

Abstract
Aims/Introduction
Glimepiride is a sulfonylurea known to have unique insulin mimetic and sensitizing effects. We aimed to study the efficacy of glimepiride in a patient with type A insulin resistance syndrome.
Materials and Methods
A 15‐year‐old girl with type A insulin resistance syndrome was treated with glimpiride for 6 months. Self‐monitoring of blood glucose was recorded, and oral glucose tolerance tests on glucose and insulin were measured during the treatment. Hyperinsulinemic euglycemic clamp was used to evaluate whole‐body insulin sensitivity before and after the treatment.
Results
A novel heterozygous missense mutation at exon 19 (c.3427A>T) in the tyrosine kinase domain of the INSR gene was identified, causing an amino acid replacement of phenylalanine for isoleucine at codon 1143 (Ile1143Phe). Before the treatment, the patient's glycated hemoglobin was 7.0%, plasma glucose during oral glucose tolerance test was 6.7, 12.8 and 17.3 mmol/L, and simultaneous serum insulin was 80.7, 137.5 and >300 μU/mL. There were no significant differences between self‐monitored blood glucose measured at each time‐point among different glimepiride dosages, or during the 14 weeks when glimepiride was used at its maximal dosage (6 mg/day). Oral glucose tolerance test showed little change in plasma glucose and serum insulin. Glycated hemoglobin decreased by 0.8% after the treatment. However, a euglycemic clamp study showed that the M value decreased from 5.25 to 2.90 mg/kg/min, showing increased insulin resistance.
Conclusions
Treatment with glimepiride did not improve insulin sensitivity in a patient with type A insulin resistance syndrome carrying Ile1143Phe heterozygous mutation in the INSR gene. Large‐scale long‐term studies assembled worldwide are required to optimize treatment algorithms for patients with type A insulin resistance syndrome.
Keywords: Glimepiride, Novel mutation, Type A insulin resistance syndrome
Introduction
Type A insulin resistance syndrome is on the right side (from severe to mild) of the congenital severe insulin resistance syndrome spectrum due to mutations in the insulin receptor (INSR) gene. On the left side are Donohue syndrome and Rabson–Mendenhall syndrome caused by homozygous mutation or compound heterozygous mutations involving the α subunit of the INSR gene, and patients usually do not survive to adulthood. In contrast, type A insulin resistance syndrome is relatively mild. It is usually diagnosed in lean female patients complaining of hirsutism, oligomenorrhea, acne and acanthosis nigricans during puberty, which results from hyperandrogenism due to severe insulin resistance and polycystic ovaries. Genetic screening usually identifies a single heterozygous mutation involving the tyrosine kinase domain in the β subunit of the INSR gene.1 The mutations in this area cause insulin resistance in a dominant‐negative fashion.2, 3, 4 In heterozygous mutation carriers, only the symmetrical wild‐type receptor (a2[βwt]2) is active as a tyrosine kinase, whereas the hybrid α2βwtβmut and the symmetrical a2(βmut)2 receptors are inactive.5 Thus, the autophosphorylation at multiple sites in the tyrosine kinase domain in response to insulin binding is blocked in the defective receptor, so thus the subsequent signaling transduction pathway that induced GLUT4 translocation to the membrane, as well as the synthesis of glycogen and lipid.1
There are not many reports in the literature about the treatment of type A insulin resistance syndrome. Meformin and thiazolidinediones, two traditional insulin sensitizers, were the initial choices of medication in sporadic case reports. Glimepiride, a potent insulin secretogogoue to treat type 2 diabetes, has been shown to have unique insulin mimetic/sensitizing effects by acting on the caveolin/detergent‐insoluble glycolipid‐ enriched rafts (DIG) areas, causing the activation of non‐receptor tyrosine kinase (non‐RTK), such as pp59Lyn, to phosphorylates insulin receptor substrate proteins, which are downstream of the insulin receptor, initiating the metabolic insulin‐mimetic signaling to the lipid and glycogen synthesis pathways, and the GLUT4 translocation.6 A recent study7 also showed that glimepiride had peroxisome proliferator‐activated receptor‐γ (PPARγ) agonist activity, whose potencies were 16–25% of the maximum level achieved by pioglitazone.
In the present study, we aimed to investigate the efficacy of glimepiride in a patient with type A insulin resistance syndrome due to a novel heterozygous missense mutation of Ile1143Phe in exon 19 of the INSR gene. Hyperinsulinemic euglycemic clamp was used to evaluate the change in insulin sensitivity before and after the treatment.
Methods
Case report
The participant was a 15‐year‐old girl who was born at full‐term from non‐consanguineous parents. Her birthweight was 2.2 kg (P3). She was found to be hirsute since the age of 5 years. Secondary sex characters and accelerated growth occurred from the age of 13 years, but no menarche occurred. Her body mass index (BMI) was 18.4 kg/m2. Plasma glucose during a 5‐h oral glucose tolerance test was 7.2, 17.1, 18.6, 14.2, 12.9 and 6.8 mmol/L at 0, 1‐h, 2‐h, 3‐h, 4‐h and 5‐h time‐points. Simultaneous measurements of serum insulin were 96.1, 256.8, >300, >300, >300 and 258.9 μU/mL. The patient's glycated hemoglobin (HbA1c) was 6.8%. Ultrasound showed polycystic ovaries. Her serum testosterone was mildly elevated to 0.97 ng/mL (normal range 0.1–0.75 ng/mL). Diagnosis of type A insulin resistance syndrome was made, and the patient was prescribed pioglitazone 15 mg/day for 6 consecutive months, but no improvement in plasma glucose nor serum insulin was evident. Serum fasting insulin was 80.7 μU/mL, and HbA1c was 7.0% after the suspension of pioglitazone. No menstrual onset was recorded during or after the treatment.
Genetic analysis
Peripheral blood (5 mL) of the patient was sent to BGI Diagnosis Co., Shenzhen, Guangdong province, China (certified with ISO 15189), for next generation sequencing, targeting a panel of 11 genes (INSR, ABCC8, KCNJ11, GCK, HADH, GLUD1, HNF4A, HNF1A, UCP2, FOXA2, SLC16A1) known to be associated with congenital hyperinsulinism.8 Genomic DNA was extracted using QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany), then sheared by Covaris LE220 (Woburn, MA, USA) to generate a paired‐end library (200–250 bp). The library was enriched by array hybridization, followed by elution and post‐capture amplification. The products were subjected to Agilent 2100 Bioanalyzer (Agilent, CA, USA) and ABI StepOne (Thermo Fisher Scientific, Waltham, MA, USA) to estimate the magnitude of enrichment. After quality control, captured library sequencing was carried out on Illumina HiSeq2500 Analyzers (Illumina, San Diego, CA, USA) for 90 cycles per read to generate paired‐end reads. Image analysis, error estimation and base calling were carried out using Illumina Pipeline software (version 1.3.4; Illumina, San Diego, CA, USA) to generate raw data.
To detect potential variants, bioinformatics processing and data analysis was carried out. Filtering criteria to generate “clean reads” was used as reported in the literature.9 The “clean reads” (with a length of 90 bp) derived from targeted sequencing and filtering were then aligned to the human genome reference (hg19) using the Burrows Wheeler Aligner Multi‐Vision software package (available from: http://maq.sourceforge.net). After alignment, the output files were used to carry out sequencing coverage and in‐depth analysis of the target region, single‐ nucleotide variants and insertion/deletion calling.10
SOAPsnp software (available from http://soap.genomics.org.cn under GPL license) and Samtools (available from: http://samtools.sourceforge.net) was used to detect single‐ nucleotide variants and insertion/deletions. All single‐nucleotide variants and insertion/deletions were filtered and estimated through multiple databases, including NCBI dbSNP, HapMap, 1,000 human genome dataset and database of 100 Chinese healthy adults.11
To predict the effect of missense variants, dbNSFP was used containing well established in silico prediction programs (Polyphen2, MutationTaster, Scale‐Invariant Feature Transform, LRT and PhyloP). Pathogenic variants were assessed under the protocol issued by the American College of Medical Genetics.12 The Human Gene Mutation Database was used to screen mutations reported in published studies. All mutations and potential pathogenic variants were validated using conventional Sanger sequencing methods.
Hyperinsulinemic euglycemic clamp study
The hyperinsulinemic euglycemic clamp was carried out before and after the treatment with glimepiride. After a 12‐h overnight fast, an intravenous cannula was inserted into the antecubital vein of both arms. Blood from the right was arterialized by placing the arm continuously in an electric warmer set at approximately 50°C, and sampled for plasma glucose and serum insulin. First‐phase insulin secretion was determined by an intravenous glucose tolerance test before the initiation of insulin infusion. Serum insulin concentration was sampled at 0, 1, 2, 4, 6 and 10 min after intravenous injection of 50% glucose 50 mL within 1 min. Then a primed (3 mU/kg/min) and continuous (1 mU/kg/min) infusion of regular insulin was started, and plasma glucose was measured with a glucose analyzer (Beckman Instruments, Brea, CA, USA) at 5‐min intervals throughout the clamp study. A variable infusion of 20% dextrose was used to maintain euglycemia (4.4–5.0 mmol/L). Steady‐state was achieved approximately 120 min after insulin infusion. Whole‐body glucose uptake was estimated as the metabolic rate (M value), defined as the infusion rate of exogenous glucose, corrected for urinary glucose losses and the glucose space correction. The clamp data were compared with those of six normal glucose tolerance (NGT) young (aged 22.8 ± 0.4 years) women who had matched BMI (18.8 ± 1.3 kg/m2) and no familial history of diabetes, as well as 13 newly diagnosed patients with type 2 diabetes and 17 individuals with impaired glucose tolerance (IGT).
Treatment protocol with glimepiride
Glimepiride was started the next day after hyperinsulinemic euglycemic clamp from a small dose (1 mg/d), and titrated every 2 weeks by 1 mg each time, until 6 mg/day was achieved as the maximum, and maintained the dosage thereafter. The whole treatment period lasted for a total of 6 months. Self‐monitored blood glucose during fasting, 2 h after each meal, and that before dinner and bedtime was recorded on 1 day every week. Hypoglycemia and other adverse events were carefully monitored. Oral glucose tolerance tests were carried out during the first 3 months of the titration for the measurement of plasma glucose and serum insulin concentration. One week after the suspension of glimepiride, hyperinsulinemic euglycemic clamp study was repeated.
The study was approved by the Medical Research and Ethics Committee of the First Affiliated Hospital of Sun Yat‐sen University. Written informed consent was obtained from all the participants.
Results
Novel heterozygous missense mutation (Ile1143Phe) identified in the INSR gene
There were in total 17 variants identified in either the exons or the flanking intron areas in eight out of the 11 screened genes, as listed in Table 1. Most of the variants were documented in the 1,000 Genome project database with varying frequencies, and showed benign changes or polymorphisms. Four heterozygous mutations occurred in the INSR gene, two of which were silent mutations in the coding region (Ala550Ala in exon 8 and His1085His in exon 17). A novel heterozygous missense mutation at exon 19 (c.3427A>T) in the catalytic group of the tyrosine kinase domain in the β subunit of the INSR gene was identified, causing an amino acid replacement of phenylalanine for isoleucine, which has never been reported in the literature (Figure 1a). Prediction of the impact of the altered amino acid using Polyphen2 and MutationTaster programs and so on showed that the missense mutation was deleterious. The isoleucine 1,143 is exquisitely conserved among orthologs in different species, thus the mutation is probably pathogenic (Figure 1b). Another novel polymorphism (c.653‐5_‐4 delTC) in intron 2 was also identified, the significance of which was illusive.
Table 1.
Variants identified in the coding regions as well as the flanking intron regions in genes related to severe insulin resistance
| No. | Gene | Transcript | Nucleotide change | Amino acid change | Chromosome | Exon/intron | Heterozygosity | Frequency |
|---|---|---|---|---|---|---|---|---|
| 1 | SLC16A1 | NM_003051 | c.1470T>A | p.Asp490Glu | chr1:113456546 | Exon 5 | Hom | 0.4533 |
| 2 | GLUD1 | NM_005271 | c.942A>G | p.Leu314Leu | chr10:88820789 | Exon 7 | Hom | 0.1062 |
| 3 | KCNJ11 | NM_001166290 | c.748G>A | p.Val250Ile | chr11:17408630 | Exon 2 | Hom | 0.5321 |
| 4 | KCNJ11 | NM_001166290 | c.309C>T | p.Ala103Ala | chr11:17409069 | Exon 2 | Het | 0.3544 |
| 5 | ABCC8 | NM_000352 | c.4105G>T | p.Ala1369Ser | chr11:17418477 | Exon 33 | Hom | 0.5256 |
| 6 | ABCC8 | NM_000352 | c.3819G>A | p.Arg1273Arg | chr11:17419279 | Exon 31 | Het | 0.4002 |
| 7 | ABCC8 | NM_000352 | c.1947G>A | p.Lys649Lys | chr11:17449929 | Exon 14 | Het | 0.2729 |
| 8 | ABCC8 | NM_000352 | c.207T>C | p.Pro69Pro | chr11:17496516 | Exon 2 | Het | 0.5256 |
| 9 | UCP2 | NM_003355 | c.164C>T | p.Ala55Val | chr11:73689104 | Exon 4 | Het | 0.5009 |
| 10 | INSR | NM_000208 | c.3427A>T | p.Ile1143Phe | chr19:7122727 | Exon 19 | Het | 0 |
| 11 | INSR | NM_000208 | c.3255C>T | p.His1085His | chr19:7125297 | Exon 17 | Het | 0.3782 |
| 12 | INSR | NM_000208 | c.1650G>A | p.Ala550Ala | chr19:7166376 | Exon 8 | Het | 0.3416 |
| 13 | FOXA2 | NM_153675 | c.1188A>G | p.Gln396Gln | chr20:22562674 | Exon 3 | Hom | 0.881 |
| 14 | GCK | NM_000162 | c.1253 + 8C>T | – | chr7:44185088 | Intron 9 | Het | 0.3791 |
| 15 | ABCC8 | NM_000352 | c.2117‐3C>T | – | chr11:17448704 | Intron 15 | Het | 0.4451 |
| 16 | INSR | NM_000208 | c.653‐5_‐4 delTC | – | chr19:7184652..718 | Intron 2 | Het | 0 |
| 17 | HNF4A | NM_175914 | c.50‐5C>T | – | chr20:43034693 | Intron 1 | Het | 0.3095 |
Het, heterozygous; Hom, homozygous.
Figure 1.
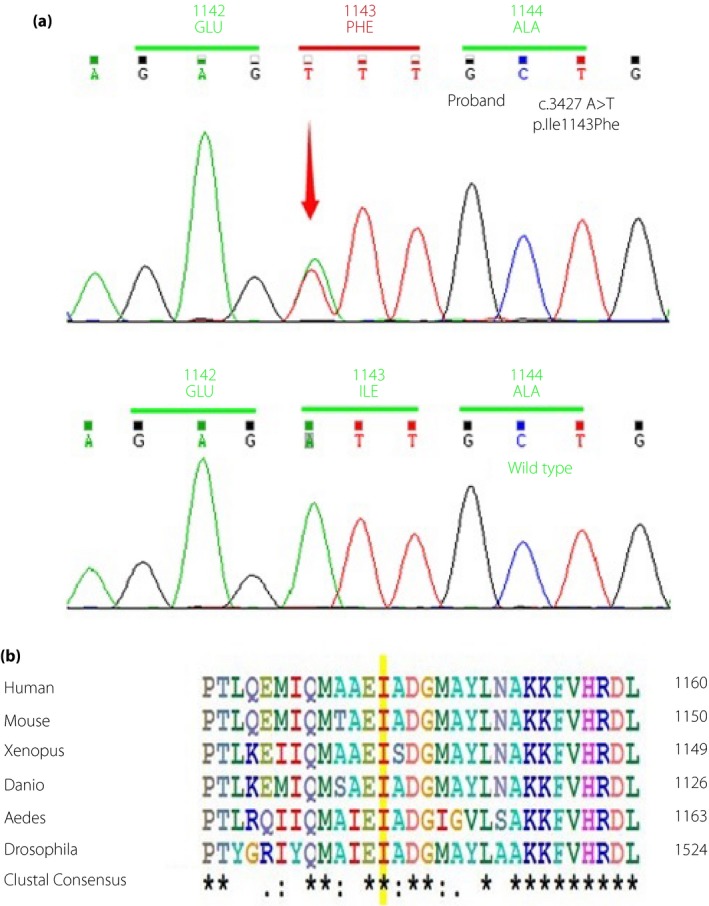
(a) Sequencing of the insulin receptor (INSR) gene in the patient and wild‐type control. The arrow shows the nucleotide position at 3,427, where the original A was replaced by T, resulting in a heterozygous missense mutation of phenylalanine for isoleucine at codon 1,143. (b) Alignment of the insulin receptor amino acid sequences from different species. The isoleucine 1,143 residue is highlighted in yellow for each sequence.
Direct sequencing of the INSR gene was also carried out in both parents and the patient's younger brother, none of who were hyperinsulinemic (Table2). The Ile1143Phe missense mutation was not identified in any of them. Microsatellite analysis was suggested to confirm the family relationship, but rejected by the parents. Thus, it was not certain whether the mutation had arisen de novo. Genetic screening was also declined outside of the family.
Table 2.
Plasma glucose and serum Insulin levels in family members
| Father | Mother | Brother | |
|---|---|---|---|
| Age (years) | 48 | 48 | 13 |
| BMI (kg/m2) | 24.6 | 18.2 | 18.9 |
| HbA1c (%) | 6.6 | 5.3 | 4.8 |
| OGTT glucose (mmol/L) | |||
| 0 min | 8.4 | 5.3 | 5.2 |
| 30 min | 15.0 | 8.4 | 11.0 |
| 120 min | 12.2 | 9.1 | 7.2 |
| OGTT insulin (μU/mL) | |||
| 0 min | 8.5 | 1.5 | 2.6 |
| 30 min | 51.8 | 21.1 | 64.3 |
| 120 min | 64.4 | 64.3 | 51.7 |
BMI, body mass index; Hba1c, glycated hemoglobin; OGTT, oral glucose tolerance test.
Clinical characteristics of the patient in comparison with NGT, IGT and type 2 diabetes controls
The anthropometric values, glucose and insulin concentration during a 75‐g oral glucose tolerance test, lipid profiles and M value during the hyperinsulinemic euglycemic clamp of the patient was compared with NGT and glucose intolerant controls (Table 3). The values of the patient that were out of the 95% confidence intervals of the control groups were denoted. As indicated, the patient had much lower percentage of body fat as compared with NGT participants with similar BMI, waist circumference and waist‐to‐hip ratio. The HbA1c of the patient was comparable with that of the individuals with diabetes, whereas serum insulin levels were significantly higher. However, serum lipid profiles of the patient were not deranged in contrast to those of the IGT and diabetes controls. The M value was the highest in the NGT participants, and decreased with the impairment of glucose tolerance. The M value of the patient was comparable with that of the individuals with IGT and diabetes before the treatments.
Table 3.
Clinical characteristics of the patient and that of the control individuals with normal glucose tolerance, impaired glucose tolerance and type 2 diabetes
| Patient Ile1143Phe | NGT control | IGT | T2DM | |
|---|---|---|---|---|
| n | – | 6 | 17 | 13 |
| Age (years) | 15 | 22.8 ± 0.4* | 53.1 ± 6.6** | 50.9 ± 7.3 *** |
| Sex (female/male) | Female | Female | 14/3 | 7/6 |
| BMI (kg/m2) | 18.9 | 18.8 ± 1.3 | 26.2 ± 2.7** | 26.4 ± 2.8 *** |
| Waist (cm) | 62.0 | 65.0 ± 4.9 | 84.8 ± 6.7** | 89.8 ± 6.9 *** |
| Waist/hip ratio | 0.72 | 0.72 ± 0.05 | 0.88 ± 0.06** | 0.94 ± 0.07 *** |
| Fat% | 10.9 | 20.3 ± 5.6* | 31.7 ± 6.3** | 30.6 ± 9.5 *** |
| OGTT glucose (mmol/L) | ||||
| 0 min | 6.7 | 4.8 ± 0.3* | 5.1 ± 0.4** | 7.5 ± 1.6 |
| 30 min | 12.8 | 7.4 ± 0.8* | 10.2 ± 1.7** | 13.6 ± 2.2 |
| 120 min | 17.3 | 5.0 ± 1.3* | 8.9 ± 1.0** | 15.7 ± 2.9 |
| OGTT insulin (μU/mL) | ||||
| 0 min | 80.7 | 4.9 ± 2.4* | 8.4 ± 6.4** | 11.7 ± 5.8 *** |
| 30 min | 137.5 | 76.0 ± 91.8* | 93.4 ± 44.4** | 139.0 ± 74.8 |
| 120 min | >300 | 30.3 ± 17.0* | 87.9 ± 43.5** | 99.1 ± 33.4 *** |
| HbA1c (%) | 7.0 | 5.3 ± 0.2* | 6.0 ± 0.4** | 7.3 ± 1.2 |
| Cholesterol (mmol/L) | 4.8 | – | 6.0 ± 1.0** | 5.4 ± 1.1 *** |
| Triglyceride (mmol/L) | 1.6 | – | 1.8 ± 0.7 | 2.5 ± 1.2 *** |
| LDL‐c (mmol/L) | 2.9 | – | 4.3 ± 1.0** | 3.8 ± 1.0 *** |
| HDL‐c (mmol/L) | 1.3 | – | 1.2 ± 0.3 | 1.1 ± 0.2 *** |
| M value (mg/kg/min) | ||||
| Clamp 1 | 5.25 | 10.69 ± 5.25* | 5.91 ± 1.95 | 4.83 ± 2.81 |
| Clamp 2 | 2.90 | 10.69 ± 5.25* | 5.91 ± 1.95** | 4.83 ± 2.81*** |
*Patient's value outside the range of the 95% confidence interval of the individuals with normal glucose tolerance (NGT). **Patient's value outside the range of the 95% confidence interval of the individuals with impaired glucose tolerance (IGT). ***Patient's value outside the range of the 95% confidence interval of the type 2 diabetes patients. BMI, body mass index; Hba1c, glycated hemoglobin; HDL‐c, high‐density lipoprotein cholesterol; LDL‐c, low‐density lipoprotein cholesterol; OGTT, oral glucose tolerance test.
Intravenous glucose tolerance tests in the NGT participants and patients with heterozygous INSR gene mutations (Ile1143Phe vs Arg1174Trp)
First‐phase insulin secretion was evaluated in the patient and six individuals with NGT using an intravenous glucose tolerance test before the initiation of insulin infusion during the hyperinsulinemic euglycemic clamp. The acute insulin secretion profile was also compared with another patient diagnosed with type A insulin resistance syndrome with a heterozygous missense mutation (Arg1174Trp) in exon 20 of the INSR gene, as we had described previously, and followed for 14 years up till now.13, 14
Briefly, the patient with the Arg1174Trp mutation was aged 16 years at her first visit. She was slim (BMI 17.8 kg/m2), and complained of intermittent hunger, fatigue and dizziness before lunch or bedtime. She also presented with hirsutism, oligomenorrhea and polycystic ovaries. She was found to have hyperinsulinemic hypoglycemia, severe insulin resistance and diabetes. No pharmaceutical intervention was given to the patient other than yearly follow up on her glucose, insulin profiles and menstruation. With the passage of her puberty, insulin hypersecretion, glucose deterioration and hypoglycemic symptoms tended to relieve. Her menstruation period was somehow regular without hormonal replacement therapy. Her HbA1c in this year's follow up (at the age of 30 years) was 5.3%, and her fasting insulin concentration dropped to 14.3 μU/mL, much lower than 210 μU/mL when she was aged 16 years.
As shown in Figure 2a, the fasting serum insulin of the individuals with NGT was relatively low (4.93 ± 2.02 μU/mL), then gained an averaged 17‐fold increment at 2 min after glucose injection (85.0 ± 60.5 μU/mL). In contrast, the patient with the Ile1143Phe mutation had severe fasting hyperinsulinemia, but lost first‐phase insulin secretion, which was similar to that seen in patients with type 2 diabetes. However, the patient with the Arg1174Trp mutation maintained a sharp peak of insulin secretion 17‐fold of the fasting state (240.6 vs 14.3 μU/mL), and the increment fold was comparable with the individuals with NGT.
Figure 2.
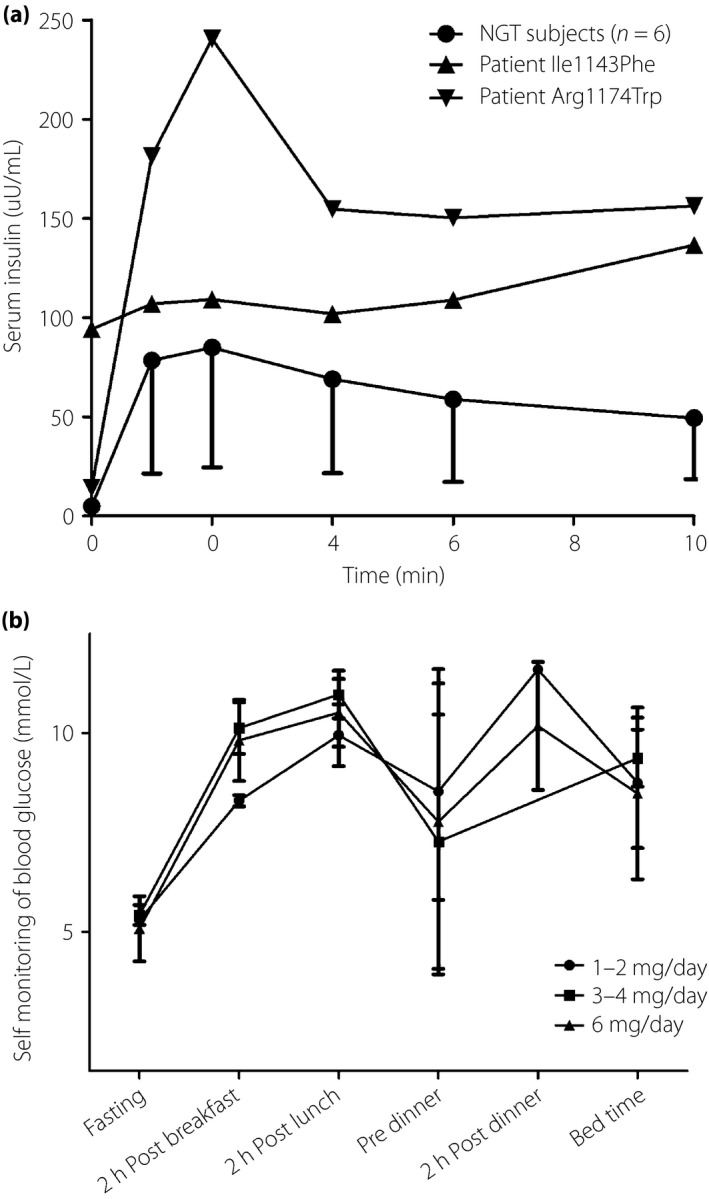
(a) Comparison of first‐phase insulin secretion induced by intravenous glucose tolerance test in the six individuals with normal glucose tolerance (NGT; circle) and that of the patient with the Ile1143Phe mutation (inverse triangle) and patient with the Arg1174Trp mutation (triangle). (b) Self‐monitored blood glucose in the patient carrying the Ile1143Phe mutation during the treatment with glimepiride by different dosages.
Efficacy and safety of glimepiride treatment in the patient with the Ile1143Phe mutation
Figure 2b shows the self‐monitored blood glucose of the patient during the titration of glimepiride from 1 mg/day to 6 mg/day. There was no significant difference between glucose concentration measured at each time‐point among different glimepiride dosages. When the maximal dosage (6 mg/day) was achieved and maintained for a total of 14 weeks, there was no significant difference between glucose concentrations measured at each time‐point between each week. Oral glucose tolerance test during the titration period showed that there were few changes in plasma glucose and serum insulin levels (Table 4). HbA1c at the end of the treatment was 6.2%, decreased by 0.8%, as compared with that (7%) before the treatment. However, hyperinsulinemic euglycemic clamp study showed that the M value further decreased to 2.90 mg/kg/min, indicating increased insulin resistance after the treatment.
Table 4.
Oral glucose tolerance test during glimepiride treatment
| Glimepiride dosage | Plasma glucose (mmol/L) | Serum insulin (μU/mL) | ||||
|---|---|---|---|---|---|---|
| 0 | 30 min | 120 min | 0 | 30 min | 120 min | |
| 2 mg/day | 5.8 | 12.4 | 16.8 | 75.69 | 154.12 | 818.19 |
| 4 mg/day | 5.1 | 11.8 | 17.3 | 70.10 | 192.03 | 858.40 |
| 6 mg/day | 5.7 | 10.9 | 15.0 | 68.02 | 135.22 | 625.75 |
Mild‐to‐moderate hypoglycemic symptoms (hunger and trembling) occurred several times (a total of 5 times in 6 months) during the treatment, especially when meals were delayed. The nadir value was 2.2 mol/L, and occurred just once before bedtime at the dosage of 6 mg/day. The symptoms were relieved rapidly with food ingestion. The patient gained 1 kg during the whole treatment period. No other adverse event was recorded during or after the treatment.
Discussion
In the present study, we reported a 15‐year‐old girl diagnosed with type A insulin resistance syndrome carrying a novel heterozygous missense mutation of Ile1143Phe in exon 19 of the INSR gene. Even though we did not carry out detailed experiments to check for the functional consequence of the mutation, multiple well‐established in silico prediction programs showed that the mutation is hazardous. Furthermore, the phenotype of the patient (lean female, small for gestational age, hirsutism, amenorrhea, polycystic ovaries, severe hyperinsulinemia) matched the common genotypes of the mutation occurring in the tyrosine kinase domain of the INSR gene. Thus, we believe that the mutation is pathogenic and is responsible for the severe insulin resistance in this patient. We treated the patient with glimepiride for 6 months. Even though her HbA1c decreased by 0.8% after the treatment, her serum insulin during oral glucose tolerance test, or insulin resistance as evaluated by hyperinsulinemic euglycemic clamp showed that glimepiride did not improve insulin sensitivity in this patient.
Glimepiride is a potent insulin secretogogoue, the efficacy and safety of which has long been validated in clinical practice for the treatment of type 2 diabetes. Thus, it might be disputable to use glimepiride in patients with severe hyperinsulinemia. Our assumption was based on the two unique insulin‐sensitizing aspects of glimepiride. First, Müller et al.6 proposed the working model for direct insulin mimetic and sensitizing signaling of glimepiride based on rigorous in vitro experiments. Glimepiride partitions directly into the caveolin/DIG areas of the adipocyte or myocyte plasma membranes in a non‐saturable and time‐dependent fashion, thereby causing redistribution of specific DIG/caveolin/non‐RTK components. An acylated non‐RTK, such as pp59Lyn, is thus freed from inhibition, and dissociates from caveolin and migrates into the non‐DIG areas of the plasma membrane. The activated non‐RTK (pp59Lyn) then bypasses the insulin receptor and phosphorylates insulin receptor substrate proteins directly, and initiates downstream signaling of lipid and glycogen synthesis, as well as GLUT4 translocation through the phosphatidylinositol‐4,5‐bisphosphate 3‐kinase pathway.15, 16 In contrast, Fukuen et al.7 found that glimepiride specifically induced the transcriptional activity of PPARγ in luciferase reporter assays. It directly bound to PPARγ in a manner competitive with rosiglitazone.17 In 3T3‐L1 adipocytes, glimepiride stimulated the transcriptional activity of the gene promoter containing PPAR‐responsive element and altered messenger ribonucleic acid levels of PPARγ target genes including aP2, leptin and adiponectin. The data strongly suggested that glimepiride had PPARγ agonist activity, whose potency was approximately 16–25% of the maximum level achieved by pioglitazone.7 All these data suggested that glimepiride improves insulin sensitivity through multiple signaling pathways. Unfortunately, we could not validate its insulin‐sensitizing effect in this patient. Adipocyte membrane is the major site that glimepiride acts on. As the patient had relatively less fatty mass, we could not exclude it to be partially responsible for the null effect of glimepiride. In contrast, the proportion of glimepriride under routine treatment concentration that initiates the activation of the DIGs–caveolin–pp59Lyn pathway is elusive, thus it is not clear whether longer treatment duration or a higher dose of glimepiride could improve insulin sensitivity. Further study of constructing cell lines or animal models with the corresponding defective insulin receptor, followed by experiments on the optimal concentration of the insulin‐sensitizing effect of glimepiride is warranted.
Type A insulin resistance syndrome is usually diagnosed in lean adolescent girls complaining of hirsutism, oligomenorrhea, acne and acanthosis nigricans, which results from hyperandrogenism due to severe insulin resistance and polycystic ovaries. However, the phenotypes usually vary in patients with different gene mutations; for example, the degree of hyperinsulinemia. The fasting and post‐load serum insulin level in the patient with the Arg1174Trp mutation (at the age of 16 years) was 3–4‐fold higher than the patient with the Ile1143Phe mutation in the present study. Hyperinsulinemic hypoglycemia was the major clinical feature in the former, but absent in the latter. The first‐phase insulin secretion remained intact in the patient with the Arg1174Trp mutation during the 14 years of follow up. However, the patient with the Ile1143Phe mutation lost the acute insulin response to glucose injection at the early stage of diagnosis, and the impact on glucose metabolism in the future remains to be determined. Therefore, we could infer from this observation that the efficacies of insulin sensitizers, besides glimepiride, might also vary among different INSR gene mutation carriers. Thus, the null effect in the present case is not necessarily the same in other patients with type A insulin resistance syndrome.
Treatment of severe insulin resistance syndrome due to insulin receptoropathy is usually empirical and lacks evidence.18 Metformin and thiazolidinediones were used even in infants with Donohue syndrome and Rabson–Mendenhall syndrome in addition to a huge amount of daily insulin and rhIGF‐1, but were not consistently effective.19, 20 Treatment with leptin was reported in multiple cases of Rabson–Mendenhall syndrome.21, 22 Brown et al 22 studied the effect of metreleptin in five patients for >12 months, two of which were treated for 10 years. HbA1c declined with each cycle of metreleptin and rose with each withdrawal. Glycemic improvement in these patients was partly explained by weight loss associated with the treatment.22 Acarbose was recommended in patients with type A insulin resistance syndrome who showed postprandial hyperglycemia and delayed hypoglycemia.18 The efficacies of thiazolidinediones on type A insulin resistance syndrome varied. Hattori et al.23 reported a female patient with heterozygous deletion mutation of Leu1026Del at exon 17 in the INSR gene, and was treated with pioglitazone for 10 months. Serum insulin, glucose levels and hyperandrogenism were ameliorated, and the patient resumed menstruation 2 months after the treatment.23 In contrast, Vestergaard et al.24 reported two brothers with biallelic deletion mutation at exon 17 of the INSR gene treated with rosiglitazone for 180 days. No improvement in plasma glucose, serum insulin and lipid profiles were evident.24 In the present study, the patient with the Ile1143Phe mutation was treated with pioglitazone 15 mg/day for 6 months before glimepiride treatment, but this did not prove effective. Thus, the efficacies of thiazolidnediones in the treatment of type A insulin resistance syndrome remained to be determined. In contrast, the patient with the Arg1174Trp mutation did not receive any insulin‐sensitizing agent during the 14 years of follow up. However, hyperinsulinemic hypoglycemia and menstruation problems were somehow relieved with the passage of her puberty, and no diabetic microvascular or macrovascular complications were evident. Type A insulin resistance syndrome is relatively rare, and the correlation between genotype and phenotype varies.25 There are not enough large‐scale, long‐term, follow‐up studies focusing on the treatment and prognosis in the literature to guide our treatment choices. There is a pressing need to assemble genotype and phenotype data of type A insulin resistance syndrome worldwide to optimize treatment algorithms.
There were some limitations to the present study. For example, we identified a novel missense mutation (Ile1143Phe) in the INSR gene. We did not carry out detailed experiments on the alteration of insulin receptor expression, insulin binding or autophosphorylation capacity of the tyrosine kinase domain. We assumed the mutation was responsible based on the fact that the phenotype of the patient matched the common genotypes of the mutation occurring in the tyrosine kinase domain of the INSR gene. Furthermore, the Ile1143 amino acid is highly conserved in multiple species. Replacement with a phenylalanine at this site is probably hazardous. In addition, multiple powerful programs, each with >80% accuracy, unanimously predicted that the mutation is deleterious. Thus, there is still some strength for the assumption that the mutation is responsible for severe insulin resistance in this patient. In contrast, we did not examine the changes in the activity of the post‐insulin receptor signaling pathway, nor did we check for its PPARγ agonist activity before and after the treatment; therefore, we could not possibly explain the null effect of glimepiride in the study. Finally, expanding investigation of the genetic background and the associated phenotype outside the family was declined. Thus, analysis of the linkage and segregation of the mutation was not possible.
In summary, we reported a female patient with type A insulin resistance syndrome carrying a novel heterozygous missense mutation of Ile1143Phe in exon 19 of the INSR gene. Treatment with glimepiride for 6 months did not improve insulin sensitivity. Genotype and phenotype correlation varies in type A insulin resistance syndrome. Large‐scale, long‐term, follow‐up studies assembled worldwide are required to optimize treatment algorithms and prognosis.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
The research was supported by Guangdong Medical Science Research Fund (2012B060300010), the 5010 Project (No. 2010002), Industrial Technology Research and Development funding projects (No. 2012A030400006), Science and Technology Program of Guangzhou (No. 2014Y2‐00127), Funding of Key Medical Laboratory of Guangdong Province, and National Key Clinical Discipline, China.
J Diabetes Investig 2018; 9: 1075–1083
References
- 1. Taylor SI. Lilly Lecture: molecular mechanisms of insulin resistance. Lessons from patients with mutations in the insulin‐receptor gene. Diabetes 1992; 41: 1473–1490. [DOI] [PubMed] [Google Scholar]
- 2. Odawara M, Kadowaki T, Yamamoto R, et al Human diabetes associated with a mutation in the tyrosine kinase domain of the insulin receptor. Science 1989; 245: 66–68. [DOI] [PubMed] [Google Scholar]
- 3. Moller DE, Flier JS. Detection of an alteration in the insulin‐receptor gene in a patient with insulin resistance, acanthosis nigricans, and the polycystic ovary syndrome (type A insulin resistance). N Engl J Med 1988; 319: 1526–1529. [DOI] [PubMed] [Google Scholar]
- 4. Cama A, de la Luz Sierra M, Ottini L, et al A mutation in the tyrosine kinase domain of the insulin receptor associated with insulin resistance in an obese woman. J Clin Endocrinol Metab 1991; 73: 894–901. [DOI] [PubMed] [Google Scholar]
- 5. Treadway JL, Morrison BD, Soos MA, et al Transdominant inhibition of tyrosine kinase activity in mutant insulin/insulin‐ like growth factor I hybrid receptors. Proc Natl Acad Sci USA 1991; 88: 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Müller G. The molecular mechanism of the insulin‐mimetic/sensitizing activity of the antidiabetic sulfonylurea drug Amaryl. Mol Med 2000; 6: 907–933. [PMC free article] [PubMed] [Google Scholar]
- 7. Fukuen S, Iwaki M, Yasui A, et al Sulfonylurea agents exhibit peroxisome proliferator‐activated receptor gamma agonistic activity. J Biol Chem 2005; 280: 23653–23659. [DOI] [PubMed] [Google Scholar]
- 8. Wei X, Ju X, Yi X, et al Identification of sequence variants in genetic disease‐causing genes using targeted next‐generation sequencing. PLoS ONE 2011; 6: e29500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 2009; 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li R, Li Y, Fang X, et al SNP detection for massively parallel whole‐genome resequencing. Genome Res 2009; 19: 1124–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li H, Handsaker B, Wysoker A, et al; 1000 Genome Project Data Processing Subgroup . The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Richards S, Aziz N, Bale S, et al; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang Z, Li Y, Tang T, et al Hyperinsulinaemic hypoglycaemia associated with a heterozygous missense mutation of R1174W in the insulin receptor (IR) gene. Clin Endocrinol (Oxf) 2009; 71: 659–665. [DOI] [PubMed] [Google Scholar]
- 14. Huang Z, Liu J, Ma L, et al Glucose metabolism, insulin sensitivity and β‐cell function in type A insulin resistance syndrome around puberty: a 9‐year follow‐up. Horm Metab Res 2014; 46: 65–72. [DOI] [PubMed] [Google Scholar]
- 15. Müller G, Geisen K. Characterization of the molecular mode of action of the sulfonylurea, glimepiride, at adipocytes. Horm Metab Res 1996; 28: 469–487. [DOI] [PubMed] [Google Scholar]
- 16. Mastick CC, Brady MJ, Saltiel AR. Insulin stimulates the tyrosine phosphorylation of caveolin. J Cell Biol 1995; 129: 1523–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee KW, Ku YH, Kim M, et al Effects of sulfonylureas on peroxisome proliferator‐activated receptor γ activity and on glucose uptake by thiazolidinediones. J Diabetes Metab 2011; 35: 340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Semple RK, Williams RM, Dunger DB. What is the best management strategy for patients with severe insulin resistance? Clin Endocrinol (Oxf) 2010; 73: 286–290. [DOI] [PubMed] [Google Scholar]
- 19. Cochran E, Musso C, Gorden P. The use of U‐500 in patients with extreme insulin resistance. Diabetes Care 2005; 28: 1240–1244. [DOI] [PubMed] [Google Scholar]
- 20. McDonald A, Williams RM, Regan FM, et al IGF‐I treatment of insulin resistance. Eur J Endocrinol 2007; 157(Suppl 1): S51–S56. [DOI] [PubMed] [Google Scholar]
- 21. Cochran E, Young JR, Sebring N, et al Efficacy of recombinant methionyl human leptin therapy for the extreme insulin resistance of the Rabson‐Mendenhall syndrome. J Clin Endocrinol Metab 2004; 89: 1548–1554. [DOI] [PubMed] [Google Scholar]
- 22. Brown RJ, Cochran E, Gorden P. Metreleptin improves blood glucose in patients with insulin receptor mutations. J Clin Endocrinol Metab 2013; 98: E1749–E1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hattori Y, Satoh H, Namatame T, et al A patient with extreme insulin resistance syndrome treated with pioglitazone. Diabetes Care 2006; 29: 2328–2329. [DOI] [PubMed] [Google Scholar]
- 24. Vestergaard H, Lund S, Pedersen O. Rosiglitazone treatment of patients with extreme insulin resistance and diabetes mellitus due to insulin receptor mutations has no effects on glucose and lipid metabolism. J Intern Med 2001; 250: 406–414. [DOI] [PubMed] [Google Scholar]
- 25. Longo N, Wang Y, Smith SA, et al Genotype‐ phenotype correlation in inherited severe insulin resistance. Hum Mol Genet 2002; 11: 1465–1475. [DOI] [PubMed] [Google Scholar]