

Abstract
Familial lecithin-cholesterol acyltransferase deficiency (FLD) is a rare recessive disorder of cholesterol metabolism, caused by loss-of-function mutations in the human LCAT gene, leading to alterations in the lipid/lipoprotein profile, with extremely low HDL levels.
The classical FLD phenotype is characterized by diffuse corneal opacification, haemolytic anaemia and proteinuric chronic kidney disease (CKD); an incomplete form, only affecting the corneas, has been reported in a few families worldwide.
We describe an intermediate phenotype of LCAT deficiency, with CKD preceding the development of corneal clouding, in two Portuguese brothers apparently homozygous for a novel missense LCAT gene mutation. The atypical phenotype, the diagnosis of membranous nephropathy in the proband’s native kidney biopsy, the late-onset and delayed recognition of the corneal opacification, the co-segregation with Gilbert syndrome and the late recurrence of the primary disease in kidney allograft all contributed to obscure the diagnosis of an LCAT deficiency syndrome for many years.
A major teaching point is that on standard light microscopy examination the kidney biopsies of patients with LCAT deficiency with residual enzyme activity may not show significant vacuolization and may be misdiagnosed as membranous nephropathy. The cases of these two patients also illustrate the importance of performing detailed physical examination in young adults presenting with proteinuric CKD, as the most important clue to the diagnosis of FLD is in the eyes.
Keywords: Corneal clouding, Familial lecithin-cholesterol acyltransferase deficiency, Familial lecithin-cholesterol acyltransferase deficiency-associated nephropathy, Gilbert syndrome, Kidney transplantation
Introduction
Lecithin-cholesterol acyltransferase (LCAT) is a plasma enzyme essential for the esterification of free cholesterol (Kuivenhoven et al. 1997; Kunnen and Van Eck 2012). LCAT reacts with preβ1-HDL containing apolipoprotein A-I (apoA1), where it esterifies free cholesterol via α-LCAT activity; a lesser amount of the enzyme circulates bound to apolipoprotein B in low-density lipoproteins (LDL) and very low-density lipoproteins (VLDL), where it esterifies cholesterol via β-LCAT activity. LCAT also plays an important role in the reverse cholesterol transport pathway.
The LCAT gene is located on chromosome 16q22.1 and comprises 6 exons (OMIM*606967; http://omim.org/entry/606967). LCAT mutations cause two very rare autosomal recessive disorders: Familial LCAT Deficiency (FLD or Norum disease; OMIM#245900; http://omim.org/entry/245900), and Fish-Eye Disease (FED; OMIM#136120; http://omim.org/entry/136120). The Human Gene Mutation Database (HGMD®) currently compiles 102 functionally relevant LCAT variants, including 77 missense/nonsense point mutations (http://www.hgmd.cf.ac.uk; last accessed on August 1, 2017).
FLD is characterized by diffuse corneal opacification, haemolytic anaemia, proteinuria and chronic kidney disease (CKD) (Kuivenhoven et al. 1997; Santamarina-Fojo et al. 2001). Corneal opacities arise in childhood and worsen with age, eventually causing severe sight impairment, sometimes requiring corneal transplantation. Proteinuria develops early, but azotaemia is usually detected only after the second decade of life, progressing to nephrotic proteinuria and end-stage renal disease (ESRD) by the fourth decade. The FLD-associated nephropathy also recurs in kidney allografts (Panescu et al. 1997; Strom et al. 2011; Hui Liew et al. 2016). In FLD, α- and β-LCAT activities are suppressed, leading to high levels of unesterified cholesterol and extremely low levels of high-density lipoproteins (HDL). LDL levels are also low, with normal or increased triglyceride levels. Cholesterol-laden foam-cells and membrane-bound vesicles accumulate in the corneas, kidneys, liver, spleen, bone marrow and arteries. However, hepatomegaly, splenomegaly and lymphadenopathy have been rarely reported and the risk for coronary heart disease is only modestly increased (Santamarina-Fojo et al. 2001; Calabresi et al. 2009).
FED presents a milder phenotype, with predominant involvement of the cornea, without anaemia or CKD. Plasma triglyceride levels are normal to increased and HDL is decreased, due to a partial deficiency of α-LCAT activity (Kuivenhoven et al. 1997). Since β-LCAT activity is preserved, the cholesterol esterification rate and the percentage of cholesteryl esters in plasma are normal. Intermediate, atypical phenotypes have also been described (Kuivenhoven et al. 1997).
The lipid/lipoprotein profile is indistinguishable between subjects classified as FLD or FED, and significantly low levels of HDL and apoA1 are a hallmark of both disorders (Calabresi et al. 2005).
Although the pathogenesis of glomerulosclerosis and progressive CKD in FLD is not well understood, renal accumulation of lipoprotein-X (Lp-X) is probably a major factor contributing to the development of glomerular basement membrane (GBM) and endothelial damage, podocyte effacement, expansion of the mesangial matrix and renal tubule vacuolation. Lp-X is a multilamellar vesicle enriched in free cholesterol, and relatively devoid of cholesterol esters, triglycerides and apolipoproteins; notably, due to the residual activity of LCAT, it does not accumulate in FED. In cell culture studies, Lp-X was found to be cytotoxic and pro-inflammatory (Lynn et al. 2001), and its chronic administration to LCAT−/− knockout (KO) mice results in accumulation of Lp-X in the kidney, recapitulating most of the renal pathological hallmarks of FLD and the onset of proteinuria (Ossoli et al. 2016).
Genotype–phenotype correlations have been difficult to establish, since affected relatives may have different clinical and biochemical manifestations (Calabresi et al. 2005). Diagnosis of LCAT deficiency may be difficult to recognize, particularly in patients with atypical phenotypes.
Herein, we report on two brothers presenting with an intermediate phenotype of LCAT deficiency-associated with a novel LCAT gene mutation, in which the interpretation of the biochemical laboratory data was confounded by co-segregation of LCAT deficiency with Gilbert syndrome.
Case Reports and Family Data
Patient 1: The Proband
Hypertension and proteinuric CKD were incidentally recognized in this patient at age 26 years, warranting referral to a nephrology clinic for further assessment. The baseline laboratory workup showed moderate normochromic/normocytic anaemia, mild thrombocytopenia, azotaemia (plasma creatinine (pCr): 1.6 mg/dL), erythrocyturia and nephrotic proteinuria (6.88 g/24 h), with normal results of protocol immunological and serological testing. Membranous nephropathy was suggested by kidney biopsy, but its evaluation had been restricted to routine light microscopy (LM). Although the proband had a mildly intellectually disabled older brother who also suffered from advanced CKD with massive proteinuria (Fig. 1), investigation for an underlying hereditary disorder was not pursued.
Fig. 1.
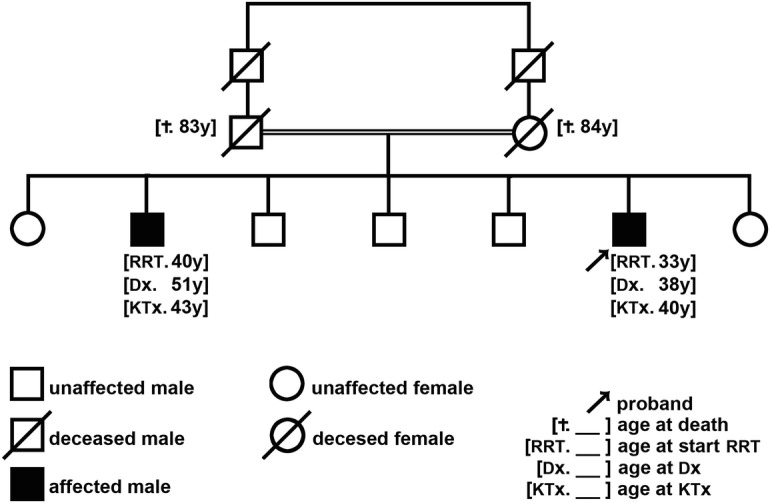
Family pedigree. The proband is the youngest male of 7 sibs, whose parents were first-cousins. Five of the sibs reported no health problems. Only the mother and the older sister accepted to be screened for manifestations of LCAT deficiency and CKD: none of them showed corneal clouding and their laboratory workups were entirely normal, including the peripheral blood counts as well as the serum lipid profile and the apolipoprotein A–I level. None of the patients’ relatives accepted genetic screening. Both parents eventually died aged over 80. Ages are reported in full years. y years, RRT renal replacement therapy, KTx kidney transplantation, Dx LCAT deficiency diagnosis, † death
Regular haemodialysis treatment was started at age 33 and, 5 years afterwards, following notice of progressive clouding of both corneas causing visual impairment and referral for ophthalmological examination (Fig. 2a), the diagnosis of LCAT deficiency (Fig. 2b) was eventually established. The patient’s lipid and lipoprotein profile was characterized by low levels of HDL and apoA1, with a normal ratio of free to esterified cholesterol (Fig. 2c). In addition, persistent unconjugated hyperbilirubinemia with normal plasma levels of other liver function tests, lactate dehydrogenase and haptoglobin, an unremarkable peripheral blood smear and negative Coombs test, was suggestive of Gilbert syndrome. The liver and spleen were not palpable by physical examination and presented normal sizes on the ultrasound scan.
Fig. 2.

(a) Dystrophic corneal opacity; (b) DNA electropherogram of the relevant sequence showing apparent homozygosity for a c.803G>T transition in exon 6, leading to a novel non-conservative substitution of arginine by leucine on amino acid position 268 of the LCAT protein – i.e., p.(Arg268Leu); (c) Summary of the lipid profiles of the patients and some of their relatives
Sequence analysis of all the exons and corresponding exon–intron boundaries of the LCAT gene revealed a c.803G>T transition in exon 6, leading to a novel non-conservative substitution of arginine by leucine on amino acid position 268 of the LCAT protein – i.e., p.(Arg268Leu) – apparently in homozygosity. In addition, genetic analysis of the 5′ promoter region of the uridine 5′-diphospho-glucuronosyltransferase gene (UGT1A1) showed (apparent) homozygosity for the longer A(TA)7TAA variant of the TATAA element, confirming the diagnosis of Gilbert syndrome.
At age 40, the patient received a deceased-donor kidney allograft. The post-transplant period was uneventful for several years, with stable graft function (pCr: 1 mg/dL) and without anaemia, proteinuria or microscopic haematuria, under maintenance triple-drug immunosuppression with prednisolone, tacrolimus and mycophenolate mofetil. The abnormal lipid profile and unconjugated hyperbilirubinemia did not resolve after kidney transplantation and a bone marrow aspirate, obtained for the investigation of unremitting thrombocytopenia, did not show any relevant abnormalities, including foam cells or sea-blue histiocytes. Noncontact in vivo confocal laser scanning microscopy for high resolution imaging of all corneal layers revealed diffuse hyperreflectivity of the corneal stroma, due to coalescence of multiple hyperreflective spots (Fig. 3a, b).
Fig. 3.

(a, b) Multiple dark striae and diffuse hyperreflectivity; (c) Enlargement of the mesangium, slightly thickened GBM and foci of interstitial fibrosis with atrophic tubules (LM, by haematoxylin and eosin stain (HES) 100×); (d) Deposits of heterogeneous electrondense lipid material were located on the subendothelial side of the GBM and in the mesangium (EM, 4,000×); (e) Thickening and double contour of the capillary walls of the glomeruli, with vacuolated mesangial matrix (LM, HES 100×); (f) Subendothelial, intramembranous and mesangial heterogeneous electrondense lipid material, surrounded by an electro-lucent zone (EM, 4,000×)
Recurrence of haematuria and proteinuria (1.7 g/24 h) supervened during the fifth year post-transplant, prompting an allograft biopsy, whilst the pCr concentration remained within normal range. The transplant kidney biopsy showed the typical histopathological and ultrastructural features of FLD-associated nephropathy (Fig. 3c, d). Treatment with a renin-angiotensin-aldosterone system (RAAS) inhibitor led to partial resolution of proteinuria, but the haematuria persisted and mild azotaemia (pCr: 1.4 mg/dL) was detected for the first time.
Case 2: The Proband’s Older Brother
When the diagnosis of LCAT deficiency was established in the proband, his older brother, then aged 51, was on the eighth year post-kidney transplantation, undergoing maintenance triple-drug immunosuppression with prednisolone, cyclosporine and mycophenolate mofetil.
The patient’s previous renal history was relevant for high blood pressure known since his mid-thirties followed, at age 38, by diagnosis of CKD (pCr: 2.5 mg/dL) with nephrotic syndrome. Kidney biopsy was not performed due to the advanced renal disease. Additional relevant baseline laboratory findings included moderate normochromic/normocytic anaemia and mild thrombocytopenia. Bilateral, slight corneal clouding had already been noticed, but did not prompt appropriate ophthalmological evaluation. Regular haemodialysis treatment was initiated 2 years after, and at the age of 43, the patient received a deceased-donor kidney allograft.
The diagnosis of LCAT deficiency in his younger brother led to a targeted review of the patient’s medical records, which showed a serum lipid profile similar to that of the proband (Fig. 1c), as well as persistent mild thrombocytopenia and unconjugated hyperbilirubinemia, without other blood cytopaenias, evidence of chronic liver disease or of chronic haemolysis. Sequencing analyses of the LCAT exon 6 and of the UGT1A1 5′ promoter region revealed the same genetic makeup as in his brother.
During the 12th post-transplantation year, the new development of microscopic haematuria and proteinuria (3.69 g/24 h), without allograft dysfunction (pCr: 0.97 mg/dL), prompted a diagnostic allograft biopsy, which documented the recurrence of FLD-associated nephropathy (Fig. 3e, f), even before than in his younger brother.
The patient’s subsequent clinical course was marked by severe, badly controlled hypertension; persistent proteinuria, despite upward dose titration of the RAAS inhibitor; many hospitalizations due to infectious complications; and rapid decline of the allograft function, reaching a pCr of 1.62 mg/dL at 14th year post-transplantation.
Discussion
We describe an intermediate clinical phenotype of FLD in two brothers who are (apparently) homozygous for a novel LCAT missense variant, concordantly predicted to be deleterious on bioinformatic analyses using the online software tools MutationTaster (http://www.mutationtaster.org), Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org/). Although parental heterozygosity for the p.(Arg268Leu) LCAT variant could not be directly demonstrated, their consanguineous condition and the rarity of LCAT deficiency in the general population are strong indirect evidence in favour of homozygosity. A further argument against the alternative explanation of compound heterozygosity with a large deletion involving the exon 6 of LCAT, not identifiable on routine DNA sequencing, is that no such type of mutation has so far been reported to the HGMD®. Three other missense mutations, all of them described in patients presenting with FLD, have been reported to affect the same LCAT codon – p.(Arg268Gly) (Skretting et al. 1992), p.(Arg268His) (Calabresi et al. 2005) and p.(Arg268Cys) (Charlton-Menys et al. 2007).
Our patients presented with clinical features of renal involvement that are typical of FLD. The significant age difference at the beginning of haemodialysis illustrates the phenotypic variation of LCAT deficiency within affected families (Skretting et al. 1992). However, the late presentation of corneal opacification and the absence of haemolytic anaemia are not typical of FLD and instead suggest an attenuated phenotype. Moreover, the normal serum ratio of free to esterified cholesterol observed in the proband is indicative of a significant level of residual LCAT activity. The mild, nonspecific intellectual disability manifested by his brother is not attributable to the enzyme deficiency (Santamarina-Fojo et al. 2001).
In vivo confocal microscopy demonstrated the presence of the characteristic corneal opacities in LCAT deficiency: multiple dark striae and hyperreflective deposits diffusely present throughout the stroma, corresponding to excessive extracellular deposition of cholesterol. These findings allow the differential diagnosis with other metabolic corneal dystrophies (Palmiero et al. 2009).
Persistence of unconjugated hyperbilirubinemia despite the complete recovery of anaemia observed in both patients after kidney transplantation, and the lack of any biochemical or haematological markers of chronic haemolysis, led us to consider the differential diagnosis of Gilbert syndrome. This was indeed confirmed by molecular analysis of the 5′ promoter region of UGT1A1, as the two sibs were found to carry the most common genotype associated with Gilbert syndrome in the Caucasian population.
Thrombocytopenia was the only haematological abnormality that did not correct after kidney transplantation and, in the absence of hypersplenism or bone marrow abnormalities, its cause remained elusive. Although thrombocytopenia is not a feature of the classical phenotype of FLD, it has been reported in association with partial deficiency of plasma LCAT activity, in a boy with a peculiar disease phenotype (De Buyzere et al. 1992), without noticeable corneal involvement up to the age of 20 years.
On LM examination of kidney biopsies of FLD patients, the typical presentation is a glomerulopathy with prominent accumulation of lipid-containing, vacuolated foam cells in capillaries and the mesangium (Hirashio et al. 2014). The mesangial matrix is often expanded, and segmental sclerosis is present in more advanced cases. The glomerular basement membrane (GBM) is thickened, with segmental areas of vacuolated appearance; on silver-stained sections, GBM resembles late-stage membranous nephropathy (Hirashio et al. 2014). Unfortunately, the proband’s native kidney biopsy was not examined by electron microscopy (EM), and the paraffin-embedded fragments were no longer available for review. However, as it is quite unlikely that the striking glomerular foam cell infiltrate characteristic of FLD was overlooked on the original LM examination, we hypothesize that in patients with residual LCAT activity and kidney involvement, the major histopathological feature might be a membranous nephropathy. This is in line with the report of a patient presenting with nephrotic syndrome caused by immune-mediated acquired LCAT deficiency (Takahashi et al. 2013), whose baseline kidney biopsy exhibited glomerular lesions similar to those of FLD, together with changes of membranous nephropathy. A follow-up kidney biopsy, obtained 5 months after the initiation of steroids and clinical improvement with serum LCAT activity normalization, showed a marked reduction in the glomerular foam cells, and improvement of the mesangial lesions, but persistence of diffuse GBM thickening.
Lp-X stimulates monocyte infiltration of the glomeruli via a mechanism involving mesangial monocyte chemoattractant protein-1 (MCP-1/CCL2) expression (Lynn et al. 2001). The upregulation of MCP-1 mRNA expression and the increased activity of the proinflammatory nuclear factor kappa B (NF-κB) transcription factor in mesangial cells suggest that Lp-X induces inflammatory response in those cells (Lynn et al. 2001). In the LCAT knockout mouse model, only those in which Lp-X was detected developed proteinuria and glomerulosclerosis (Lambert et al. 2001). Therefore, the immunosuppressive and anti-inflammatory actions of steroids, including the repression of the activity of NF-κB, might antagonize the proinflammatory action of Lp-X in glomerular cells and the pathogenesis of the FLD nephropathy. Combined treatment with nicotinic acid and fenofibrate has been reported to decrease plasma Lp-X levels and the urine albumin/creatinine ratio in a patient with FLD (Yee et al. 2009). In a report of a patient with FLD followed-up for 5 years, lipid-lowering drugs and angiotensin II receptor blockers showed benefit in blood pressure, lipid profile, proteinuria and kidney function (Aranda et al. 2008).
The role of LCAT in the development of atherosclerosis and cardiovascular disease remains incompletely understood. The paradoxical low atherosclerotic risk in LCAT deficiency (Calabresi et al. 2005, 2009), particularly in FDL, has been partly explained by kinetic studies that demonstrated an increased catabolism of LDL and by an up-regulation of the LDL receptor pathway (Nishiwaki et al. 2006).
Currently, there is no specific treatment available for LCAT deficiency but the infusion of recombinant human LCAT (rhLCAT) into mouse models of LCAT deficiency rapidly restored the normal lipoprotein phenotype in LCAT-KO mice and increased cholesterol efflux, suggesting that it might be used as an enzyme replacement therapy (ERT) agent for LCAT deficiency (Rousset et al. 2010). Furthermore, incubation of plasma obtained from FLD subjects and from healthy controls with rhLCAT led to normalization of the lipid/lipoprotein profile in the former, while no major changes were observed in the latter (Simonelli et al. 2013).
A phase 1b, open-label, single-dose escalation study of rhLCAT therapy in subjects with stable CHD and low HDL demonstrated favourable, dose-dependent pharmacodynamic effects on HDL metabolism, with acceptable safety and tolerability (Shamburek et al. 2016a, b). Also encouraging were the beneficial changes in clinical, biochemical and lipoprotein parameters observed in a single patient with FLD and advanced CKD, during an 8-month course of ERT, supporting continued development of rhLCAT therapy (Shamburek et al. 2016a, b) and highlighting the role of Lp-X as a possible biomarker for its monitoring (Shamburek et al. 2016a, b).
Whether rhLCAT therapy is effective to halt CKD progression if started at an earlier stage of the disease will have to be carefully assessed in future clinical trials. Until then, and since ESRD is the major cause of morbidity and mortality in patients with FLD (Myhre et al. 1977), renoprotective therapy by optimal control of blood pressure and reduction of proteinuria using drugs acting on the RAAS should be instituted as soon as the diagnosis of FLD is established. Although anecdotal evidence (Miarka et al. 2011) suggests that corticosteroid treatment may delay the progression of the FLD-associated nephropathy in the native kidneys, its recurrence in the allograft has been consistently reported in kidney-transplanted FLD patients (Panescu et al. 1997; Strom et al. 2011; Hui Liew et al. 2016), even though corticosteroids are standard therapy in kidney transplantation.
In conclusion, we describe an incomplete clinical and biochemical FLD phenotype, followed by histologically confirmed recurrence of the primary kidney disease in the renal allograft, in two sibs who are most probably homozygous for a novel LCAT missense variant. The atypical phenotype, the delayed recognition and semiological valuing of the corneal opacification, the coexistence of Gilbert syndrome, the diagnosis of membranous nephropathy in the proband’s native kidney biopsy, as well as late allograft recurrence of the primary disease, all contributed to obscure the diagnosis of an LCAT deficiency syndrome for many years.
A major teaching point is that on standard LM examination the kidney biopsies of patients with LCAT deficiency and residual enzyme activity, presenting with nephrotic proteinuria and CKD, may not show significant vacuolization and may be misdiagnosed as membranous nephropathy. The cases of these two patients also illustrate the importance of performing detailed physical examination in young adults presenting with proteinuric CKD, as the most important clue to the diagnosis of FLD is in the eyes (Kettritz et al. 2009). Pending the availability of ERT, the mainstay of treatment is intensive blood pressure control and minimization of proteinuria, using RAAS blocking drugs.
Acknowledgements
We thank the patients and their mother and older sister for having participated in this study and consented to its publication.
The LCAT gene mutational analysis of the proband was performed at GENDIA (GENetic DIAgnostic Network; http://www.gendia.net/), purchased as an outsource laboratory service.
The UGT1A1 promoter sequence analysis was performed at the “Instituto de Genética Médica Jacinto Magalhães” (Porto, Portugal), purchased as an outsource laboratory service.
Take Home Message
LCAT deficiency diagnosis may be misleading, particularly in atypical phenotypes, highlighting the importance of a detailed physical examination in patients presenting with proteinuric CKD, as the most important clue to the diagnosis of FLD is in the eyes.
Details of the Contributions of Individual Authors
- Dr. Inês Castro Ferreira
provided clinical data and drafted the manuscript;
- Dr. Rute Carmo
performed the kidney biopsy and drafted the manuscript;
- Dr. Sérgio Estrela Silva
performed the ophthalmological examinations and provided clinical data and the relevant photographs;
- Dr. Otília Corrêa
performed the lipid and lipoprotein analyses;
- Dr. Susana Fernandes
performed the LCAT genetic analysis and provided the electropherogram;
- Dr. Susana Sampaio
performed histopathological evaluation of kidney allograft biopsies and provided the relevant illustrations/photographs;
- Dr. Pedro Rodrigues Pereira
performed histopathological evaluation of kidney allograft biopsies and provided the relevant illustrations/photographs;
- Dr. Augusta Praça
provided clinical data;
- Prof. Dr. João Paulo Oliveira
provided clinical data and drafted the manuscript and is the guarantor for the chapter.
Corresponding Author
I. Castro-Ferreira.
E-mail: inescastroferreira@sapo.pt.
Details of Funding
None.
Compliance with Ethics Guidelines
Conflict of Interest
Inês Castro Ferreira, Rute Carmo, Sérgio Estrela Silva, Otília Corrêa, Susana Fernandes, Susana Sampaio, Pedro Rodrigues Pereira, Augusta Praça and João Paulo Oliveira declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients for being included in the study.
Ethical Standards
There are no identifiable patient’s personal data on this manuscript.
The publication of this manuscript was approved by the Committee on Ethics for Health of the Centro Hospitalar São João/Faculdade de Medicina da Universidade do Porto.
Contributor Information
I. Castro-Ferreira, Email: inescastroferreira@sapo.pt
Rute Carmo, Email: rute.carvalho.carmo@gmail.com.
Sérgio Estrela Silva, Email: sestrelasilva@gmail.com.
Otília Corrêa, Email: Otilia.correa@labco.eu.
Susana Fernandes, Email: sf@med.up.pt.
Susana Sampaio, Email: susana.sampaio@sapo.pt.
Rodrigues-Pereira Pedro, Email: pe_r_pereira@hotmail.com.
Augusta Praça, Email: augustapraca@gmail.com.
João Paulo Oliveira, Email: jpo@med.up.pt.
Collaborators: Matthias Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
Reference
- Aranda P, Valdivielso P, Pisciotta L, et al. Therapeutic management of a new case of LCAT deficiency with a multifactorial long-term approach based on high doses of angiotensin II receptor blockers (ARBs) Clin Nephrol. 2008;69:213–218. doi: 10.5414/CNP69213. [DOI] [PubMed] [Google Scholar]
- Calabresi L, Pisciotta L, Costantin A, et al. The molecular basis of lecithin-cholesterol acyltransferase deficiency syndromes: a comprehensive study of molecular and biochemical findings in 13 unrelated Italian families. Arterioscler Thromb Vasc Biol. 2005;25:1972–1978. doi: 10.1161/01.ATV.0000175751.30616.13. [DOI] [PubMed] [Google Scholar]
- Calabresi L, Baldassarre D, Castelnuovo S, et al. Functional lecithin-cholesterol acyltransferase is not required for efficient atheroprotection in humans. Circulation. 2009;120:628–635. doi: 10.1161/CIRCULATIONAHA.108.848143. [DOI] [PubMed] [Google Scholar]
- Charlton-Menys V, Pisciotta L, Durrington PN, et al. Molecular characterization of two patients with severe LCAT deficiency. Nephrol Dial Transplant. 2007;22:2379–2382. doi: 10.1093/ndt/gfm311. [DOI] [PubMed] [Google Scholar]
- De Buyzere M, Delanghe J, Labeur C, et al. Acquired hypolipoproteinemia. Clin Chem. 1992;38:776–781. [PubMed] [Google Scholar]
- Hirashio S, Ueno T, Naito T, Masaki T. Characteristic kidney pathology, gene abnormality and treatments in LCAT deficiency. Clin Exp Nephrol. 2014;18:189–193. doi: 10.1007/s10157-013-0895-4. [DOI] [PubMed] [Google Scholar]
- Kettritz R, Elitok S, Koepke ML, Kuchenbecker J, Schneider W, Luft FC. The case: the eyes have it! Kidney Int. 2009;76:465–466. doi: 10.1038/ki.2009.235. [DOI] [PubMed] [Google Scholar]
- Kuivenhoven JA, Pritchard H, Hill J, et al. The molecular pathology of lecithin-cholesterol acyltransferase (LCAT) deficiency syndromes. J Lipid Res. 1997;38:191–205. [PubMed] [Google Scholar]
- Kunnen S, Van Eck M. Lecithin-cholesterol acyltransferase: old friend or foe in atherosclerosis? J Lipid Res. 2012;53:1783–1799. doi: 10.1194/jlr.R024513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert G, Sakai N, Vaisman BL, et al. Analysis of glomerulosclerosis and atherosclerosis in lecithin-cholesterol acyltransferase deficient mice. J Biol Chem. 2001;276:15090–15098. doi: 10.1074/jbc.M008466200. [DOI] [PubMed] [Google Scholar]
- Liew H, Simpson I, Kanellis J, Mulley WR. Recurrent glomerulopathy in a renal allograft due to lecithin-cholesterol acyltransferase deficiency. Nephrology (Carlton) 2016;21(1):73–74. doi: 10.1111/nep.12554. [DOI] [PubMed] [Google Scholar]
- Lynn EG, Siow YL, Frohlich J, Cheung GT, O K. Lipoprotein-X stimulates monocyte chemoattractant protein-1 expression in mesangial cells via nuclear factor-kappa B. Kidney Int. 2001;60:520–532. doi: 10.1046/j.1523-1755.2001.060002520.x. [DOI] [PubMed] [Google Scholar]
- Miarka P, Idzior-Waluœ B, KuŸniewski M, Waluœ-Miarka M, Klupa T, Sułowicz W. Corticosteroid treatment of kidney disease in a patient with familial lecithin-cholesterol acyltransferase deficiency. Clin Exp Nephrol. 2011;15:424–429. doi: 10.1007/s10157-011-0409-1. [DOI] [PubMed] [Google Scholar]
- Myhre E, Gjone E, Flatmark A, Hovig T. Renal failure in familial lecithin-cholesterol acyltransferase deficiency. Nephron. 1977;18:239–248. doi: 10.1159/000180835. [DOI] [PubMed] [Google Scholar]
- Nishiwaki M, Ikewaki K, Bader G, et al. Human lecithin-cholesterol acyltransferase deficiency: in vivo kinetics of low-density lipoprotein and lipoprotein-X. Arterioscler Thromb Vasc Biol. 2006;26:1370–1375. doi: 10.1161/01.ATV.0000217910.90210.99. [DOI] [PubMed] [Google Scholar]
- Ossoli A, Neufeld EB, Thacker SG, et al. Lipoprotein X causes renal disease in LCAT deficiency. PLoS One. 2016;11(2):e0150083. doi: 10.1371/journal.pone.0150083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmiero P-M, Sbeity Z, Liebmann J, Ritch R. In vivo imaging of the cornea in a patient with lecithin-cholesterol acyltransferase deficiency. Cornea. 2009;29:1061–1064. doi: 10.1097/ICO.0b013e31819839ae. [DOI] [PubMed] [Google Scholar]
- Panescu V, Grignon Y, Hestin D, et al. Recurrence of lecithin-cholesterol acyltransferase deficiency after kidney transplantation. Nephrol Dial Transplant. 1997;12:2430–2432. doi: 10.1093/ndt/12.11.2430. [DOI] [PubMed] [Google Scholar]
- Rousset X, Vaisman B, Auerbach B, et al. Effect of recombinant human lecithin-cholesterol acyltransferase infusion on lipoprotein metabolism in mice. J Pharmacol Exp Ther. 2010;335(1):140–148. doi: 10.1124/jpet.110.169540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamarina-Fojo S, Hoeg J, Assmann G, Brewer HBJ. Lecithin-cholesterol acyltransferase deficiency and fish eye disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 2817–2833. [Google Scholar]
- Shamburek RD, Bakker-Arkema R, Auerbach BJ, et al. Familial lecithin-cholesterol acyltransferase deficiency: first-in-human treatment with enzyme replacement. J Clin Lipidol. 2016;10(2):356–367. doi: 10.1016/j.jacl.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamburek RD, Bakker-Arkema R, Shamburek AM, et al. Safety and tolerability of ACP-501, a recombinant human lecithin-cholesterol acyltransferase, in a phase 1 single-dose escalation study. Circ Res. 2016;118(1):73–82. doi: 10.1161/CIRCRESAHA.115.306223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonelli S, Tinti C, Salvini L, et al. Recombinant human LCAT normalizes plasma lipoprotein profile in LCAT deficiency. Biologicals. 2013;41:446–449. doi: 10.1016/j.biologicals.2013.09.007. [DOI] [PubMed] [Google Scholar]
- Skretting G, Blomhoff JP, Solheim J, Prydz H. The genetic defect of the original Norwegian lecithin-cholesterol acyltransferase deficiency families. FEBS Lett. 1992;309:307–310. doi: 10.1016/0014-5793(92)80795-I. [DOI] [PubMed] [Google Scholar]
- Strom EH, Sund S, Reier-Nilsen M, Dorje C, Leren TP. Lecithin-cholesterol acyltransferase (LCAT) deficiency: renal lesions with early graft recurrence. Ultrastruct Pathol. 2011;35:139–145. doi: 10.3109/01913123.2010.551578. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Hiromura K, Tsukida M, et al. Nephrotic syndrome caused by immune-mediated acquired LCAT deficiency. J Am Soc Nephrol. 2013;24:1305–1312. doi: 10.1681/ASN.2012090913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee MS, Pavitt DV, Richmond W, et al. Changes in lipoprotein profile and urinary albumin excretion in familial LCAT deficiency with lipid lowering therapy. Atherosclerosis. 2009;205:528–532. doi: 10.1016/j.atherosclerosis.2008.11.033. [DOI] [PubMed] [Google Scholar]