediated by a diverse group of bacteria, even though some of them are not the most abundant taxa. Moreover, anthropogenic and agricultural activities resulted in large nitrate amounts entering Xiangshan Bay, and HABs may significantly influence the removal of nitrate from the ecosystem.
In the present study, we studied the microbial metabolic function potential in the HAB process, based on 16S rRNA gene sequencing and PICRUSt, and we realized that there might be some bias in this method. Gene ontology in the PICRUSt script is based on the GreenGenes v13.5 database, which is not as accurate as the newest SILVA database. Moreover, the annotation is implemented based on well-characterized human-pathogenic species, which may not be suitable for environmental samples. A more accurate input metagenome would require a newer database and more annotated species from environmental samples. In addition, quantitative data, such as absolute bacterial abundance, and functional genes should be integrated. Therefore, more reliable analytical techniques, including quantitative PCR, metagenomic sequencing, and metatranscriptomic sequencing, will be applied to study metabolic function potential in future work.
Overall, microbial communities in this early-spring bloom exhibited changes in diversity and composition during the bloom process. In particular, the 30 most abundant OTUs responded significantly to the algal bloom, exhibiting clear successional patterns in distinct stages. Moreover, our exploration of the diatom bloom in the Xiangshan Bay also provided novel insights about the relative roles of the deterministic and stochastic processes in driving microbial community structures. Both ecological processes played important roles in the assembly of microbial communities, but the deterministic process was of greater importance during the bloom process. In addition, metabolic potential prediction suggested that the outbreak of diatoms might have a significant impact on the denitrification process. In the future, detailed information about the interaction between microbial communities and phytoplankton, as well as metabolic functional characteristics, is essential to better understand the mechanisms influencing the formation of HABs.
MATERIALS AND METHODS
Sample collection.
Samples were collected in February and March 2017 from 5 stations in Xiangshan Bay during the diatom bloom process, in the ES, MS, LS, and AB stage (Fig. 1). For the ES, samples were collected on 10 February and 15 February. For the MS, samples were collected on 19 February and 23 February. For the LS and the AB stage, samples were collected on 27 February and 8 March, respectively. Twenty-nine water samples were collected from the surface layer (0.5 m deep), using a 5-liter water sampler. For phytoplankton quantification, 1 liter of each water sample was fixed with 1% Lugol's solution. After filtration with a 100-μm mesh, approximately 600 ml of seawater was filtered using 0.2-μm filters (47-mm-diameter polycarbonate; Millipore, USA); the filters were then stored at −80°C prior to DNA extraction. To prevent contamination between samples, the water sampler and filtration systems were washed carefully with sterile water before water collection and filtration.
After homogenization by gentle shaking, phytoplankton samples were characterized and enumerated using a counting chamber with a light microscope (Axioplan; Carl Zeiss, Jena, Germany). Water temperature, salinity, and pH were measured on board. DO, Chl a, chemical oxygen demand (COD), phosphate, silicate, nitrate, nitrite, and ammonium levels were measured using standard methods (52).
DNA extraction and PCR amplification.
DNA extraction was performed using the Power Soil DNA isolation kit (Mo Bio, USA), according to the manufacturer's instructions. DNA concentrations were quantified using a Nanodrop 2000 instrument (Thermo Fisher Scientific, Wilmington, DE, USA). The V4 hypervariable region of the 16S rRNA gene was amplified in triplicate PCRs (20-μl reaction volume, with 5× FastPfu buffer, 2.5 mM deoxynucleoside triphosphates [dNTPs], 5 μM combined primers, and FastPfu polymerase), using 10 ng DNA template and the primer pair 515f and 806r (53). PCR cycling conditions were as follows: denaturation at 95°C for 3 min, 27 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 45 s, and a final extension at 72°C for 10 min. Triplicate PCR amplicons were mixed after purification using a TaKaRa purification kit (TaKaRa, Japan). Libraries were then generated using the TruSeq DNA sample preparation kit for Illumina (Illumina, San Diego, CA, USA), following the manufacturer's instructions. The libraries were sequenced using a MiSeq platform (Illumina) in a paired-end 250-bp sequence read run at MajorBio Co. Ltd. (Shanghai, China).
Data processing.
QIIME v1.9 (54) and USEARCH v6.1 (55) pipelines were used to perform quality control and to cluster sequences into OTUs. Overlapping reads were merged using FLASH, with default parameters (56). Raw sequences were demultiplexed and filtered to remove low-quality sequences using QIIME. Chimeras were detected and removed with USEARCH using the SILVA 128 database (57). OTUs were clustered at 97% sequence similarity using UCLUST (58). Taxonomic assignments were achieved by UCLUST with the SILVA 128 reference database. OTUs affiliated with chloroplast, mitochondrion, unassigned, and unclassified sequences and singletons were removed from the data set before downstream analysis. The numbers of final clean reads in each sample ranged from 16,618 to 82,467 reads, yielding a total of 3,529 OTUs (see Data Set S1 in the supplemental material).
Statistical analyses.
For estimation of α-diversity, sequences in each sample were normalized at the lowest sequence depth and rarefied at 16,610 reads per sample. The different α-diversity estimations (richness, Shannon index, phylogenetic diversity, and evenness) were calculated using QIIME and visualized using the ggplot2 package (59). The relationships between α-diversity indices and environmental variables were calculated by Spearman's rank correlation using the psych package (60). The MCCs were analyzed at the class (relative abundance of >0.5%) and family (relative abundance of >5%) levels. The 30 most abundant OTUs were selected for study of the shifts in MCCs at distinct bloom stages, and the abundance of each OTU was scaled by color in a heat map using the pheatmap package (61). Random forest analysis was performed to test whether the 30 most abundant OTUs could predict the distinct bloom stages using the randomForest package (62). To track the successional trajectories of microbial communities in the bloom process, β-diversity was calculated using the Bray-Curtis distance and visualized by a two-dimensional NMDS plot. The db-RDA (63) was calculated using the capscale function in vegan (64), to explore the effects of environmental variables on MCCs. Mantel tests (65) and PERMANOVA (66) were used to verify the explanation of environmental variables in the succession of MCCs. To determine environmental heterogeneity, the homogeneity of multivariate dispersions, applied to the environmental variables (67), was calculated using the betadisper function in vegan. All of the environmental variables were standardized before the calculation of environmental heterogeneity. All statistical analyses were performed with R statistical software (https://www.r-project.org).
Analysis of assembly processes of microbial communities.
The phylogenetic turnover (phylogenetic β-diversity) was quantified using the βNTI (12). Based on the random shuffling of OTU labels across the tips of the phylogeny, the βNTI was computed as the number of standard deviations that observed β-mean nearest taxon distance (βMNTD) values deviated from the mean of the null distribution (999 null replicates) (12, 68). The absolute magnitude of the βNTI reflects the influence of the deterministic process (∣βNTI∣ of >2); the greater the magnitude is, the greater the influence of the deterministic process is. Values of ∣βNTI∣ of <2 indicate that the stochastic process may play an important role in structuring microbial communities during the bloom process.
Metagenomic prediction of microbial communities.
Functional metagenomes during the bloom stages were predicted from clean reads from 16S rRNA sequencing using PICRUSt (43). Briefly, OTUs were picked based on the closed reference OTU-picking method, at 97% sequence similarity, against the GreenGenes v13.5 database in QIIME. The OTU table was submitted to local PICRUSt v1.1.2 for metagenome prediction, which then created the final metagenomic functional predictions based on KEGG pathways, at classification levels 2 and 3. To ensure the accuracy of metagenomic predictions, the NSTI value, which indicates the distance of the 16S rRNA-sequenced microbes from the genome-sequenced microbes, was calculated. Lower NSTI values reflect a shorter distance between our sequenced microbes and the genome-sequenced microbes in the reference database (43).
A db-RDA was used to estimate the relationships between metabolic gene ontologies (Bray-Curtis distances from predicted gene abundance) and environmental variables, including nitrate, nitrite, ammonium, phosphate, and silicate levels. The genes predicted to be associated with nitrogen metabolism were clustered (genes with total abundance of <7,000 copies in all samples were discarded) and visualized using the pheatmap package in R (61). The contribution of OTUs to nitrogen metabolism-related genes was computed using metagenome_contributions.py in PICRUSt. Bacterial families with contributions to nitrogen metabolism of >1% were selected and graphed.
Accession number(s).
Raw sequences were submitted to the NCBI Sequence Read Archive with the accession number SRP130859.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the National Natural Science Foundation of China (grant 41706132), the Zhejiang Provincial Natural Science Foundation of China (grant LQ17D060001), the Natural Science Foundation of Ningbo (grant 2016A610094), the Open Fund of Key Laboratory of Integrated Marine Monitoring and Applied Technologies for Harmful Algal Blooms, State Oceanic Administration (grant MATHAB201708), and the K. C. Wong Magna Fund of Ningbo University.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01000-18.
REFERENCES
- 1.Teeling H, Fuchs BM, Bennke CM, Kruger K, Chafee M, Kappelmann L, Reintjes G, Waldmann J, Quast C, Glockner FO, Lucas J, Wichels A, Gerdts G, Wiltshire KH, Amann RI. 2016. Recurring patterns in bacterioplankton dynamics during coastal spring algae blooms. Elife 5:e11888. doi: 10.7554/eLife.11888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson DM, Cembella AD, Hallegraeff GM. 2012. Progress in understanding harmful algal blooms: paradigm shifts and new technologies for research, monitoring, and management. Annu Rev Mar Sci 4:143–176. doi: 10.1146/annurev-marine-120308-081121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Teeling H, Fuchs BM, Becher D, Klockow C, Gardebrecht A, Bennke CM, Kassabgy M, Huang S, Mann AJ, Waldmann J. 2012. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336:608–611. doi: 10.1126/science.1218344. [DOI] [PubMed] [Google Scholar]
- 4.Amin SA, Hmelo LR, van Tol HM, Durham BP, Carlson LT, Heal KR, Morales RL, Berthiaume CT, Parker MS, Djunaedi B, Ingalls AE, Parsek MR, Moran MA, Armbrust EV. 2015. Interaction and signalling between a cosmopolitan phytoplankton and associated bacteria. Nature 522:98–101. doi: 10.1038/nature14488. [DOI] [PubMed] [Google Scholar]
- 5.Taylor JD, Cottingham SD, Billinge J, Cunliffe M. 2014. Seasonal microbial community dynamics correlate with phytoplankton-derived polysaccharides in surface coastal waters. ISME J 8:245–248. doi: 10.1038/ismej.2013.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wemheuer B, Gullert S, Billerbeck S, Giebel HA, Voget S, Simon M, Daniel R. 2014. Impact of a phytoplankton bloom on the diversity of the active bacterial community in the southern North Sea as revealed by metatranscriptomic approaches. FEMS Microbiol Ecol 87:378–389. doi: 10.1111/1574-6941.12230. [DOI] [PubMed] [Google Scholar]
- 7.Landa M, Blain S, Christaki U, Monchy S, Obernosterer I. 2016. Shifts in bacterial community composition associated with increased carbon cycling in a mosaic of phytoplankton blooms. ISME J 10:39–50. doi: 10.1038/ismej.2015.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berry MA, Davis TW, Cory RM, Duhaime MB, Johengen TH, Kling GW, Marino JA, Den Uyl PA, Gossiaux D, Dick GJ, Denef VJ. 2017. Cyanobacterial harmful algal blooms are a biological disturbance to western Lake Erie bacterial communities. Environ Microbiol 19:1149–1162. doi: 10.1111/1462-2920.13640. [DOI] [PubMed] [Google Scholar]
- 9.Chen H, Zhang H, Xiong J, Wang K, Zhu J, Zhu X, Zhou X, Zhang D. 2016. Successional trajectories of bacterioplankton community over the complete cycle of a sudden phytoplankton bloom in the Xiangshan Bay, East China Sea. Environ Pollut 219:750–759. doi: 10.1016/j.envpol.2016.07.035. [DOI] [PubMed] [Google Scholar]
- 10.Albright MBN, Martiny JBH. 2018. Dispersal alters bacterial diversity and composition in a natural community. ISME J 12:296–299. doi: 10.1038/ismej.2017.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teurlincx S, Velthuis M, Seroka D, Govaert L, van Donk E, Van de Waal DB, Declerck SAJ. 2017. Species sorting and stoichiometric plasticity control community C:P ratio of first-order aquatic consumers. Ecol Lett 20:751–760. doi: 10.1111/ele.12773. [DOI] [PubMed] [Google Scholar]
- 12.Stegen JC, Lin X, Konopka AE, Fredrickson JK. 2012. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J 6:1653–1664. doi: 10.1038/ismej.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chase JM, Myers JA. 2011. Disentangling the importance of ecological niches from stochastic processes across scales. Philos Trans R Soc Lond B Biol Sci 366:2351–2363. doi: 10.1098/rstb.2011.0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, Shen J, Wu Y, Tu C, Soininen J, Stegen JC, He J, Liu X, Zhang L, Zhang E. 2013. Phylogenetic beta diversity in bacterial assemblages across ecosystems: deterministic versus stochastic processes. ISME J 7:1310–1321. doi: 10.1038/ismej.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graham EB, Knelman JE, Andreas S, Steven S, Marc B, Anthony Y, Beman JM, Guy A, Laurent P, James P. 2016. Microbes as engines of ecosystem function: When does community structure enhance predictions of ecosystem processes? Front Microbiol 7:214. doi: 10.3389/fmicb.2016.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morrison-Whittle P, Goddard MR. 2015. Quantifying the relative roles of selective and neutral processes in defining eukaryotic microbial communities. ISME J 9:2003–2011. doi: 10.1038/ismej.2015.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huo Y, Wu H, Chai Z, Xu S, Han F, Dong L, He P. 2012. Bioremediation efficiency of Gracilaria verrucosa for an integrated multi-trophic aquaculture system with Pseudosciaena crocea in Xiangshan Harbor, China. Aquaculture 326-329:99–105. doi: 10.1016/j.aquaculture.2011.11.002. [DOI] [Google Scholar]
- 18.Bird DF, Kalff J. 1984. Empirical relationships between bacterial abundance and chlorophyll concentration in fresh and marine waters. Can J Fish Aquat Sci 41:1015–1023. doi: 10.1139/f84-118. [DOI] [Google Scholar]
- 19.Pinhassi J, Hagström Å. 2000. Seasonal succession in marine bacterioplankton. Aquat Microb Ecol 21:245–256. doi: 10.3354/ame021245. [DOI] [Google Scholar]
- 20.Suzuki MT, Preston CM, Chavez FP, DeLong EF. 2001. Quantitative mapping of bacterioplankton populations in seawater: field tests across an upwelling plume in Monterey Bay. Aquat Microb Ecol 24:117–127. doi: 10.3354/ame024117. [DOI] [Google Scholar]
- 21.Pinhassi J, Sala MM, Havskum H, Peters F, Guadayol O, Malits A, Marrase C. 2004. Changes in bacterioplankton composition under different phytoplankton regimens. Appl Environ Microbiol 70:6753–6766. doi: 10.1128/AEM.70.11.6753-6766.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eigemann F, Hilt S, Salka I, Grossart HP. 2013. Bacterial community composition associated with freshwater algae: species specificity vs. dependency on environmental conditions and source community. FEMS Microbiol Ecol 83:650–663. doi: 10.1111/1574-6941.12022. [DOI] [PubMed] [Google Scholar]
- 23.Okie JG, Van Horn DJ, Storch D, Barrett JE, Gooseff MN, Kopsova L, Takacs-Vesbach CD. 2015. Niche and metabolic principles explain patterns of diversity and distribution: theory and a case study with soil bacterial communities. Proc R Soc B 282:20142630. doi: 10.1098/rspb.2014.2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Héry M, Volant A, Garing C, Luquot L, Elbaz PF, Gouze P. 2014. Diversity and geochemical structuring of bacterial communities along a salinity gradient in a carbonate aquifer subject to seawater intrusion. FEMS Microbiol Ecol 90:922–934. doi: 10.1111/1574-6941.12445. [DOI] [PubMed] [Google Scholar]
- 25.Buchan A, LeCleir GR, Gulvik CA, Gonzalez JM. 2014. Master recyclers: features and functions of bacteria associated with phytoplankton blooms. Nat Rev Microbiol 12:686–698. doi: 10.1038/nrmicro3326. [DOI] [PubMed] [Google Scholar]
- 26.Croft MT, Lawrence AD, Raux-Deery E, Warren Amp MJ, Smith AG. 2005. Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature 438:90–93. doi: 10.1038/nature04056. [DOI] [PubMed] [Google Scholar]
- 27.Helliwell KE, Wheeler GL, Leptos KC, Goldstein RE, Smith AG. 2011. Insights into the evolution of vitamin B12 auxotrophy from sequenced algal genomes. Mol Biol Evol 28:2921–2933. doi: 10.1093/molbev/msr124. [DOI] [PubMed] [Google Scholar]
- 28.Sañudo-Wilhelmy S, Gómez-Consarnau L, Suffridge C, Webb E. 2014. The role of B vitamins in marine biogeochemistry. Annu Rev Mar Sci 6:339–367. doi: 10.1146/annurev-marine-120710-100912. [DOI] [PubMed] [Google Scholar]
- 29.Eiler A, Hayakawa DH, Church MJ, Karl DM, Rappé MS. 2009. Dynamics of the SAR11 bacterioplankton lineage in relation to environmental conditions in the oligotrophic North Pacific subtropical gyre. Environ Microbiol 11:2291–2300. doi: 10.1111/j.1462-2920.2009.01954.x. [DOI] [PubMed] [Google Scholar]
- 30.West NJ, Lepère C, Manes CLDO, Catala P, Scanlan DJ, Lebaron P. 2016. Distinct spatial patterns of SAR11, SAR86, and Actinobacteria diversity along a transect in the ultra-oligotrophic South Pacific Ocean. Front Microbiol 7:234. doi: 10.3389/fmicb.2016.00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turner J, Good B, Cole D, Lipp E. 2009. Plankton composition and environmental factors contribute to Vibrio seasonality. ISME J 3:1082–1092. doi: 10.1038/ismej.2009.50. [DOI] [PubMed] [Google Scholar]
- 32.Asplund ME, Rehnstam-Holm AS, Atnur V, Raghunath P, Saravanan V, Härnström K, Collin B, Karunasagar I, Godhe A. 2011. Water column dynamics of Vibrio in relation to phytoplankton community composition and environmental conditions in a tropical coastal area. Environ Microbiol 13:2738–2751. doi: 10.1111/j.1462-2920.2011.02545.x. [DOI] [PubMed] [Google Scholar]
- 33.Landa M, Kirchman DL, Cottrell MT, Obernosterer I, Blain S. 2013. Changes in bacterial diversity in response to dissolved organic matter supply in a continuous culture experiment. Aquat Microb Ecol 69:157–168. doi: 10.3354/ame01632. [DOI] [Google Scholar]
- 34.Andersson AF, Riemann L, Bertilsson S. 2010. Pyrosequencing reveals contrasting seasonal dynamics of taxa within Baltic Sea bacterioplankton communities. ISME J 4:171–181. doi: 10.1038/ismej.2009.108. [DOI] [PubMed] [Google Scholar]
- 35.Kristyanto S, Chaudhary DK, Lee S-S, Kim J. 2017. Characterization of Marinomonas algicida sp. nov., a novel algicidal marine bacterium isolated from seawater. Int J Syst Evol Microbiol 67:4777–4784. doi: 10.1099/ijsem.0.002374. [DOI] [PubMed] [Google Scholar]
- 36.Pokrzywinski KL, Place AR, Warner ME, Coyne KJ. 2012. Investigation of the algicidal exudate produced by Shewanella sp. IRI-160 and its effect on dinoflagellates. Harmful Algae 19:23–29. doi: 10.1016/j.hal.2012.05.002. [DOI] [Google Scholar]
- 37.Liu J, Fu B, Yang H, Zhao M, He B, Zhang XH. 2015. Phylogenetic shifts of bacterioplankton community composition along the Pearl Estuary: the potential impact of hypoxia and nutrients. Front Microbiol 6:64. doi: 10.3389/fmicb.2015.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lauber C, Hamady M, Knight R, Fierer N. 2009. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl Environ Microbiol 75:5111–5120. doi: 10.1128/AEM.00335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stumpf RP, Tomlinson MC, Calkins JA, Kirkpatrick B, Fisher K, Nierenberg K, Currier R, Wynne TT. 2009. Skill assessment for an operational algal bloom forecast system. J Mar Syst 76:151–161. doi: 10.1016/j.jmarsys.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao D, Cao X, Huang R, Zeng J, Shen F, Xu H, Wang S, He X, Yu Z. 2017. The heterogeneity of composition and assembly processes of the microbial community between different nutrient loading lake zones in Taihu Lake. Appl Microbiol Biotechnol 101:5913–5923. doi: 10.1007/s00253-017-8327-0. [DOI] [PubMed] [Google Scholar]
- 41.Jiang ZB, Chen QZ, Zeng JN, Liao YB, Shou L, Liu J. 2012. Phytoplankton community distribution in relation to environmental parameters in three aquaculture systems in a Chinese subtropical eutrophic bay. Mar Ecol Prog 446:73–89. doi: 10.3354/meps09499. [DOI] [Google Scholar]
- 42.Cram JA, Chow CE, Sachdeva R, Needham DM, Parada AE, Steele JA, Fuhrman JA. 2015. Seasonal and interannual variability of the marine bacterioplankton community throughout the water column over ten years. ISME J 9:563–580. doi: 10.1038/ismej.2014.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Langille MGI, Zaneveld J, Caporaso JG, Mcdonald D, Dan K, Reyes JA, Clemente JC, Burkepile DE, Thurber RLV, Knight R. 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lopes LD, Andreote FD. 2016. Bacterial abilities and adaptation toward the rhizosphere colonization. Front Microbiol 7:1341. doi: 10.3389/fmicb.2016.01341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koo H, Mojib N, Hakim JA, Hawes I, Tanabe Y, Andersen DT, Bej AK. 2017. Microbial communities and their predicted metabolic functions in growth laminae of a unique large conical mat from Lake Untersee, East Antarctica. Front Microbiol 8:1347. doi: 10.3389/fmicb.2017.01347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mills HJ, Hunter E, Humphrys M, Kerkhof L, McGuinness L, Huettel M, Kostka EJ. 2008. Characterization of nitrifying, denitrifying, and overall bacterial communities in permeable marine sediments of the northeastern Gulf of Mexico. Appl Environ Microbiol 74:4440–4453. doi: 10.1128/AEM.02692-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin SY, Hameed A, Liu YC, Hsu YH, Hung MH, Lai WA, Young CC. 2017. Ruficoccus amylovorans gen. nov., sp. nov, an amylolytic and nitrate-reducing diazotroph of the family Puniceicoccaceae. Int J Syst Evol Microbiol 67:956–962. doi: 10.1099/ijsem.0.001723. [DOI] [PubMed] [Google Scholar]
- 48.Musat F, Harder J, Widdel F. 2006. Study of nitrogen fixation in microbial communities of oil-contaminated marine sediment microcosms. Environ Microbiol 8:1834–1843. doi: 10.1111/j.1462-2920.2006.01069.x. [DOI] [PubMed] [Google Scholar]
- 49.Chen Y, Zhou W, Li Y, Zhang J, Zeng G, Huang A, Huang J. 2014. Nitrite reductase genes as functional markers to investigate diversity of denitrifying bacteria during agricultural waste composting. Appl Microbiol Biotechnol 98:4233–4243. doi: 10.1007/s00253-014-5514-0. [DOI] [PubMed] [Google Scholar]
- 50.Straub KL, Schönhuber WA, Buchholz-Cleven BEE, Schink B. 2004. Diversity of ferrous iron-oxidizing, nitrate-reducing bacteria and their involvement in oxygen-independent iron cycling. Geomicrobiol J 21:371–378. doi: 10.1080/01490450490485854. [DOI] [Google Scholar]
- 51.Ntougias S, Zervakis GI, Fasseas C. 2007. Halotalea alkalilenta gen. nov., sp. nov., a novel osmotolerant and alkalitolerant bacterium from alkaline olive mill wastes, and emended description of the family Halomonadaceae Franzmann et al. 1989, emend. Dobson and Franzmann 1996. Int J Syst Evol Microbiol 57:1975–1983. doi: 10.1099/ijs.0.65078-0. [DOI] [PubMed] [Google Scholar]
- 52.General Administration of Quality Supervision, Inspection, and Quarantine. 2007. The specification for marine monitoring of China, part 4: seawater analysis. GB 17378.4-2007. General Administration of Quality Supervision, Inspection, and Quarantine, Beijing, People's Republic of China. [Google Scholar]
- 53.Parada AE, Needham DM, Fuhrman JA. 2016. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ Microbiol 18:1403–1414. doi: 10.1111/1462-2920.13023. [DOI] [PubMed] [Google Scholar]
- 54.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Magoč T, Salzberg SL. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 59.Wickham H. 2009. ggplot2: elegant graphics for data analysis. Springer-Verlag, New York, NY. [Google Scholar]
- 60.Revelle W. 2018. psych: procedures for personality and psychological research. Northwestern University, Evanston, IL. [Google Scholar]
- 61.Kolde R. 2015. pheatmap: pretty heatmaps. R Package version 1.0.10. https://cran.r-project.org/web/packages/pheatmap/index.html.
- 62.Liaw A, Wiener M. 2002. Classification and regression by randomForest. R News 2:18–22. [Google Scholar]
- 63.Legendre P, Anderson MJ. 1999. Distance-based redundancy analysis: testing multispecies responses in multifactorial ecological experiments. Ecol Monogr 69:1–24. doi: 10.1890/0012-9615(1999)069[0001:DBRATM]2.0.CO;2. [DOI] [Google Scholar]
- 64.Oksanen J, Kindt R, Legendre P, O'Hara B, Stevens MHH, Oksanen MJ, Suggests M. 2007. The vegan package: community ecology package 10. https://cran.r-project.org/web/packages/vegan/index.html.
- 65.Diniz-Filho JAF, Soares TN, Lima JS, Dobrovolski R, Landeiro VL, Telles MPDC, Rangel TF, Bini LM. 2013. Mantel test in population genetics. Genet Mol Biol 36:475–485. doi: 10.1590/S1415-47572013000400002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anderson MJ. 2001. A new method for non-parametric multivariate analysis of variance. Austral Ecol 26:32–46. doi: 10.1111/j.1442-9993.2001.01070.pp.x. [DOI] [Google Scholar]
- 67.Anderson MJ, Ellingsen KE, McArdle BH. 2006. Multivariate dispersion as a measure of beta diversity. Ecol Lett 9:683–693. doi: 10.1111/j.1461-0248.2006.00926.x. [DOI] [PubMed] [Google Scholar]
- 68.Fine PVA, Kembel SW. 2011. Phylogenetic community structure and phylogenetic turnover across space and edaphic gradients in western Amazonian tree communities. Ecography 34:552–565. doi: 10.1111/j.1600-0587.2010.06548.x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.