

Abstract
Objective:
Mitochondrial dysfunction and oxidative stress in insulin responsive tissues is implicated in the pathogenesis of type 2 diabetes. Whether these perturbations extend to other tissues and contribute to their pathophysiology is less well established. The objective of this study was to investigate platelet mitochondria to evaluate whether type 2 diabetes associated mitochondrial dysfunction is evident in circulating cells.
Method:
A pilot study of mitochondrial respiratory function and proteomic changes comparing platelets extracted from insulin sensitive (n=8) and type 2 diabetic subjects (n=7).
Results:
In-situ platelet mitochondria show diminished oxygen consumption and lower oxygen-dependent ATP synthesis in diabetic versus control subjects. Mass spectrometric identification and confirmatory immunoblot analysis identifies induction of the mitochondrial anti-oxidant enzymes superoxide dismutase 2 and thioredoxin-dependent peroxide reductase 3 in platelets of diabetic subjects. As oxidative stress upregulates anti-oxidant enzymes we assessed mitochondrial protein carbonylation as an index of oxidative-stress. Platelets of diabetic subjects exhibit significantly increased protein carbonylation compared to controls.
Conclusions:
As platelets are anuclear fragments of megakaryocytes, our data suggest that the bone marrow compartment in type 2 diabetic subjects is exposed to increased mitochondrial oxidative stress with upregulation of nuclear-encoded antioxidant mitochondrial enzymes. This ‘stress-signature’ in platelets of diabetic subjects is associated with a diminution of their mitochondrial contribution to energy production and support that mitochondrial perturbations in type 2 diabetes extends beyond the classical insulin responsive tissues. Platelets, as “accessible human tissue”, may be useful to measure the mitochondrial modulatory effects of emerging anti-diabetic therapeutics.
Keywords: Mitochondria, platelets, reactive oxygen species, respiration, diabetes
Introduction
Type 2 diabetes mellitus (T2DM) is an evolving global pandemic with deleterious health consequences. Although the pathophysiology associated with the development of T2DM is heterogeneous, mitochondrial perturbations are shown to contribute to disease progression in the pancreas and in insulin responsive metabolic tissues [1,2]. Whether mitochondrial dysfunction contributes to T2DM pathophysiology in other tissues is unclear.
Although platelet dysfunction is evident in diabetes [3], whether mitochondrial perturbations contribute to this pathophysiology is similarly unknown. Furthermore, as platelets are anuclear viable fragments of bone marrow megakaryocytes the study of platelet mitochondria should provide a unique ‘window’ into the bone marrow. This concept is particularly pertinent to mitochondria as intergenomic regulation between the nucleus and mitochondria is required to maintain mitochondrial homeostasis [4]. Hence, the investigation of functional and protein changes in platelet mitochondria probably reflect, in large part, the ‘historical’ nuclear-mitochondrial regulation within the intact megakaryocytes.
This pilot study evaluated mitochondrial respiration and the ‘oxidative-stress profile’ in platelets comparing healthy insulin sensitive to T2DM subjects. We hypothesized that platelets from subjects with T2DM would exhibit altered mitochondrial characteristics.
Research Design and Methods
All subjects were enrolled after informed consent, approved by the NHLBI Internal Review Board. Eighteen subjects were enrolled into this pilot study at the NIH Clinical Research Center - ClinicalTrials.gov Identifier: NCT00833846. Three subjects were excluded from analysis as their glucose tolerance tests showed an intermediate phenotype of impaired glucose tolerance. Exclusion criteria included the use of insulin or thiazolidinediones due to their known mitochondrial effects [5]. Additional exclusion criteria included pregnancy, concurrent acute/chronic illnesses or age greater than 60 years.
Fresh blood was collected and subjected to centrifugation, after which the upper 2/3 of the platelet-rich plasma was separated and re-centrifuged. The platelet pellet was washed three-times with 0.9% NaCl. The purified platelets were then resuspended and platelet respiration was measured using a Seahorse XF24 Extracellular Flux Analyzer (Seahorse BioScience) [6]. Platelets were adhered to 24-well respiration plates using Cell-Tak (BD Biosciences) and uniform plating was confirmed by direct microscopic visualization. As previously described [6], maximal oxidative capacity was calculated as the difference in oxygen consumption between dinitrophenol (200 μM) administration and the basal oxygen consumption rate (OCR). ATP synthesis was calculated as the difference in OCR between basal and oligomycin (1 μM) mediated inhibition of the ATP synthase. The mitochondrial proton leak was calculated as the OCR difference between administration of oligomycin and rotenone (1 μM) plus antimycin A (1 μM). The cytosolic utilization is represented as the residual oxygen consumption following rotenone/antimycin A administration.The platelet respiratory profile was repeated in triplicate and data normalized to protein content.
Platelet isolation for protein analysis was performed similarly to that described for respiratory studies. Platelet purity was confirmed using CD61 positive immunofluorescence labeling antibody (BD Biosciences) by flow cytometry (Data not shown). The isolation of the crude mitochondrial fraction was performed using differential centrifugation as described [7].
The crude mitochondrial pellet was suspended in lysis buffer containing 15 mM Tris-HCl pH 8.5, 7 M urea, 2 M thiourea, and 4% CHAPS. Two-dimensional differential in-gel electrophoresis (2D-DIGE) and mass spectrometric (MS) analysis was performed as previously described [8]. Twenty µg of crude mitochondrial protein from each subject was then employed for Western Blot analysis. Antibodies directed against manganese superoxide dismutase (SOD2; Santa Cruz Biotechnology), thioredoxin-dependent peroxide reductase 3 (PRDX3; Abcam) and β-actin (Sigma) were employed. Protein carbonylation (PC) modification on protein samples was determined using a PC test kit (BioCell Corp). The results expressed as total ELISA absorption for each participant.
Experimental data was analyzed by unpaired 2-tailed t-Test and the subject gender distribution using the Fisher’s exact test. Results are expressed as mean±SEM with differences significant for p-value <0.05.
Results
Participants were matched for age, gender, body mass index and lipid profiles. Significant elevations in fasting glucose levels (6.4±0.2 versus 5±0.2 mmol/L), glucose levels after the 2-hour oral glucose tolerance test (12.1±1.8 versus 5.3±0.5 mmol/L) and HbA1c levels (7.1±0.6 versus 5.6±0.1%) were evident in T2DM participants versus controls.
Representative tracings of oxygen consumption in a paired control and diabetic subject at baseline and in response to electron transfer chain site-specific inhibitors are shown in Fig. 1. The composite data of all subjects is depicted in Fig. 1C. The results show that: i) basal mitochondrial oxygen consumption is reduced by ≈ 40% in diabetic versus control subjects (p=0.02); ii) the relative contribution of oxygen consumption for ATP production is similarly reduced in the diabetic subjects (p=0.03) and iii) interestingly, the diabetic platelet mitochondria exhibited an ≈ 50% reduction in proton leak (p=0.01). The relative utilization of cytosolic oxygen is similar in platelets from control and diabetic subjects.
Fig. 1.
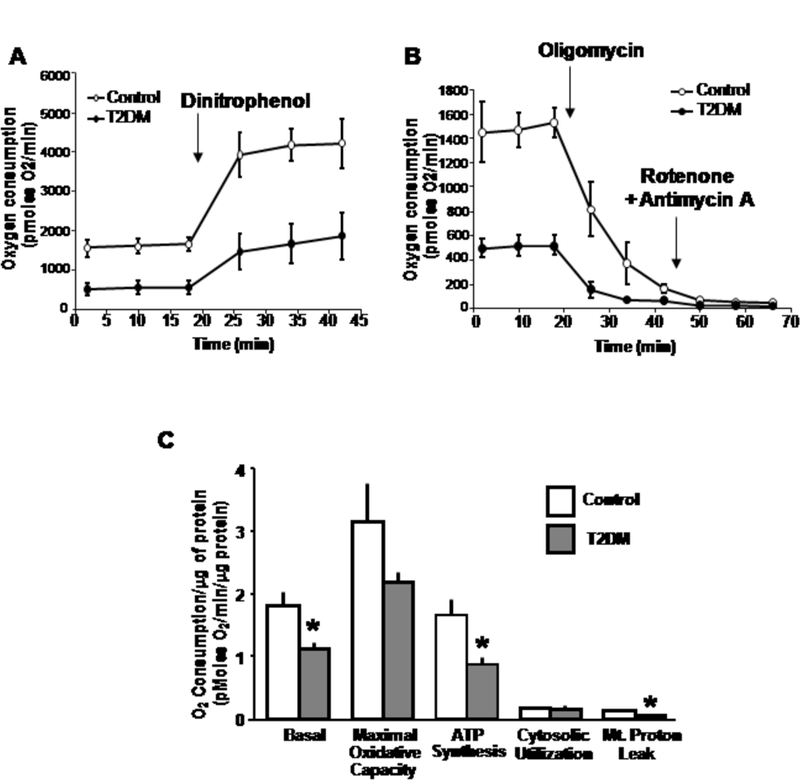
Mitochondrial respiration is perturbed in diabetic subject platelets. A. Representative tracing of basal oxygen consumption and maximal oxygen consumption induced by the uncoupler dinitrophenol comparing platelets from a control and diabetic subject. B. Representative tracing of oxygen consumption in response to inhibition of the F1/Fo ATP synthase with oligomycin, and in response to the subsequent inhibition of electron transfer chain activity with the inhibitors rotenone and antimycin A. The reduction in oxygen consumption following oligomycin represents oxygen utilization for ATP production. The difference in oxygen consumption between the two pharmacologic interventions represents the proton leak, and the residual oxygen utilization represents cytosolic oxygen consumption. C. Histograms showing the absolute differences in oxygen consumption comparing groups normalized to total protein content. The asterisk represents a p<0.05 versus respective control subjects. Abbreviation: Mt - Mitochondria
Using fluorescent labeled 2D-DIGE, multiple differences in mitochondrial protein expression was evident comparing control to T2DM subjects (data not shown). In keeping with the known association between redox stress and mitochondrial dysfunction in diabetes [9], an intriguing finding was the induction of protein levels of two mitochondrial anti-oxidant enzymes, SOD2 and PRDX3 in diabetic subjects as shown by 2D-DIGE (Fig. 2A) and additional post-translational modifications of SOD2 are similarly evident comparing control to T2DM subjects (Fig. 2B).
Fig. 2.
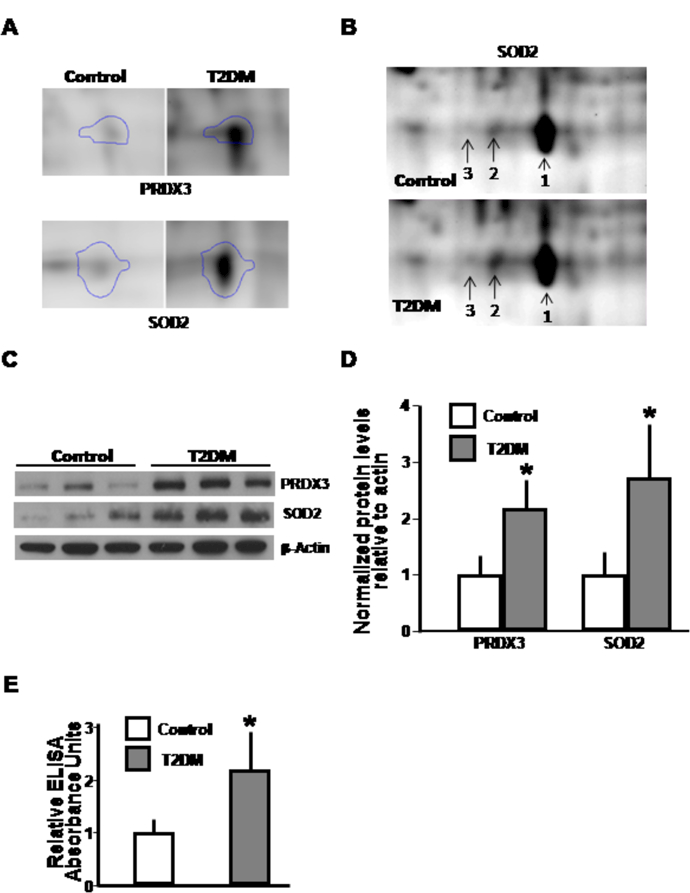
Protein analyses showing altered reactive oxygen species regulatory proteins and consequences in diabetic subject platelets. A. Magnification of 2D-DIGE with Progenesis Samespot software after mass spectrometry which located and analyzed protein spots assigning statistical confidence and difference in anti-oxidant protein levels. B. Post-translation modification of SOD2 on 2D-DIGE that would be compatible with change in the phosphorylation status between control and diabetic samples. C. Immunoblot confirmation of expression of anti-oxidant proteins. β-actin shows protein loading control. D. Histogram shows protein expression levels from all subjects. E. Relative carbonylation levels from platelet mitochondrial proteins from each subject. Asterisks highlight p<0.05 versus relative control subjects.
To confirm the relative abundance of these anti-oxidant proteins identified, immunoblot analyses using mitochondrial protein was performed. There were a greater than 2-fold induction in the levels of SOD2 (p=0.01) and PRDX3 (p=0.001) in T2DM compared to controls (Fig. 2C and 2D). To assess whether these reflect changes in association with increased redox stress, we measured protein carbonyl content as a marker of oxidative modification of proteins. T2DM subjects had ≈ 1.6 times higher carbonyl levels than controls (p=0.0002, Fig. 2E).
Conclusion
The data from this pilot study shows blunted mitochondrial respiration with upregulation of mitochondrial anti-oxidant proteins and concomitant evidence of increased oxidative stress. Additionally, post-translational modifications evident in the two-dimensional gel analysis of SOD2 similarly may reflect alterations in the antioxidant regulatory program [10]. These data suggest more widespread disruption in mitochondrial function beyond the classical insulin responsive metabolic organs and support the oxidative stress hypothesis in the pathogenesis of T2DM [9].
The attenuation of mitochondrial oxygen consumption in insulin responsive tissues and in the pancreas during diabetes is well established and this is often associated with the downregulation of oxidative phosphorylation proteins [11]. Although we show that this ‘respiratory profile’ extends to platelets we did not see changes in mitochondrial oxidative phosphorylation protein levels by 2-D-DIGE (data not shown). This may represent a sensitivity issue due to the low number of mitochondria in platelet fragments or could reflect a change in respiratory function due to post-translational modifications not identified by 2D-DIGE. An intriguing respiratory observation in this study is the attenuation of proton leak in the diabetic subjects’ platelet mitochondria. This has been previously shown in skeletal muscle mitochondria from older insulin resistant versus young insulin sensitive rats [12]. Whether a reduction in inner mitochondrial membrane proteins in diabetes changes the membrane physicochemical properties to attenuate this leak is an intriguing concept that would need to be validated. Additionally, whether a smaller proton leak exacerbates mitochondrial ROS generation is similarly plausible [13].
Mitochondrial anti-oxidant enzymes are nuclear-encoded and the integration of an intact nuclear-mitochondrial regulatory axis would be necessary to upregulate mitochondrial protective programming. Redox-stress itself initiates the upregulation of genes encoding mitochondrial anti-oxidant proteins to facilitate mitochondrial attenuation of subsequent oxidative damage [14]. In this study we show upregulation of anti-oxidant mitochondrial defenses against increased levels of oxidative stress in platelet mitochondria in T2DM subjects compared to controls. Specifically we note that T2DM subjects have increased levels of mitochondrial SOD2 and PRDX3. Although, we do not have data to dissect out the temporal course of events, the absence of the nuclear-mitochondrial intergenomic system in platelets supports that oxidative stress evoked upregulation of mitochondrial antioxidant enzymes preceded anucleation during platelet release into the vasculature. The increased carbonylation modifications confirm redox-stress although we cannot exclude extra-mitochondrial sources. Interestingly, oxidative stress is hypothesized to contribute to end-organ damage in diabetes and to mitochondria damage during the development of diabetes [9]. A unifying hypothesis of our findings would be that the oxidative stress evident in the platelets of the diabetic subjects contributes to subsequent mitochondrial respiratory perturbations.
In conclusion, the findings in this pilot study generate a hypothesis suggesting that T2DM is associated with an increased redox-stress milieu within the bone marrow megakaryocytes which initiates oxidative stress-damage with the concomitant induction of mitochondrial anti-oxidant regulatory programs. As a possible consequence platelet mitochondrial respiratory function is attenuated which, may in turn, exacerbate the generation of mitochondrial reactive oxygen species. Whether megakaryocyte redox-stress and associated platelet mitochondrial perturbations contribute to diabetic platelet dysfunction is an intriguing hypothesis that would need to be explored. Furthermore, the evaluation of mitochondrial platelet respiratory function may be a useful peripheral blood ‘signature’ to measure reversibility of diabetic sequelae in response to emerging ‘mitochondrial-modulatory’ diabetic therapies [5,15].
Acknowledgements
This research is funded by the Division of Intramural Research of the NHLBI. CA and RJH were funded by an NIH-Pfizer Clinical Research Training Fellowship. We thank the NHLBI Flow Cytometry Core Facility for assistance in the confirmation of platelet purity and Stephanie A French, Darci Phillips and Dr. Marjan Gucek of the NHBLI for helpful discussion regarding mitochondrial protein isolation and proteomic analysis.
Abbreviations:
- T2DM
Type 2 diabetes mellitus
- SOD2
Superoxide dismutase 2
- PRDX3
Thioredoxin-dependent peroxide reductase 3
- OCR
Oxygen consumption rate
- 2D-DIGE
Two-dimensional differential in-gel electrophoresis
- MS
Mass spectroscopy
Footnotes
ClinicalTrials.gov Identifier: NCT00833846
Conflict of interest The authors declare that there is no conflict of interest associated with this manuscript.
References
- 1.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science 2005;307:384–387 [DOI] [PubMed] [Google Scholar]
- 2.Pagel-Langenickel I, Bao J, Pang L, Sack MN. The role of mitochondria in the pathophysiology of skeletal muscle insulin resistance. Endocr Rev 2010;31:25–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baalbaki HA, Bell DS. Insulin resistance and thrombogenesis: recent insights and therapeutic implications. EndocrPract 2007;13:679–686 [DOI] [PubMed] [Google Scholar]
- 4.Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev 2004;18:357–368 [DOI] [PubMed] [Google Scholar]
- 5.Pagel-Langenickel I, Bao J, Joseph JJ, et al. PGC-1alpha integrates insulin signaling, mitochondrial regulation, and bioenergetic function in skeletal muscle. JBiolChem 2008;283:22464–22472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu M, Neilson A, Swift AL, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. AmJPhysiol Cell Physiol 2007;292:C125–C136 [DOI] [PubMed] [Google Scholar]
- 7.Pallotti F, Lenaz G Isolation and subfractionation of mitochondria from animal cells and tissue culture lines. Methods Cell Biol 2007;80:3–44 [DOI] [PubMed] [Google Scholar]
- 8.Wong R, Aponte AM, Steenbergen C, Murphy E. Cardioprotection leads to novel changes in the mitochondrial proteome. Am J Physiol Heart Circ Physiol 2010;298:H75–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonnard C, Durand A, Peyrol S, et al. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. JClinInvest 2008;118:789–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hopper RK, Carroll S, Aponte AM, et al. Mitochondrial matrix phosphoproteome: effect of extra mitochondrial calcium. Biochemistry 2006;45:2524–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. NEnglJMed 2004;350:664–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iossa S, Mollica MP, Lionetti L, et al. A possible link between skeletal muscle mitochondrial efficiency and age-induced insulin resistance. Diabetes 2004;53:2861–2866 [DOI] [PubMed] [Google Scholar]
- 13.Sack MN. Mitochondrial depolarization and the role of uncoupling proteins in ischemia tolerance. CardiovascRes 2006;72:210–219 [DOI] [PubMed] [Google Scholar]
- 14.Fernandez V, Tapia G, Varela P, et al. Redox up-regulated expression of rat liver manganese superoxide dismutase and Bcl-2 by thyroid hormone is associated with inhibitor of kappaB-alpha phosphorylation and nuclear factor-kappaB activation. JEndocrinol 2005;186:539–547 [DOI] [PubMed] [Google Scholar]
- 15.Milne JC, Lambert PD, Schenk S, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 2007;450:712–716 [DOI] [PMC free article] [PubMed] [Google Scholar]