

Abstract
Purpose
Gemcitabine is currently the standard first-line chemotherapeutic drug for treating pancreatic cancer. However, many factors can contribute to gemcitabine resistance. One of the most important reasons is the low hENT1 expression. In this study, we tested the antitumor effect of gemcitabine-loaded human serum albumin nanoparticle (GEM-HSA-NP) on gemcitabine-resistant pancreatic cancer induced by low hENT1 expression.
Materials and methods
S-(4-nitrobenzyl)-6-thioinosine was utilized to inhibit the activity of hENT1 and simulate low hENT1 expression. Growth inhibition assays and cell cycle and apoptosis analyses were performed on human pancreatic cancer cell lines such as BxPC-3 and SW1990. The in vivo antitumor effect was studied by using patient-derived xenograft (PDX) models. The in vivo toxicity assessment was performed on healthy Kunming mice.
Results
In in vitro studies, GEM-HSA-NP showed its ability to inhibit cell proliferation, arrest cell cycle and induce apoptosis when tumor cells were resistant to gemcitabine. In in vivo studies, GEM-HSA-NP was more effective than gemcitabine on inhibiting tumor growth whether the expression levels of hENT1 were high or low in PDX models. The in vivo toxicity assessment showed that the biotoxicity of GEM-HSA-NP did not increase compared with gemcitabine.
Conclusion
GEM-HSA-NP can overcome gemcitabine resistance induced by low hENT1 expression, which suggests its potential role for the clinical application.
Keywords: pancreatic cancer, gemcitabine resistance, albumin nanoparticles, low hENT1 expression, PDX model
Introduction
Pancreatic cancer, commonly referred to as pancreatic ductal adenocarcinoma (PDAC), is one of the most lethal human malignancies in the world with a mortality rate almost equals its incidence rate.1 It is the seventh leading cause of cancer-related death globally and the fourth in the USA.2,3 Despite the progress in uncovering pathogenesis, diagnosis, staging and surgical treatment have been made in the past decades, the overall median survival of this devastating disease is <1 year,4 and the 5-year survival rate is <5%,5 which is the lowest among all kinds of cancer. Surgical resection is still the only approach that may achieve curative treatment. However, about 80% of patients present with a locally advanced or distant metastatic disease that cannot be resected when diagnosed.6 Thus, chemotherapy is the most important treatment for most patients with pancreatic cancer.
Gemcitabine (2,2-difluoro-2-deoxycytidine, GEM) is currently the standard first-line option for treating PDAC in neoadjuvant chemotherapy and postoperative chemotherapy as well as single-agent and combination chemotherapy.7 However, many factors can contribute to GEM resistance in PDAC, such as low expression of human nucleoside transporters (hNTs)8 or deoxycytidine kinase (dCK),9 overexpression of cytosolic 5′-nucleotidase (5′-NT)10 or cytidine deaminase (CDA)11 and so forth. Among all these factors, lack of adequate drug transport is considered as the key reason for chemoresistance.12,13
As a nucleoside analog, GEM is naturally hydrophilic and would not permeate the cellular lipid bilayer by passive diffusion readily. Instead, its uptake into cells needs the presence of hNTs.14 Human NT is a family of integral membrane proteins and consists of two major classes. One is sodium independent, mediating a bidirectional equilibrative activity, and is termed as human equilibrative nucleoside transporter (hENT). The other is sodium dependent, mediating an inwardly directed concentrative activity against the concentration gradient, and is termed as human concentrative nucleoside transporter (hCNT).15,16 It has been demonstrated that hENT1, hCNT1 and hCNT3 play important roles in the transport of GEM across the cell membrane.17 Among these three kinds of transporters, hENT1 mediates the most of GEM uptake, and hENT1-deficient cells are highly resistant to this nucleoside analog.18 Clinical researches have confirmed that low hENT1 expression results in GEM resistance by showing a positive correlation between the expression levels of hENT1 and the survival in patients with GEM-treated PDAC.19,20 Therefore, it is important to find novel therapeutic strategies to overcome GEM resistance induced by low hENT1 expression.
Our team has previously reported on coupling myristoyl with the 4-amino group of GEM to synthesize 4-N-myristoyl-GEM (GEM-C14) to enhance the lipophilic property of GEM, and using nanoparticle albumin-bound technology to encapsulate GEM-C14 into human serum albumin nanoparticle to produce GEM-loaded human serum albumin nanoparticle (GEM-HSA-NP). This kind of nanoparticles had an average size of 150 ± 27 nm and showed improved targeted cytotoxicity and prolonged drug release characteristics.21 Studies have shown that particles up to 100–200 nm could be internalized by receptor-mediated endocytosis.22,23 Therefore, we assumed that, with the help of this novel drug carrier, GEM resistance induced by low hENT1 expression can be overcome.
In this study, we used S-(4-nitrobenzyl)-6-thioinosine (NBMPR) which could inhibit hENT1 at nanomolar concentration24 to simulate low hENT1 expression and verified the effect of low hENT1 expression on GEM chemosensitivity in PDAC. Furthermore, we validated the antitumor effect of GEM-HSA-NP on GEM-resistant PDAC induced by low hENT1 expression.
Materials and methods
Chemicals and reagents
GEM (crude drug) was obtained from Eli Lilly and Company (Indianapolis, IN, USA). GEM-C14 and GEM-HSA-NP were prepared as reported previously.21 NBMPR was purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Normal saline (NS) was purchased from Shanghai Baxter Healthcare Co., Ltd. (Shanghai, China). Roswell Park Memorial Institute (RPMI)-1640 culture medium, fetal bovine serum and Dulbecco’s phosphate-buffered saline (DPBS) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Cell-Counting Kit-8 (CCK-8) was purchased from Dojindo (Kyushu, Japan). Annexin V-fluorescein isothiocyanate (FITC)/7-aminoactinomycin D (7-AAD) kit and propidium iodide (PI) kit were purchased from BD Biosciences (San Jose, CA, USA). RNeasy Mini kit was purchased from Qiagen NV (Venlo, the Netherlands). Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) kit was purchased from Hoffman-La Roche Ltd. (Basel, Switzerland).
Cell culture
The human pancreatic cancer cell lines BxPC-3 and SW1990 were obtained from the Shanghai Branch of the Chinese Academy of Sciences (Shanghai, China). No ethics statement was required from the institutional review board for the use of these cell lines. Cells were cultured at 37°C in the presence of 5% CO2 and 95% air with 95% humidity. Growth medium was RPMI-1640, containing 10% fetal bovine serum, 100 U/mL penicillin and 100 mg/mL streptomycin.
Establishment of pancreatic cancer patient-derived xenograft (PDX) models
The study protocol for this study was approved by the ethics committee of Huashan Hospital, Fudan University. Written informed consent was obtained from each patient. All animal studies were in compliance with the approved animal protocols and the guidelines of the Institutional Animal Care and Use Committee of Fudan University. PDAC tissues from 10 consecutive patients who underwent radical pancreatic cancer surgery were obtained. Each tumor tissue was washed three times with DPBS and was finely cut into a 20–30 mm3 fragment. Then, the tumor specimen was subcutaneously implanted in the right shoulder of 6- to 8-week-old female BALB/c-nu/nu mice (Shanghai Laboratory Animal Research Center, Shanghai, China). When the size of the xenograft reached 600–800 mm3, the tumor tissue was harvested. Real-time quantitative polymerase chain reaction (qPCR) and immunohistochemistry (IHC) were performed to screen out the tumor tissue with the highest hENT1 expression and the tumor tissue with the lowest hENT1 expression. Then, the abovementioned process was repeated in BALB/c-nu/nu mice to expand the two tumor xenografts. Tumor volume was calculated using the following formula: volume (mm3) = (length in mm) × (width in mm)2/2.
Growth inhibition assays
The in vitro antitumor effects of GEM, GEM-C14 and GEM-HSA-NP on pancreatic cancer cells were evaluated by CCK-8 assays. Cells (2,000 cells/well) were seeded in 96-well plates, and 24 h later, the medium was replaced by 0.1 mL fresh medium containing 0.05 μg/mL GEM, 0.1 μg/mL GEM-C14 (which is equivalent to 0.05 μg/mL GEM), 2.083 μg/mL (which is equivalent to 0.05 μg/mL GEM) or 20.83 μg/mL GEM-HSA-NP in the presence or absence of 1 μM NBMPR. The cells were incubated at 37°C for different time. Ten microliters of CCK-8 was added per well, cells were incubated for an additional 1 h and then were quantitated by reading the dye absorption at 450 nm with an automatic multiwall spectrophotometer.
Cell cycle and apoptosis analysis
Cells were treated with 0.05 μg/mL GEM, 0.1 μg/mL GEM-C14 or 2.083 μg/mL GEM-HSA-NP with or without 1 μM NBMPR for 48 h. Cell cycle and apoptosis analyses were conducted by flow cytometry using an FACSCalibur system (BD Biosciences). Data of the cell cycle were analyzed by ModFit LT™ software (Verity Software House, Inc., Topsham, ME, USA) to determine the proportions of cells in G0–G1, S and G2–M phases of cell cycle. The cell apoptosis was evaluated using the Annexin V-FITC/7-AAD kit, and the data were analyzed by FlowJo software (BD Biosciences, San Jose, CA, USA) to show the apoptosis rate in each group.
Real-time qPCR analysis
Tumor tissues, obtained from PDAC patients, were subjected to RNA extraction. Two microliters of total RNA was used for cDNA synthesis by utilizing reverse transcription reaction. The cDNA was subjected to real-time qPCR analysis by using primers designed for the hENT1 gene (5′-GGCAAAGAGGAATCTGGAGTT-3′ and 5′-ACAGTCACGGCTGGAAACAT-3′). GAPDH gene (5′-TGACTTCAACAGCGACACCCA-3′ and 5′-CACCCTGTTGCTGTAGCCAAA-3′) was used as an internal control.
Immunohistochemistry
The IHC was conducted using a two-step protocol as described previously.25 Briefly, after antigen retrieval by microwave, the specimens were incubated with primary antibodies for 60 min at room temperature and then were incubated with secondary antibody for 30 min. The sections were dyed in diaminobenzidine solution and were counter-stained with hematoxylin.
Antitumor study in PDX mouse models
To assess the in vivo antitumor effect of GEM-HSA-NP, two groups of PDX models were established. One group of xenografts was high hENT1 expression (hENT1-H group), and the other group was low hENT1 expression (hENT1-L group). Each group of mice was randomized into three subgroups (n = 6 per subgroup) and was treated with 100 μL of NS, GEM and GEM-HSA-NP through tail vein injection on days 0, 3, 6 and 9 (GEM and the equivalent GEM-HSA-NP dosage: 40 mg/kg body weight). NS was used as control. Tumor size was measured every 2 days until day 12.
Detection of apoptosis in PDAC xenografts
On day 12, the mice were sacrificed and the tumor tissues were harvested. After fixing with paraformaldehyde for 24 h, the tumor tissues were embedded in paraffin. Apoptosis in tumor tissues was detected by TUNEL staining. Apoptotic cells were visualized by green fluorescence, and DAPI (blue) was used to stain nuclei.
Biotoxicity assessment
To assess the in vivo toxicity of GEM-HSA-NP, 18 healthy 6-week-old male Kunming (KM) mice (Shanghai Laboratory Animal Research Center) were randomized into three groups (n = 6 per group) and were treated with 200 μL of NS, GEM and GEM-HSA-NP through tail vein injection on days 0, 3, 6 and 9 (GEM and the equivalent GEM-HSA-NP dosage: 40 mg/kg body weight). NS was used as control. The body weight of each mouse was measured every 2 days until day 12. On day 12, the blood sample of each mouse was collected to analyze the hematological parameters and biochemical indices.
Statistical analyses
Data are expressed as mean ± SD. Student’s t-test was used for the analyses of differences between two groups, and multiple groups were analyzed by one-way analysis of variance test. All the statistical analyses were conducted using SPSS software (version 22.0; IBM Corporation, Armonk, NY, USA). A value of P < 0.05 was considered as statistically significant.
Results and discussion
To evaluate the in vitro antitumor effect of GEM-HSA-NP on low hENT1 expression cancer cells, we utilized NBMPR to inhibit the activity of hENT1 and simulate low hENT1 expression. The growth inhibition assays were conducted on human pancreatic cancer cell lines such as BxPC-3 and SW1990. Cell viability was measured at 0, 24, 48, 72 and 96 h after tumor cells were treated with different drugs with or without NBMPR. In the absence of NBMPR, as shown in Figure 1A and B, GEM, GEM-C14, GEM-HSA-NP and 10 × GEM-HSA-NP all caused cell growth inhibition in a time-dependent manner. When together with NBMPR, however, GEM lost its antitumor effect and tumor cells showed significant GEM resistance (P < 0.05). The growth-inhibiting ability of GEM-C14, GEM-HSA-NP and 10 × GEM-HSA-NP was not significantly influenced (Figure 1C and D). They could still potently inhibit the growth of tumor cells. The cytotoxicity of 10 × GEM-HSA-NP was higher than GEM-HSA-NP, suggesting that GEM-HSA-NP inhibited cell proliferation in a concentration-dependent manner.
Figure 1.

The growth inhibition effect of GEM-HSA-NP on BxPC-3 and SW1990 cell lines.
Notes: Cell viability was measured at 0, 24, 48, 72 and 96 h after BxPC-3 cells (A) and SW1990 cells (B) were treated with 0.05 μg/mL GEM, 0.1 μg/mL GEM-C14 (which is equivalent of 0.05 μg/mL GEM), 2.083 μg/mL (which is equivalent of 0.05 μg/mL GEM) or 20.83 μg/mL GEM-HSA-NP, with or without 1 μM NBMPR (an inhibitor of hENT1). (C) Quantitative analysis of the cell proliferation in each group of BxPC-3 cell line. *A significant difference compared with the GEM + NBMPR group. (D) Quantitative analysis of the cell proliferation in each group of SW1990 cell line.
Abbreviations: GEM, gemcitabine; GEM-C14, 4-N-myristoyl-gemcitabine; GEM-HSA-NP, gemcitabine-loaded human serum albumin nanoparticle; NBMPR, S-(4-nitrobenzyl)-6-thioinosine.
Regulating the progression of cell cycle and inducing apoptosis in cancer cells are two effective mechanisms for controlling tumor growth.26,27 In our study, cell cycle analysis demonstrated that GEM alone arrested tumor cells in G0–G1 phase and caused significant decreases in S and G2–M phases in both BxPC-3 cells (P < 0.05; Figure 2A and B) and SW1990 cells (P < 0.05; Figure 2C and D). Adding NBMPR to GEM-treated group could restore the cell cycle to a relatively normal range (P > 0.05). Similarly, GEM alone significantly increased apoptosis rates in both BxPC-3 cells (P < 0.05; Figure 3A and B) and SW1990 cells (P < 0.05; Figure 3C and D), together with NBMPR decreased the apoptosis rates to normal levels. These data confirmed that the low expression and low activity of hENT1 would lead to GEM resistance.15,28 Unlike GEM, GEM-HSA-NP and GEM-C14 had the abilities to induce cell cycle arrest and promote apoptosis whether NBMPR existed or not (P < 0.05). It was shown that these two drugs were effective when the activity of hENT1 was low. Detailed data on cell cycle and apoptosis rates in BxPC-3 and SW1990 cells are summarized in Tables S1 and S2.
Figure 2.

The effect of GEM-HSA-NP on cell cycle arrest.
Notes: (A) The proportions of cell cycle analyzed by flow cytometry after BxPC-3 cells were treated with 0.05 μg/mL GEM, 0.1 μg/mL GEM-C14 and 2.083 μg/mL GEM-HSA-NP for 48 h, with or without 1 μM NBMPR. (B) Proportion of each phase of the cell cycle in each group of BxPC-3 cell line. (C) The proportions of cell cycle analyzed by flow cytometry after SW1990 cells were treated with 0.05 μg/mL GEM, 0.1 μg/mL GEM-C14 and 2.083 μg/mL GEM-HSA-NP for 48 h, with or without 1 μM NBMPR. (D) Proportion of each phase of the cell cycle in each group of SW1990 cell line.
Abbreviations: GEM, gemcitabine; GEM-C14, 4-N-myristoyl-gemcitabine; GEM-HSA-NP, gemcitabine-loaded human serum albumin nanoparticle; NBMPR, S-(4-nitrobenzyl)-6-thioinosine.
Figure 3.

The effect of GEM-HSA-NP on cell apoptosis.
Notes: (A) The apoptosis rates in BxPC-3 cells analyzed by flow cytometry after cells were treated with 0.05 μg/mL GEM, 0.1 μg/mL GEM-C14 and 2.083 μg/mL GEM-HSA-NP for 48 h, with or without 1μM NBMPR. (B) Apoptosis rates in each group of BxPC-3 cell line. (C) The apoptosis rates in SW1990 cells analyzed by flow cytometry after cells were treated with 0.05 μg/mL GEM, 0.1 μg/mL GEM-C14 and 2.083 μg/mL GEM-HSA-NP for 48 h, with or without 1μM NBMPR. (D) Apoptosis rates in each group of SW1990 cell line.
Abbreviations: 7-AAD, 7-aminoactinomycin D; GEM, gemcitabine; GEM-C14, 4-N-myristoyl-gemcitabine; GEM-HSA-NP, gemcitabine-loaded human serum albumin nanoparticle; NBMPR, S-(4-nitrobenzyl)-6-thioinosine.
To investigate the in vivo antitumor effect of GEM-HSA-NP, we established two groups of PDX models (Figure 4A and B). Almost all PDACs contain highly heterogeneous subpopulations of tumor cells. Various subpopulations of tumor cells show different sensitivity to chemotherapeutic drugs.29 Moreover, PDAC is characterized by a dense, poorly vascularized stroma; this microenvironment contains a mixture of interacting cellular and noncellular elements.30 These elements will hinder the penetration of chemotherapeutic drugs and weaken their antitumor effect. Compared with cancer cell line xenograft models, PDX models are more accurate in recapitulating the complex tumor heterogeneity and microenvironment and can reflect the true efficacy of chemotherapeutic drugs.31,32
Figure 4.

PDX models and IHC of hENT1 in tumor tissues.
Notes: Representative images of PDX models in the hENT1-H group (A) and hENT1-L group (B). IHC of hENT1 in hENT1-H (C) and hENT1-L (D) tumor tissues. IHC was used to confirm that the hENT1 expression was higher in the hENT1-H group than in the hENT1-L group.
Abbreviations: PDX, patient-derived xenograft; IHC, immunohistochemistry; hENT1-H, high hENT1 expression; hENT1-L, low hENT1 expression.
First, we used real-time qPCR to select the tumor specimen with the highest hENT1 expression and the specimen with the lowest hENT1 expression from 10 resected PDAC tissues. Subsequently, the two tumor specimens were subjected to IHC to confirm the expression levels of hENT1 (Figure 4C and D). Then, the two specimens were subcutaneously implanted in nude mice. GEM and GEM-HSA-NP were administrated via the tail vein on days 0, 3, 6 and 9. NS was used as control. On day 12, tumor tissues were harvested and weighed. Tumor size was measured every 2 days until day 12. As shown in Figure 5A and E, in the hENT1-H group, tumor volumes increased significantly slower in GEM-HSA-NP-treated subgroup (244.44 ± 68.35 mm3) than in GEM-treated subgroup (303.91 ± 94.87 mm3) and NS-treated subgroup (1,277.85 ± 398.34 mm3, P < 0.05). In addition, the tumor weights followed the order: GEM-HSA-NP (0.10 ± 0.04 g) < GEM (0.18 ± 0.05 g) < control (1.19 ± 0.18 g, P < 0.05; Figure 5C). The decrease in tumor volumes and weights was more marked in GEM-HSA-NP-treated subgroup, showing that GEM-HSA-NP was more effective than GEM in in vivo tumor growth inhibition. This result was consistent with our previous report. As described previously, the enhanced permeability and retention (EPR) effect and the active targeting approach of albumin nanoparticles might be the main reasons for this difference in drug efficacy.21 In the hENT1-L group, the tumor volumes also followed the order: GEM-HSA-NP (425.24 ± 216.87 mm3) < GEM (663.16 ± 226.47 mm3) < control (1,304.99 ± 313.48 mm3, P < 0.05; Figure 5B and F). Figure 5D demonstrated the tumor weights in GEM-HSA-NP-treated (0.20 ± 0.08 g), GEM-treated (0.44 ± 0.15 g) and NS-treated subgroups (0.85 ± 0.22 g, P < 0.05).
Figure 5.

Tumor weights and volume profiles in hENT1-H and hENT1-L group.
Notes: (A) Tumors harvested from each subgroup after different treatments in the hENT1-H group. (B) Tumors harvested from each subgroup after different treatments in the hENT1-L group. (C) Tumor weights of each subgroup in the hENT1-H group. (D) Tumor weights of each subgroup in the hENT1-L group. (E) Tumor volume profiles of mice treated with NS, GEM and GEM-HSA-NP in the hENT1-H group. (F) Tumor volume profiles of mice treated with NS, GEM and GEM-HSA-NP in the hENT1-L group. NS was used as control. ①, ② and ③ represent NS, GEM and GEM-HSA-NP, respectively.
Abbreviations: GEM, gemcitabine; GEM-HSA-NP, gemcitabine-loaded human serum albumin nanoparticle; NS, normal saline; hENT1-H, high hENT1 expression; hENT1-L, low hENT1 expression.
By calculating the tumor growth inhibition rate (tumor growth inhibition rate = [1 – the average tumor weight of the treated group/the average tumor weight of the control group] × 100%), we found that the tumor growth inhibition rate of the GEM-treated mice in the hENT1-L group was significantly lower than that of the hENT1-H group (47.79% ± 17.86% vs 84.77% ± 3.81%). This suggested that when the expression level of hENT1 decreased, the antitumor effect of GEM would decrease accordingly. The tumor growth inhibition rate of the GEM-HSA-NP-treated mice in the hENT1-L group was also lower than that of the hENT1-H group (76.66% ± 9.25% vs 91.23% ± 2.90%); however, this difference was not statistically significant (P > 0.05). It suggested that GEM-HSA-NP still had a remarkable antitumor effect when the expression level of hENT1 was low.
The difference of drug efficacy between GEM and GEM-HSA-NP was more significant in the hENT1-L group than in the hENT1-H group. Since the expression level of hENT1 in the hENT1-L group was lower than in the hENT1-H group, the uptake of GEM into tumor cells consequently reduced. To some extent, this resulted in tumor cells in the hENT1-L group being more insensitive to GEM. The entry of GEM-HSA-NP into tumor cells was performed by endocytosis and therefore was not influenced by the decreased hENT1 expression. Thus, in addition to the EPR effect and the active targeting approach of albumin nanoparticles, GEM insensitivity induced by lower hENT1 expression also contributed to the difference in effi-cacy between the two drugs in the hENT1-L group.
TUNEL staining was used to detect apoptotic cells in tumor tissues resected from PDX models (Figure 6). The degree of apoptosis in both the hENT1-H group and the hENT1-L group followed the order: control < GEM < GEM-HSA-NP, demonstrating that GEM-HSA-NP was more effective than GEM in inducing the apoptosis of tumor cells, no matter the hENT1 expression was high or low.
Figure 6.

The merged TUNEL staining indicated the degree of apoptosis in tumor tissues after various treatments in the hENT1-H and hENT1-L groups.
Notes: Green fluorescence represented apoptotic cells, and DAPI (blue) was used to stain nuclei.
Abbreviations: GEM, gemcitabine; GEM-HSA-NP, gemcitabine-loaded human serum albumin nanoparticle; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; hENT1-H, high hENT1 expression; hENT1-L, low hENT1 expression.
Our study showed that GEM-HSA-NP was more effective than GEM on inhibiting tumor growth, no matter the expression of hENT1 was high or low. In low hENT1 expression tumor cells, GEM lost its antitumor activity and the tumor cells showed marked GEM resistance. The expression level of hENT1 decreased, and the uptake of GEM by tumor cells reduced accordingly. However, hENT1 was not the prerequisite for GEM-HSA-NP uptake. Low hENT1 expression did not influence the antitumor effect of GEM-HSA-NP. It was still effective in suppressing tumor growth.
To assess the biotoxicity of GEM-HSA-NP, the in vivo toxicity test was performed on healthy KM mice. As shown in Figure 7, after the second administration, the average body weights of GEM-HSA-NP-treated mice and GEM-treated mice decreased (day 4), but the differences were not statistically significant compared with the NS-treated mice. In this 12-day test, there were no significant differences among the average body weights of the three groups. On day 12, we tested the hematological parameters and biochemical indices of each mouse. The results (Table 1) showed that GEM-HSA-NP had no significant effects on the hematological parameters and biochemical indices of mice compared with the NS-treated mice and GEM-treated mice. All these results suggested that GEM-HSA-NP had little biotoxicity.
Figure 7.
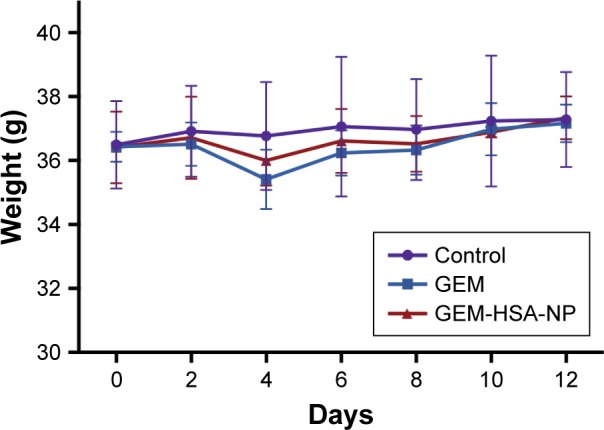
Body weights of KM mice.
Note: NS was used as control.
Abbreviations: GEM, gemcitabine; GEM-HSA-NP, gemcitabine-loaded human serum albumin nanoparticle; KM, Kunming; NS, normal saline.
Table 1.
The hematological parameters and biochemical indices of KM mice
| Index | Control | GEM | GEM-HSA-NP |
|---|---|---|---|
| WBC (109/L) | 6.48 ± 0.49 | 6.26 ± 1.88 | 6.45 ± 1.41 |
| NEUT% | 25.78 ± 9.17 | 28.11 ± 8.69 | 25.84 ± 9.06 |
| LY% | 67.28 ± 10.18 | 68.71 ± 10.43 | 66.53 ± 9.85 |
| RBC (109/L) | 10.19 ± 0.59 | 10.41 ± 1.32 | 9.69 ± 0.98 |
| Hb (g/L) | 148.84 ± 10.91 | 143.32 ± 8.77 | 146.62 ± 10.57 |
| PLT (109/L) | 565.17 ± 46.23 | 583.59 ± 52.18 | 569.78 ± 39.55 |
| ALT (U/L) | 35.09 ± 4.79 | 36.15 ± 6.18 | 35.62 ± 5.27 |
| AST (U/L) | 138.83 ± 19.54 | 135.82 ± 15.77 | 137.29 ± 17.85 |
| TP (g/L) | 65.69 ± 1.99 | 67.94 ± 1.54 | 65.75 ± 2.13 |
| ALB (g/L) | 16.34 ± 2.58 | 18.13 ± 1.86 | 18.26 ± 2.79 |
| BUN (mmol/L) | 6.57 ± 0.89 | 6.14 ± 0.54 | 6.46 ± 0.85 |
Abbreviations: ALT, alanine transaminase; ALB, albumin; AST, aspartate transaminase; BUN, blood urea nitrogen; GEM, gemcitabine; GEM-HSA-NP, gemcitabine-loaded human serum albumin nanoparticle; Hb, hemoglobin; KM, Kunming; LY%, lymphocyte %; NEUT%, neutrophil %; PLT, platelet; RBC, red blood cell; TP, total protein; WBC, white blood cell.
Conclusion
GEM is a nucleoside analog, its uptake into pancreatic cancer cells requires the mediation of hENT1. Low hENT1 expression results in GEM resistance. GEM-HSA-NP can be taken into tumor cells through endocytosis, and that will bypass the transport of hENT1. Therefore, GEM-HSA-NP can overcome GEM resistance induced by low hENT1 expression in pancreatic cancer, which suggests its potential role for the clinical application.
Supplementary materials
Table S1.
The proportion of each phase of cell cycle in BxPC-3 and SW1990 cell lines
| Group | BxPC-3
|
SW1990
|
||||
|---|---|---|---|---|---|---|
| G0–G1 phase (%) | S phase (%) | G2–M phase (%) | G0–G1 phase (%) | S phase (%) | G2–M phase (%) | |
| Control | 57.99 ± 1.22 | 30.58 ± 0.60 | 11.43 ± 0.78 | 49.93 ± 1.86 | 37.44 ± 1.46 | 12.63 ± 3.18 |
| NBMPR | 57.29 ± 0.95 | 31.55 ± 1.18 | 11.16 ± 0.94 | 50.34 ± 1.40 | 36.76 ± 1.01 | 12.89 ± 1.95 |
| GEM | 74.68 ± 1.64 | 24.12 ± 1.44 | 1.20 ± 0.24 | 62.50 ± 0.94 | 37.48 ± 0.94 | 0.02 ± 0.01 |
| GEM + NBMPR | 61.35 ± 1.30 | 28.16 ± 1.49 | 10.49 ± 0.85 | 53.57 ± 1.16 | 37.44 ± 1.25 | 8.99 ± 0.72 |
| GEM-C14 | 76.28 ± 1.05 | 22.66 ± 1.22 | 1.06 ± 0.27 | 61.77 ± 1.72 | 37.92 ± 1.41 | 0.31 ± 0.54 |
| GEM-C14 + NBMPR | 74.39 ± 1.29 | 24.67 ± 1.47 | 0.94 ± 0.28 | 61.36 ± 2.91 | 38.61 ± 2.91 | 0.03 ± 0.01 |
| GEM-HSA-NP | 75.16 ± 1.74 | 23.57 ± 1.45 | 1.27 ± 0.41 | 62.85 ± 1.29 | 37.01 ± 1.29 | 0.14 ± 0.01 |
| GEM-HSA-NP + NBMPR | 76.04 ± 1.57 | 22.92 ± 1.41 | 1.04 ± 0.26 | 66.64 ± 1.24 | 33.35 ± 1.24 | 0.01 ± 0.00 |
Note: Data are expressed as mean ± SD.
Abbreviations: GEM, gemcitabine; GEM-C14, 4-N-myristoyl-gemcitabine; GEM-HSA-NP, gemcitabine-loaded human serum albumin nanoparticle; NBMPR, S-(4-nitrobenzyl)-6-thioinosine.
Table S2.
The apoptosis rate of each group in BxPC-3 and SW1990 cell lines
| Group | BxPC-3
|
SW1990
|
||||
|---|---|---|---|---|---|---|
| Early apoptosis rate (%) | Late apoptosis rate (%) | Total apoptosis rate (%) | Early apoptosis (%) | Late apoptosis (%) | Total apoptosis (%) | |
| Control | 4.71 ± 0.44 | 10.64 ± 1.16 | 15.35 ± 0.71 | 3.10 ± 0.41 | 9.41 ± 1.16 | 12.51 ± 0.97 |
| NBMPR | 4.58 ± 0.35 | 10.90 ± 1.81 | 15.48 ± 1.55 | 3.06 ±0.21 | 9.46 ± 0.91 | 12.52 ± 0.73 |
| GEM | 6.20 ± 0.94 | 19.23 ± 1.19 | 25.43 ± 1.69 | 7.22 ± 0.31 | 29.11 ± 1.31 | 36.33 ± 1.30 |
| GEM + NBMPR | 4.18 ± 0.37 | 10.38 ± 0.86 | 14.56 ± 1.09 | 3.16 ± 0.37 | 10.61 ± 1.20 | 13.77 ± 0.93 |
| GEM-C14 | 7.29 ± 0.94 | 19.60 ± 0.78 | 26.89 ± 1.19 | 8.15 ± 0.81 | 21.95 ± 2.15 | 30.10 ± 1.34 |
| GEM-C14 + NBMPR | 8.49 ± 0.43 | 19.44 ± 1.88 | 27.93 ± 1.45 | 4.65 ± 1.19 | 21.33 ± 3.57 | 25.98 ± 2.41 |
| GEM-HSA-NP | 7.55 ± 0.69 | 21.41 ± 1.10 | 28.96 ± 1.11 | 6.54 ± 1.51 | 24.48 ± 1.26 | 31.02 ± 1.72 |
| GEM-HSA-NP + NBMPR | 8.78 ± 0.78 | 22.67 ± 0.78 | 31.45 ± 0.94 | 8.31 ± 0.97 | 24.73 ± 2.48 | 33.04 ± 1.59 |
Note: Data are expressed as mean ± SD.
Abbreviations: GEM, gemcitabine; GEM-C14, 4-N-myristoyl-gemcitabine; GEM-HSA-NP, gemcitabine-loaded human serum albumin nanoparticle; NBMPR, S-(4-nitrobenzyl)-6-thioinosine.
Acknowledgments
This study was financially supported by the National Natural Science Foundation of China (no 81772566).
Footnotes
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 4.Polireddy K, Chen Q. Cancer of the pancreas: molecular pathways and current advancement in treatment. J Cancer. 2016;7(11):1497–1514. doi: 10.7150/jca.14922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolfgang CL, Herman JM, Laheru DA, et al. Recent progress in pancreatic cancer. CA Cancer J Clin. 2013;63(5):318–348. doi: 10.3322/caac.21190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gillen S, Schuster T, Meyer ZBC, Friess H, Kleeff J. Preoperative/neoadjuvant therapy in pancreatic cancer: a systematic review and meta-analysis of response and resection percentages. PLoS Med. 2010;7(4):e1000267. doi: 10.1371/journal.pmed.1000267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taieb J, Pointet AL, Van Laethem JL, et al. What treatment in 2017 for inoperable pancreatic cancers. Ann Oncol. 2017;28(7):1473–1483. doi: 10.1093/annonc/mdx174. [DOI] [PubMed] [Google Scholar]
- 8.Mori R, Ishikawa T, Ichikawa Y, et al. Human equilibrative nucleoside transporter 1 is associated with the chemosensitivity of gemcitabine in human pancreatic adenocarcinoma and biliary tract carcinoma cells. Oncol Rep. 2007;17(5):1201–1205. [PubMed] [Google Scholar]
- 9.Saiki Y, Yoshino Y, Fujimura H, et al. DCK is frequently inactivated in acquired gemcitabine-resistant human cancer cells. Biochem Biophys Res Commun. 2012;421(1):98–104. doi: 10.1016/j.bbrc.2012.03.122. [DOI] [PubMed] [Google Scholar]
- 10.Cividini F, Filoni DN, Pesi R, Allegrini S, Camici M, Tozzi MG. IMP-GMP specific cytosolic 5′-nucleotidase regulates nucleotide pool and prodrug metabolism. Biochim Biophys Acta. 2015;1850(7):1354–1361. doi: 10.1016/j.bbagen.2015.03.017. [DOI] [PubMed] [Google Scholar]
- 11.Serdjebi C, Seitz JF, Ciccolini J, et al. Rapid deaminator status is associated with poor clinical outcome in pancreatic cancer patients treated with a gemcitabine-based regimen. Pharmacogenomics. 2013;14(9):1047–1051. doi: 10.2217/pgs.13.93. [DOI] [PubMed] [Google Scholar]
- 12.Hung SW, Marrache S, Cummins S, et al. Defective hCNT1 transport contributes to gemcitabine chemoresistance in ovarian cancer subtypes: overcoming transport defects using a nanoparticle approach. Cancer Lett. 2015;359(2):233–240. doi: 10.1016/j.canlet.2015.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao X, Wang X, Sun W, et al. Precision design of nanomedicines to restore gemcitabine chemosensitivity for personalized pancreatic ductal adenocarcinoma treatment. Biomaterials. 2018;158:44–55. doi: 10.1016/j.biomaterials.2017.12.015. [DOI] [PubMed] [Google Scholar]
- 14.Mackey JR, Mani RS, Selner M, et al. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998;58(19):4349–4357. [PubMed] [Google Scholar]
- 15.Damaraju VL, Damaraju S, Young JD, et al. Nucleoside anticancer drugs: the role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene. 2003;22(47):7524–7536. doi: 10.1038/sj.onc.1206952. [DOI] [PubMed] [Google Scholar]
- 16.Koczor CA, Torres RA, Lewis W. The role of transporters in the toxicity of nucleoside and nucleotide analogs. Expert Opin Drug Metab Toxicol. 2012;8(6):665–676. doi: 10.1517/17425255.2012.680885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Binenbaum Y, Na’ara S, Gil Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist Updat. 2015;23:55–68. doi: 10.1016/j.drup.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 18.Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann Oncol. 2006;17(suppl 5):v7–v12. doi: 10.1093/annonc/mdj941. [DOI] [PubMed] [Google Scholar]
- 19.Spratlin J, Sangha R, Glubrecht D, et al. The absence of human equilibrative nucleoside transporter 1 is associated with reduced survival in patients with gemcitabine-treated pancreas adenocarcinoma. Clin Cancer Res. 2004;10(20):6956–6961. doi: 10.1158/1078-0432.CCR-04-0224. [DOI] [PubMed] [Google Scholar]
- 20.Yamada R, Mizuno S, Uchida K, et al. Human equilibrative nucleoside transporter 1 expression in endoscopic ultrasonography-guided fine-needle aspiration biopsy samples is a strong predictor of clinical response and survival in the patients with pancreatic ductal adenocarcinoma undergoing gemcitabine-based chemoradiotherapy. Pancreas. 2016;45(5):761–771. doi: 10.1097/MPA.0000000000000597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu X, Di Y, Xie C, et al. An in vitro and in vivo study of gemcitabine-loaded albumin nanoparticles in a pancreatic cancer cell line. Int J Nanomedicine. 2015;10:6825–6834. doi: 10.2147/IJN.S93835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaksonen M, Roux A. Mechanisms of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2018;19(5):313–326. doi: 10.1038/nrm.2017.132. [DOI] [PubMed] [Google Scholar]
- 23.Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- 24.Shimada T, Nakanishi T, Tajima H, et al. Saturable hepatic extraction of gemcitabine involves biphasic uptake mediated by nucleoside transporters equilibrative nucleoside transporter 1 and 2. J Pharm Sci. 2015;104(9):3162–3169. doi: 10.1002/jps.24498. [DOI] [PubMed] [Google Scholar]
- 25.Li T, Dong ZR, Guo ZY, et al. Aspirin enhances IFN-α-induced growth inhibition and apoptosis of hepatocellular carcinoma via JAK1/STAT1 pathway. Cancer Gene Ther. 2013;20(6):366–374. doi: 10.1038/cgt.2013.29. [DOI] [PubMed] [Google Scholar]
- 26.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432(7015):316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 27.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med. 2009;361(16):1570–1583. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H, Word BR, Lyn-Cook BD. Enhanced efficacy of gemcitabine by indole-3-carbinol in pancreatic cell lines: the role of human equilibrative nucleoside transporter 1. Anticancer Res. 2011;31(10):3171–3180. [PubMed] [Google Scholar]
- 29.Makohon-Moore A, Iacobuzio-Donahue CA. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat Rev Cancer. 2016;16(9):553–565. doi: 10.1038/nrc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362(17):1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 31.Jung J, Seol HS, Chang S. The generation and application of patient-derived xenograft model for cancer research. Cancer Res Treat. 2018;50(1):1–10. doi: 10.4143/crt.2017.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lai Y, Wei X, Lin S, Qin L, Cheng L, Li P. Current status and perspectives of patient-derived xenograft models in cancer research. J Hematol Oncol. 2017;10(1):106. doi: 10.1186/s13045-017-0470-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.
The proportion of each phase of cell cycle in BxPC-3 and SW1990 cell lines
| Group | BxPC-3
|
SW1990
|
||||
|---|---|---|---|---|---|---|
| G0–G1 phase (%) | S phase (%) | G2–M phase (%) | G0–G1 phase (%) | S phase (%) | G2–M phase (%) | |
| Control | 57.99 ± 1.22 | 30.58 ± 0.60 | 11.43 ± 0.78 | 49.93 ± 1.86 | 37.44 ± 1.46 | 12.63 ± 3.18 |
| NBMPR | 57.29 ± 0.95 | 31.55 ± 1.18 | 11.16 ± 0.94 | 50.34 ± 1.40 | 36.76 ± 1.01 | 12.89 ± 1.95 |
| GEM | 74.68 ± 1.64 | 24.12 ± 1.44 | 1.20 ± 0.24 | 62.50 ± 0.94 | 37.48 ± 0.94 | 0.02 ± 0.01 |
| GEM + NBMPR | 61.35 ± 1.30 | 28.16 ± 1.49 | 10.49 ± 0.85 | 53.57 ± 1.16 | 37.44 ± 1.25 | 8.99 ± 0.72 |
| GEM-C14 | 76.28 ± 1.05 | 22.66 ± 1.22 | 1.06 ± 0.27 | 61.77 ± 1.72 | 37.92 ± 1.41 | 0.31 ± 0.54 |
| GEM-C14 + NBMPR | 74.39 ± 1.29 | 24.67 ± 1.47 | 0.94 ± 0.28 | 61.36 ± 2.91 | 38.61 ± 2.91 | 0.03 ± 0.01 |
| GEM-HSA-NP | 75.16 ± 1.74 | 23.57 ± 1.45 | 1.27 ± 0.41 | 62.85 ± 1.29 | 37.01 ± 1.29 | 0.14 ± 0.01 |
| GEM-HSA-NP + NBMPR | 76.04 ± 1.57 | 22.92 ± 1.41 | 1.04 ± 0.26 | 66.64 ± 1.24 | 33.35 ± 1.24 | 0.01 ± 0.00 |
Note: Data are expressed as mean ± SD.
Abbreviations: GEM, gemcitabine; GEM-C14, 4-N-myristoyl-gemcitabine; GEM-HSA-NP, gemcitabine-loaded human serum albumin nanoparticle; NBMPR, S-(4-nitrobenzyl)-6-thioinosine.
Table S2.
The apoptosis rate of each group in BxPC-3 and SW1990 cell lines
| Group | BxPC-3
|
SW1990
|
||||
|---|---|---|---|---|---|---|
| Early apoptosis rate (%) | Late apoptosis rate (%) | Total apoptosis rate (%) | Early apoptosis (%) | Late apoptosis (%) | Total apoptosis (%) | |
| Control | 4.71 ± 0.44 | 10.64 ± 1.16 | 15.35 ± 0.71 | 3.10 ± 0.41 | 9.41 ± 1.16 | 12.51 ± 0.97 |
| NBMPR | 4.58 ± 0.35 | 10.90 ± 1.81 | 15.48 ± 1.55 | 3.06 ±0.21 | 9.46 ± 0.91 | 12.52 ± 0.73 |
| GEM | 6.20 ± 0.94 | 19.23 ± 1.19 | 25.43 ± 1.69 | 7.22 ± 0.31 | 29.11 ± 1.31 | 36.33 ± 1.30 |
| GEM + NBMPR | 4.18 ± 0.37 | 10.38 ± 0.86 | 14.56 ± 1.09 | 3.16 ± 0.37 | 10.61 ± 1.20 | 13.77 ± 0.93 |
| GEM-C14 | 7.29 ± 0.94 | 19.60 ± 0.78 | 26.89 ± 1.19 | 8.15 ± 0.81 | 21.95 ± 2.15 | 30.10 ± 1.34 |
| GEM-C14 + NBMPR | 8.49 ± 0.43 | 19.44 ± 1.88 | 27.93 ± 1.45 | 4.65 ± 1.19 | 21.33 ± 3.57 | 25.98 ± 2.41 |
| GEM-HSA-NP | 7.55 ± 0.69 | 21.41 ± 1.10 | 28.96 ± 1.11 | 6.54 ± 1.51 | 24.48 ± 1.26 | 31.02 ± 1.72 |
| GEM-HSA-NP + NBMPR | 8.78 ± 0.78 | 22.67 ± 0.78 | 31.45 ± 0.94 | 8.31 ± 0.97 | 24.73 ± 2.48 | 33.04 ± 1.59 |
Note: Data are expressed as mean ± SD.
Abbreviations: GEM, gemcitabine; GEM-C14, 4-N-myristoyl-gemcitabine; GEM-HSA-NP, gemcitabine-loaded human serum albumin nanoparticle; NBMPR, S-(4-nitrobenzyl)-6-thioinosine.