

Abstract
Ornithine transcarbamylase deficiency (OTCD) is an X-linked urea cycle disorder with variable expressivity in heterozygous females. While liver function testing is often abnormal in patients with OTCD, liver failure is uncommon on presentation. A 13-month-old female with no significant past medical history presented with irritability, right arm weakness, and decreased appetite. Initial workup revealed hepatic dysfunction with an INR of 3.4, ammonia level of 75 μmol/L, and abnormal brain MRI with gyral edema with restricted diffusion, and patchy signal abnormality in basal ganglia. The MRI findings led to a putative diagnosis of acute disseminated encephalomyelitis prompting corticosteroid treatment. As steroid treatment was begun, she developed significant hepatocellular dysfunction with ALT 2,222 U/L, AST 630 U/L, prolonged INR, and elevated ammonia (213 μmol/L). Neurologic signs resolved and her ammonia level decreased (43 μmol/L) without further intervention; however, she had ongoing acute liver failure with coagulopathy and episodic irritability, managed as seronegative autoimmune hepatitis with partial response to corticosteroid therapy. At 18 months of age she presented with severe irritability with markedly increased ammonia (417 μmol/L). Plasma amino acids obtained several days prior to this acute episode demonstrated elevation in glutamine (2,725 μmol/L) and alanine (1,459 μmol/L). Biochemical testing demonstrated elevation of urine orotic acid (>240.6 mmol/mol creatinine). Genetic testing confirmed a heterozygous nonsense mutation in the OTC gene (c.958C>T, R320X). After treatment with ammonia scavengers and a protein-restricted diet, hepatic function normalized and irritability resolved. The diagnosis of a urea cycle disorder should be considered in patients with unexplained hepatic dysfunction.
Keywords: Hyperammonemia, Liver failure, Ornithine transcarbamylase deficiency, Urea cycle disorder
Introduction
Ornithine transcarbamylase (OTC) deficiency (OTCD) is an X-linked urea cycle disorder that affects both males and females (Brusilow and Horwich 2014). OTCD is caused by mutations in the gene encoding OTC on chromosome Xp11, and it is the most frequent urea cycle disorder (Tuchman et al. 2008). The urea cycle disorders have overlapping presentations that are primarily the consequence of hyperammonemia, including vomiting, lethargy, progressive somnolence, irritability, agitation, disorientation, and ataxia; however, the age of presentation and prognosis can vary from fatality in the neonatal period to variable severity appearing any time thereafter (Brusilow and Horwich 2014). Variable clinical expressivity can be marked in individuals with residual OTC enzyme activity, particularly heterozygous females (Batshaw et al. 1986). Late presentations in childhood may be associated with weaning from breast milk to higher protein formula or cow’s milk, high-protein meals, or catabolism related to infection (Brusilow and Horwich 2014).
OTCD may be suspected based on an elevated blood ammonia concentration, elevated serum glutamine concentration, low serum citrulline concentration, and elevated urine orotic acid; a definitive diagnosis is based on the identification of a pathogenic variant in the OTC gene, a pedigree analysis, elevated baseline urinary orotic acid level or after an allopurinol challenge test, and/or reduced OTC enzyme activity in liver or intestinal tissue (Tuchman et al. 2008). OTC enzyme activity has low sensitivity for diagnosis in female patients because of the X-chromosome inactivation pattern (Yorifuji et al. 1998). Liver involvement is increasingly recognized as a common complication of OTCD, which is present in more than 50% of patients presenting with symptomatic OTCD (Gallagher et al. 2014; Laemmle et al. 2016). The pathophysiology of the liver dysfunction in OTCD is poorly understood, and primary hepatic presentations are uncommonly reported.
Clinical Report
A 13-month-old female with no significant past medical history presented with irritability, right arm weakness, and decreased appetite. The report of her newborn screening was normal, and amino acid details were unknown. Her parents were healthy and the family history was negative for metabolic disorders, other than a maternal history of hypothyroidism. There was, however, a strong history of autoimmune disorders, including scleroderma in the paternal grandmother, Crohn’s disease, rheumatoid arthritis, and colitis in the paternal cousins, and hypothyroidism and alopecia in the maternal aunts. Initial workup revealed significant hepatic dysfunction with an INR of 3.4, plasma ammonia level of 75 μmol/L, and abnormal brain MRI with gyral edema of both hemispheres with restricted diffusion, and patchy signal abnormality in basal ganglia with diffusion abnormality (Fig. 1). Differential diagnoses at the time of initial presentation included cytomegalovirus infection, metabolic disorder, autoimmune hepatitis, and acute disseminated encephalomyelitis (ADEM). The MRI findings led to a putative diagnosis of ADEM, which prompted treatment with high dose intravenous corticosteroids. Shortly after steroid treatment was initiated, she was found to have significant hepatic dysfunction with ALT 2,222 U/L, AST 630 U/L, prolonged INR (3.4), and elevated plasma ammonia (213 μmol/L). Metabolic studies performed 4 days after her initial presentation included a plasma amino acid quantitation of glutamine (1,248 μmol/L; reference range 303–1,459), alanine (691 μmol/L; reference range 119–523), and citrulline (15 μmol/L; reference range 4–50). Apparently, since the glutamine and citrulline levels were within the reference range at an outside laboratory facility, these values were not investigated further. Additionally, as the plasma sample had to be sent out to another outside facility, the results were not returned until 3 weeks after the initial presentation. A urine organic acid analysis was also performed yielding elevations in lactate, 3-hydroxybutyrate, acetoacetate, 4-hydroxyphenyllactate, and 4-hydroxyphenylpyruvate.
Fig. 1.
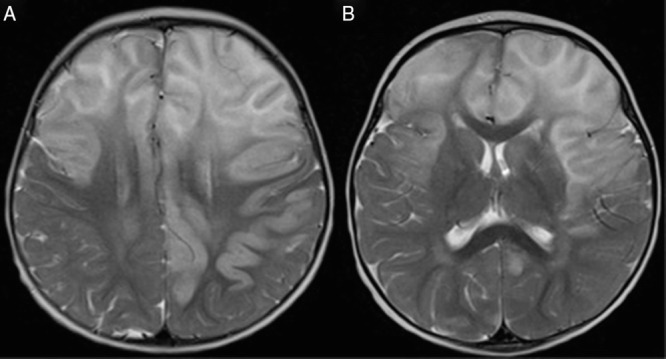
Axial T2 brain MRI on initial presentation at 13 months old demonstrated increased T2 signal intensity (a, b) throughout the frontal hemispheres extending posteriorly without enhancement or restricted diffusion (not shown)
Neurologic symptoms resolved and her ammonia level decreased (55 μmol/L) with intravenous fluids with 10% glucose and intralipids, but she continued to have ongoing acute liver failure with coagulopathy as her INR was >1.5, but without hyperbilirubinemia. Her liver dysfunction was managed as seronegative autoimmune hepatitis demonstrating partial response to corticosteroid therapy. Liver biopsy revealed mild lymphocytic inflammation with focal mild ballooning of hepatocytes. She had episodic irritability that correlated with episodes of aminotransferase elevations, though ammonia levels were not measured with these episodes.
At 18 months of age, she had a subacute presentation of severe irritability with decreased speech, and was found to have markedly increased ammonia on admission (443 μmol/L). Her family had noted unusual odors and recrudescence of her right-sided weakness for several days prior to presentation. Plasma amino acids obtained 3 days prior to this acute episode demonstrated marked elevation in glutamine (2,725 μmol/L) and alanine (1,459 μmol/L). These amino acids also demonstrated a citrulline level of 15 μmol/L (reference range 3–45 μmol/L) and arginine level of 30 μmol/L (reference range 29–116 μmol/L). She was admitted for treatment of hyperammonemia and further metabolic diagnostic evaluation for possible urea cycle disorder. The hyperammonemia resolved after treatment with ammonia scavengers and a protein-restricted diet supplemented with essential amino acids and increased calories. Brain MRI demonstrated diffuse parenchymal volume loss (Fig. 2). Hepatic function gradually normalized (Figs. 3 and 4) and irritability resolved.
Fig. 2.

Axial T2 brain MRI at 18 months old (a, b) demonstrated interval increase in abnormal white matter T2 prolongation in frontal lobes, parietal lobes, posterior temporal lobes, and centrum semiovale as well as diffuse parenchymal volume loss. MR spectroscopy (c) with a glutamine–glutamate (Glx) complex peak
Fig. 3.

Laboratory trends from time of initial presentation at 13 months old. Reference ranges AST 2–40 mmol/L, ALT 3–30 mmol/L; INR 0.87–1.13
Fig. 4.

Laboratory trends from time of initial presentation at 13 months old. Reference ranges ammonia 50–80 mmol/L; glutamine 427–907 mmol/L, alanine 122–578 mmol/L
Biochemical testing demonstrated marked elevation of urine orotic acid (>240.6 mmol/mol creatinine). Genetic testing confirmed a heterozygous nonsense pathogenic variant in the OTC gene (c.958C>T, R320X) (Demmer et al. 1996; Kim et al. 2006; Caldovic et al. 2015). This known pathogenic variant is predicted to eliminate the protein through nonsense-mediated mRNA decay. The same mutation was originally identified in a symptomatic Korean female patient with several male relatives who had died of presumptive OTCD (Yoo et al. 1996). Maternal testing for the R320X variant was negative.
On follow up at 20 months of age, she was making good developmental progress. Assessment with the Bayley Scales of Infant and Toddler Development (Third Edition) showed cognitive abilities within the average range, although she was mildly delayed in all areas. On the Bayley Cognitive Index, she attained a composite score of 85, with skills at approximately a 17-month level. On the Language Composite scale, she received a composite score of 89, with skills at the 16-month level in receptive communication and at the 19-month level in expressive communication. On the Bayley Motor Composite, she received a score of 73, with gross motor skills at the 14-month level and fine motor skills at the 12-month level. At 3 years and 4 months of age, assessment with the Wechsler Preschool and Primary Scale of Intelligence (Fourth Edition) composite scores showed average cognitive abilities (scores 85–115 represent average). Her composite score for full scale IQ was 109 with a verbal comprehension score of 114, a visual spatial score of 112, and a working memory score of 87.
Discussion
The variable clinical expressivity of male and female OTCD patients can make diagnosis challenging. Although liver failure is a known complication of OTCD, acute liver failure as the initial presentation of OTCD is uncommon, with a few reports in the medical literature presenting as young as 14 months old (Mustafa and Clarke 2006; Mira and Boles 2012). Liver involvement is present in more than half of the patients who present with symptomatic OTCD. In a historical cohort study of 49 patients with symptomatic OTCD at two centers in the United States with symptomatic OTC, 29% met criteria for acute liver failure, and three patients presented with acute liver failure (Gallagher et al. 2014). In another study cohort of Swiss patients with OTCD, 6 of 15 symptomatic female patients were found to have acute liver failure which was recurrent in 2 patients, and 6 of 9 male patients had acute liver failure (Laemmle et al. 2016).
The pathogenesis of liver injury in OTCD is unknown. Liver biopsy in previous cases has demonstrated acute hepatocellular injury with mild lobular necrosis (Gallagher et al. 2014). Reports of hepatocellular carcinoma in some patients with OTCD and other urea cycle disorders have raised concern that urea cycle disorders increase the risk of liver cancer due to chronic liver damage by toxic metabolites, and perhaps to the toxicity of excess carbamoyl phosphate, which leads to either high energy phosphate toxicity or to diversion into the pyrimidine synthesis pathway, resulting in dysregulation of the nucleotide pool (Wilson et al. 2012). In the recent report from Laemmle et al. the authors hypothesized that ammonia toxicity contributes directly to the development of acute liver failure by impairing hepatic protein synthesis with in vitro studies of hepatocytes from unaffected donors. The cell culture studies demonstrated reduced production rate of albumin upon ammonium chloride (NH4Cl) treatment, and a moderate increase of AST indicating a negative effect of ammonia on mitochondrial integrity (Laemmle et al. 2016). Our patient’s treatment with steroids may have precipitated her crisis with hyperammonemia (Gascon-Bayarri et al. 2015; Lipskind et al. 2011).
Our patient’s initial presentation with right-arm weakness was also unusual. Acute focal neurologic deficits are atypical in urea cycle disorders, but have been described in urea cycle disorders with neurologic symptoms occurring both with and without acute hyperammonemia (Keegan et al. 2003). Keegan et al. presented a 6-year-old male with partial OTCD who presented with ataxia and dysarthria that resolved after treatment. In a review of neurologic outcomes of 28 patients with OTCD, 7 patients had long-term neurologic involvement on examination; 2 with spastic quadriparesis, 4 with mixed dystonic/spastic hemiplegia, and 1 with spastic diplegia (Nicolaides et al. 2002).
The diagnosis of a urea cycle disorder should be considered in patients with unexplained hepatic dysfunction. Older infants may even present with liver failure. Under these circumstances, any patient who has had an appropriate but unsuccessful medical evaluation for common etiologies of liver failure should undergo a systematic metabolic investigation including measurement of plasma amino acid levels. Ongoing unexplained hepatic dysfunction should prompt consideration of genetic evaluation for urea cycle disorders. Recognition of a urea cycle disorder as a component of the differential diagnosis can prompt early metabolic workup and diagnosis to initiate specific lifesaving metabolic treatment. Elevated glutamine associated with an increased waste nitrogen burden may precede clinical symptoms of hyperammonemia (Brusilow and Horwich 2014). The increase in glutamine and alanine associated with hyperammonemia can be an important diagnostic clue since elevation in these amino acids may precede hyperammonemia and the onset of clinical signs and symptoms.
Synopsis
Urea cycle disorders can present with hepatic failure, and early recognition of a urea cycle disorder in the differential of hepatic dysfunction can prompt early metabolic diagnosis to initiate specific lifesaving metabolic treatment.
Contributions of Individual Authors
Farrah Rajabi drafted the initial manuscript and approved the final manuscript as submitted.
Lance H. Rodan and Gerard T. Berry assisted in drafting, and revised the manuscript and approved the final manuscript as submitted.
Maureen M. Jonas, Janet Soul, Nicole J. Ullrich, Ann Wessel, Susan E. Waisbren, and Wen-Hann Tan reviewed and revised the manuscript and approved the final manuscript as submitted.
Compliance with Ethics Guidelines
Farrah Rajabi, Lance Rodan, Maureen M. Jonas, Janet Soul, Nicole J. Ullrich, Ann Wessel, Susan E. Waisbren, Wen-Hann Tan, and Gerard T. Berry declare that they have no conflict of interest.
This is a descriptive report, and all patient information has been anonymized to ensure patients are not identifiable. All patients have provided informed consent to genetic testing. This chapter does not contain any studies with human or animal subjects performed by any of the authors.
Contributor Information
Gerard T. Berry, Email: Gerard.Berry@childrens.harvard.edu
Collaborators: Matthias Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Batshaw ML, Msall M, Beaudet AL, Trojak J. Risk of serious illness in heterozygotes for ornithine transcarbamylase deficiency. J Pediatr. 1986;108:236–241. doi: 10.1016/S0022-3476(86)80989-1. [DOI] [PubMed] [Google Scholar]
- Brusilow SW, Horwich AL. Urea cycle enzymes. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G, editors. The online metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2014. [Google Scholar]
- Caldovic L, Abdikarim I, Narain S, Tuchman M, Morizono H. Genotype-phenotype correlations in ornithine transcarbamylase deficiency: a mutation update. J Genet Genomics. 2015;42:181–194. doi: 10.1016/j.jgg.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demmer LA, Kim JM, de Martinville B, Dowton SB. A novel missense mutation in the exon containing the putative ornithine-binding domain of the OTC enzyme in a female. Hum Mutat. 1996;7:279. doi: 10.1002/(SICI)1098-1004(1996)7:3<279::AID-HUMU16>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Gallagher RC, Lam C, Wong D, Cederbaum S, Sokol RJ. Significant hepatic involvement in patients with ornithine transcarbamylase deficiency. J Pediatr. 2014;164:720–725.e6. doi: 10.1016/j.jpeds.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascon-Bayarri J, Campdelacreu J, Estela J, Rene R. Severe hyperammonemia in late-onset ornithine transcarbamylase deficiency triggered by steroid administration. Case Rep Neurol Med. 2015;2015:453752. doi: 10.1155/2015/453752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan CE, Martin DM, Quint DJ, Gorski JL. Acute extrapyramidal syndrome in mild ornithine transcarbamylase deficiency: metabolic stroke involving the caudate and putamen without metabolic decompensation. Eur J Pediatr. 2003;162:259–263. doi: 10.1007/s00431-002-1135-1. [DOI] [PubMed] [Google Scholar]
- Kim G-H, Choi J-H, Lee H-H, Park S, Kim S-S, Yoo H-W. Identification of novel mutations in the human ornithine transcarbamylase (OTC) gene of Korean patients with OTC deficiency and transient expression of the mutant proteins in vitro. Hum Mutat. 2006;27:1159. doi: 10.1002/humu.9465. [DOI] [PubMed] [Google Scholar]
- Laemmle A, Gallagher RC, Keogh A, Stricker T, Gautschi M, Nuoffer J-M, Baumgartner MR, Häberle J. Frequency and pathophysiology of acute liver failure in ornithine transcarbamylase deficiency (OTCD) PLoS One. 2016;11:e0153358. doi: 10.1371/journal.pone.0153358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipskind S, Loanzon S, Simi E, Ouyang D. Hyperammonemic coma in an ornithine transcarbamylase mutation carrier following antepartum corticosteroids. J Perinatol. 2011;31:682–684. doi: 10.1038/jp.2011.23. [DOI] [PubMed] [Google Scholar]
- Mira V, Boles RG. Liver failure with coagulopathy, hyperammonemia and cyclic vomiting in a toddler revealed to have combined heterozygosity for genes involved with ornithine transcarbamylase deficiency and Wilson disease. JIMD Rep. 2012;3:1–3. doi: 10.1007/8904_2011_70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa A, Clarke JTR. Ornithine transcarbamoylase deficiency presenting with acute liver failure. J Inherit Metab Dis. 2006;29:586. doi: 10.1007/s10545-006-0303-2. [DOI] [PubMed] [Google Scholar]
- Nicolaides P, Liebsch D, Dale N, Leonard J, Surtees R. Neurological outcome of patients with ornithine carbamoyltransferase deficiency. Arch Dis Child. 2002;86:54–56. doi: 10.1136/adc.86.1.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuchman M, Lee B, Lichter-Konecki U, Summar ML, Yudkoff M, Cederbaum SD, Kerr DS, Diaz GA, Seashore MR, Lee H-S, McCarter RJ, Krischer JP, Batshaw ML. Cross-sectional multicenter study of patients with urea cycle disorders in the United States. Mol Genet Metab. 2008;94:397–402. doi: 10.1016/j.ymgme.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JM, Shchelochkov OA, Gallagher RC, Batshaw ML. Hepatocellular carcinoma in a research subject with ornithine transcarbamylase deficiency. Mol Genet Metab. 2012;105:263–265. doi: 10.1016/j.ymgme.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo HW, Kim GH, Lee DH. Identification of new mutations in the ornithine transcarbamylase (OTC) gene in Korean families. J Inherit Metab Dis. 1996;19:31–42. doi: 10.1007/BF01799346. [DOI] [PubMed] [Google Scholar]
- Yorifuji T, Muroi J, Uematsu A, Tanaka K, Kiwaki K, Endo F, Matsuda I, Nagasaka H, Furusho K. X-inactivation pattern in the liver of a manifesting female with ornithine transcarbamylase (OTC) deficiency. Clin Genet. 1998;54:349–353. doi: 10.1034/j.1399-0004.1998.5440415.x. [DOI] [PubMed] [Google Scholar]