

Abstract
Glutaric acidemia type 1 (GA-1, OMIM no. 231670) is an autosomal recessive disorder caused by the deficiency of glutaryl-CoA dehydrogenase (GCDH). The subsequent accumulation of the amino acids lysine, hydroxylysine, and tryptophan and their breakdown intermediates can be neurotoxic and particularly cause injury to the basal ganglia.
Roughly 1 of 100,000 infants is affected with GA-1, and a common feature at birth is macrocephaly. Stress, such as in febrile illnesses, can precipitate encephalopathic crises in children generally less than 2 years with variable recovery. Many infants develop dystonia with complex movement disorders and subtle cognitive and fine motor deficits. Common neuroradiologic findings include hypoplasia of temporal and frontal lobes, striatal lesions, white matter changes, and subdural effusions.
There are three previous reports of subependymal nodules found on neuroimaging in GA-1 patients who were diagnosed as adults and untreated for GA-1. We present a unique case of an adult female who was diagnosed at age 2 months and managed prior to any metabolic decompensation. Her initial diagnosis was made based on biochemical and enzymatic analysis, and then later confirmed with genetic sequencing. She started experiencing frequent headaches at age 12 years. Neuroimaging in adulthood revealed common features seen in GA-1 in addition to the finding of subependymal nodules.
This case may provide some insight into the natural progression of the disease despite early treatment. Though subependymal nodules are typically seen in tuberous sclerosis, the significance of these lesions in GA-1 is not well understood. Disease courses of more early diagnosed and treated patients with GA-1 need to be documented.
Keywords: GA-1, GCDH, Glutaric acidemia type 1, Glutaryl-CoA dehydrogenase deficiency, Subependymal nodules
Introduction
Glutaric acidemia type 1 (GA-1, OMIM no. 231670) is an inherited autosomal recessive disorder caused by glutaryl-CoA dehydrogenase (GCDH) deficiency and impairment in the breakdown of amino acids – lysine, hydroxylysine, and tryptophan. The subsequent accumulation of these amino acids and their breakdown intermediates, glutaric acid, glutaryl-CoA, 3-hydroxyglutaric acid, and glutaconic acid, can be neurotoxic and cause striatal injury, affecting the basal ganglia. Also, secondary carnitine deficiency results as a consequence of its consumption in metabolism of glutaric acid.
Roughly 1 of 100,000 infants worldwide is affected and often born with macrocephaly. Stress, such as in dehydration, surgery, reactions to vaccinations, or febrile illnesses, can exacerbate encephalopathic crises in children less than 2 years of age with variable recovery after each episode. Many infants develop dystonia with complex movement disorders and subtle cognitive and fine motor deficits. Commonly documented neuroradiologic findings include hypoplasia of temporal and frontal lobes, striatal lesions, white matter changes, and subdural effusions (Twomey et al. 2003). Though historically this disorder has been associated with the aforementioned central nervous system involvement, recent publications have suggested other organ damage in the peripheral nervous and renal systems (Herskovitz et al. 2013; Kölker et al. 2015).
With the advancement of newborn screening and early diagnosis, initiation of a low lysine diet, carnitine supplementation, and intensified emergency management during catabolism can prevent metabolic crises. There is now expected to be a large number of asymptomatic individuals with GA-1 who have a good prognosis and are followed into adulthood. Though previous recommendations have focused on dietary treatment primarily during the first 6 years of life and relaxed management thereafter, a few documented cases of neuroradiologic abnormalities in adult-onset GA-1 and early treated GA-1 have challenged this prior doctrine (Boy et al. 2017). The natural progression of early diagnosed and treated GA-1 is not fully understood and more case studies need to be documented in the literature. We present a unique case of subependymal nodules found in an adult patient with GA-1 who was diagnosed and treated since infancy without metabolic crises.
Case Report
A Caucasian female first presented with macrocephaly (head circumference-for-age +2.1 standard deviations, Kuczmarski et al. 2000) and megalencephaly at 3 weeks of age. GA-1 was suspected and confirmed through elevated levels of glutaric acid in urine, plasma, and cerebrospinal fluid (CSF) as well as absence of GCDH activity in fibroblasts. Patient has one full sibling, an older brother, without any known symptoms, though he has not had any additional biochemical, enzymatic, or molecular assessments. The family history is significant for some paternal relatives with possible mental retardation and degenerative eye disease. The family is of Northern European descent, primarily German. There is some Native American descent on the maternal side. There are no reported birth defects or consanguinity in the family. The remainder of the family history is otherwise negative for mental retardation, birth defects, multiple pregnancy losses, or known genetic disorders.
No genetic testing was performed initially, but the patient was treated with a low protein diet, carnitine, and riboflavin and closely monitored during intercurrent illnesses. She had otherwise normal neurological presentation and did not have metabolic crises as a child. With treatment and close monitoring, she maintained a sufficient level of executive functioning but did have some learning difficulties in school. As a pre-teenager, she began having headaches. Neurology evaluation at 12 years of age determined her headaches to be migrainous and related to idiopathic intracranial hypertension (IIH) after lumbar puncture (LP) analysis showed high opening pressures.
She graduated from high school but did not attend college. The patient had two successful pregnancies at age 23 and 25 years.
At age 28 years, she presented to the hospital with 3 weeks of slurred speech as well as left facial weakness and numbness. The rest of the exam was normal. Her prior imaging was reviewed. An MRI at age 22 years showed callosal and periventricular white matter changes, age discordant parenchymal atrophy, and multifocal subependymal nodules in the lateral ventricles. These changes were stable on serial imaging at 26 years of age. CT and MRI were repeated during hospital presentation showing no acute changes. LP revealed an opening pressure of 36 cm H2O, consistent with an exacerbation of IIH. Given her unusual presentation and history of subependymal nodules not previously documented with early diagnosed and treated GA-1, she had genetic testing (TSC1 and TSC2 genes) for tuberous sclerosis (TSC) and periventricular nodular hypertopia (FLNA gene) which was negative. A CT abdomen at 24 years showed no renal cysts or angiomyolipomas often seen in TSC, and patient had no other stigmata for TSC (such as facial angiofibromas, hypopigmented macules, forehead plaques, or Shagreen patches). Further genetic testing then was done confirming her original biochemical GA-1 diagnosis.
Materials and Methods
Original biochemical and enzymatic diagnosis of GA-1 was performed during infancy. Repeated studies for continued management that also supported her diagnosis included plasma carnitine and acylcarnitines, plasma amino acids, as well as urine organic acids.
Genomic DNA was obtained from fresh blood at 28 years. Genetic testing was then performed using methods applied at a commercial laboratory, GeneDx. The following genes were specifically reviewed with the percentage of the coding region covered at >10X by exome sequencing indicated in parentheses: FLNA (100%), GCDH (100%), TSC1 (100%), and TSC2 (100%). The Agilent Clinical Research Exome kit was used to target the exonic regions and flanking splice junctions. These targeted regions were sequenced simultaneously by massively parallel (NextGen) sequencing on an Illumina HiSeq sequencing system with 100 bp paired-end reads. Bidirectional sequence was assembled, aligned to reference gene sequences based on human genome build GRCh37/UCSC hg19, and analyzed for sequence variants in the selected genes or regions of interest using a custom-developed analysis tool (Xome Analyzer). Capillary sequencing or another appropriate method was used to confirm all potentially pathogenic variants identified in this individual. Sequence alterations were reported according to the Human Genome Variation Society (HGVS) nomenclature guidelines.
Results
At initial presentation as an infant, patient had elevated glutaric acid in plasma, urine, and CSF and absence of GCDH activity in fibroblasts which established the diagnosis of GA-1.
Molecular genetic studies of FLNA, TSC1, and TSC2 were normal. However, the patient was found to be homozygous for the c.1204 C>T, p.R402W pathogenic variant in the GCDH gene in adulthood confirming her original biochemical diagnosis of GA-1. The homozygous state of the R402W variant of the GCDH gene has been reported in multiple patients with clinical features and biochemical profiles consistent with GA-1 (Biery et al. 1996; Busquets et al. 2000; Gupta et al. 2015).
MRI at 29 years showed stable callosal and periventricular white matter changes, age discordant parenchymal atrophy, and multifocal subependymal nodules in the lateral ventricles (Figs. 1 and 2).
Fig. 1.
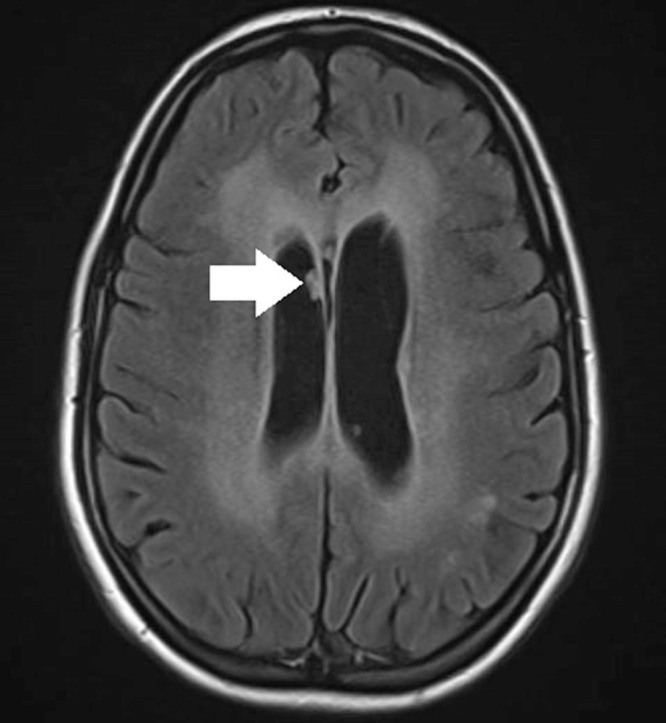
MRI brain axial flair imaging of case patient at 29 years of age. A white arrow is pointing to one of the periventricular subependymal nodules
Fig. 2.

MRI brain sagittal T2 weighted imaging of case patient at 29 years of age
Discussion
Subependymal nodules are hamartomatous forms of heterotopia, which are benign yet disorganized growths composed of elements of the adjacent tissue. These lesions line the walls of ventricles and are most common in TSC, infants with TORCH infections, and elderly patients with neoplastic growths of subependymal cells. Subependymal nodules are seen in roughly 80% of patients with TSC. They are thought to be asymptomatic and have no epileptogenic potential. They are of unknown clinical significance, though they are presumed to have theoretical potential to develop into subependymal giant cell astrocytomas, which are the most common brain tumor type in TSC (Klar et al. 2016).
Upon review of the literature, there have been no other cases of GA-1 patients who were early diagnosed and treated since infancy presenting with subependymal nodules. Three cases of subependymal nodules have been reported in patients with GA-1, but all were diagnosed in adulthood and had no other unifying diagnosis (Table 1). All three of these patients had different ethnic backgrounds, presentations, and pathogenic variants in the GCDH gene.
Table 1.
Documented cases of subependymal nodules in patients with biochemical and genetic testing confirming glutaric acidemia type 1
| Proband | Herskovitz et al. (2013) | Korman et al. (2007) | Pierson et al. (2015) | |
|---|---|---|---|---|
| Sex/background | Female, Caucasian | Male, Iraqi Jewish | Male, Palestinian Arab | Female, Hispanic Mexican |
| Age of diagnosis | 2 months old | 56 years old | 30 years old | 55 years old |
| Comorbidities | Idiopathic intracranial hypertension | None reported | None reported | Crohn’s disease |
| Neurologic symptoms | Migrainous and IIH headaches, mild learning disability, recent onset slurred speech with left facial weakness/numbness | 30-year history of pain in feet, gradual weakness in legs, speech disturbance, and incontinence | Borderline IQ, normal neurological exam; otherwise “asymptomatic”; discovered after seeking prenatal genetic counseling as he was paternal uncle of another patient in the case series | Bilateral lower extremity spasticity, numbness, and paresthesias |
| MRI findings | Callosal and periventricular white matter changes, age discordant parenchymal atrophy, and multifocal subependymal nodules (a type of heterotopia or disorganized brain tissue) in the lateral ventricles | Communicating hydrocephalous, bilateral frontotemporal atrophy, bilateral temporal arachnoid cysts, prominent periventricular and deep leukodystrophy, and subependymal cauliflower-like mass lesions | Patchy signal changes in the corpus callosum with wart-like mass lesions extending from the ependymal lining into the lateral ventricles in the upper part in the ventricular system and showing some contrast enhancement, resembling subependymal nodules found in tuberous sclerosis | Extensive bilateral white matter changes in the periventricular deep and subcortical white matter tracts; multiple subependymal nodules projecting into lateral ventricles; temporal lobe hypoplasia; normal striatum and corpus callosum |
| GCDH gene mutations | Homozygous for the c.1204 C>T, p.R402W pathogenic variant | Previously reported homozygous Gly101Arg mutation | Compound heterozygosity of a novel variant (c. 578_579 insTCA; pThr193_R194insHis) and known pathogenic mutation (c.877G>A; p.Ala293Thr) | Compound heterozygosity of a novel variant (c.1219 C>G; pLeu407Val) and known pathogenic mutation (c.848delT;pL283RfsX8) |
Our patient’s case reveals that subependymal nodules may develop as a natural progression of GA-1 despite early diagnosis and metabolic control, though their clinical significance is yet to be determined. More cases needed to be documented in literature.
Synopsis
We report the first case of subependymal nodules in an adult, without any stigmata for tuberous sclerosis, who was diagnosed and treated for glutaric acidemia type 1 since infancy without any metabolic decompensation.
Compliance with Ethics Guidelines
Conflict of Interest
All authors (Bimal Patel, Surekha Pendyal, Priya Kishnani, Marie McDonald, and Lauren Bailey) declare there are neither competing interests nor financial disclosures.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from the patient included in this study.
Animal Studies
This chapter does not contain any studies with animal subjects performed by the any of the authors.
Authorship Contributorship Statements
All authors have been involved in (a) conception and design, or analysis and interpretation of data, and (b) drafting the chapter or revising it critically for important intellectual content.
All coauthors have seen the final version of the chapter, confirm that the work has not been published/submitted elsewhere, and agree with submission.
- Bimal Patel:
conception and design, analysis, interpretation of data, writing/drafting of the manuscript, final approval of chapter.
- Surekha Pendyal:
conception and design, analysis, interpretation of data, final approval of chapter.
- Priya Kishnani:
primary physician caring for patient since early childhood, conception and design, analysis, interpretation of data, final approval of chapter.
- Marie McDonald:
conception and design, analysis, interpretation of data, final approval of chapter.
- Lauren Bailey:
genetic counselor caring for patient, conception and design, analysis, interpretation of data, final approval of chapter.
Contributor Information
Priya S. Kishnani, Email: priya.kishnani@duke.edu
Collaborators: Matthias Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Biery BJ, Stein DE, Morton DH, et al. Gene structure and mutations of glutaryl-coenzyme A dehydrogenase: impaired association of enzyme subunits that is due to an A421V substitution causes glutaric acidemia type I in the Amish. Am J Hum Genet. 1996;59:1006–1011. [PMC free article] [PubMed] [Google Scholar]
- Boy N, et al. Proposed recommendations for diagnosing and managing individuals with glutaric aciduria type I: second revision. J Inherit Metab Dis. 2017;40:75–101. doi: 10.1007/s10545-016-9999-9. [DOI] [PubMed] [Google Scholar]
- Busquets C, Coll MJ, Ribes A. Evidence of a single origin for the most frequent mutation (R402W) causing glutaryl-CoA dehydrogenase deficiency: identification of 3 novel polymorphisms and haplotype definition. Hum Mutat. 2000;15:207. doi: 10.1002/(SICI)1098-1004(200002)15:2<207::AID-HUMU15>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Gupta N, Singh PK, Kumar M, et al. Glutaric acidemia type 1-clinico-molecular profile and novel mutations in GCDH gene in Indian patients. JIMD Rep. 2015;21:45–55. doi: 10.1007/8904_2014_377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskovitz M, et al. Subependymal mass lesions and peripheral polyneuropathy in adult-onset glutaric aciduria type I. Neurology. 2013;81:849–850. doi: 10.1212/WNL.0b013e3182a2cbf2. [DOI] [PubMed] [Google Scholar]
- Klar N, Cohen B, Lin DD. Neurocutaneous syndromes. Handb Clin Neurol. 2016;135:570. doi: 10.1016/B978-0-444-53485-9.00027-1. [DOI] [PubMed] [Google Scholar]
- Kölker S, Valayannopoulos V, Burlina AB, et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 2: The evolving clinical phenotype. J Inherit Metab Dis. 2015;38:1059–1074. doi: 10.1007/s10545-015-9840-x. [DOI] [PubMed] [Google Scholar]
- Korman SH, et al. Glutaric aciduria type 1: clinical, biochemical, and molecular findings in patients from Israel. Eur J Paediatr Neurol. 2007;11:81–89. doi: 10.1016/j.ejpn.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Kuczmarski RJ, Ogden CL, Grummer-Strawn LM, et al. CDC growth charts: United States. Adv Data. 2000;314:1–27. [PubMed] [Google Scholar]
- Pierson TM, et al. Adult-onset glutaric aciduria type I presenting with white matter abnormalities and subependymal nodules. Neurogenetics. 2015;16:325–328. doi: 10.1007/s10048-015-0456-y. [DOI] [PubMed] [Google Scholar]
- Twomey EL, Naughten ER, Donoghue VB, et al. Neuroimaging findings in glutaric aciduria type 1. Pediatr Radiol. 2003;33:823–830. doi: 10.1007/s00247-003-0956-z. [DOI] [PubMed] [Google Scholar]