

Abstract
Diabetes has become a major burden of healthcare expenditure. Diabetes management following a uniform treatment algorithm is often associated with progressive treatment failure and development of diabetic complications. Recent advances in our understanding of the genomic architecture of diabetes and its complications have provided the framework for development of precision medicine to personalize diabetes prevention and management. In the present review, we summarized recent advances in the understanding of the genetic basis of diabetes and its complications. From a clinician's perspective, we attempted to provide a balanced perspective on the utility of genomic medicine in the field of diabetes. Using genetic information to guide management of monogenic forms of diabetes represents the best‐known examples of genomic medicine for diabetes. Although major strides have been made in genetic research for diabetes, its complications and pharmacogenetics, ongoing efforts are required to translate these findings into practice by incorporating genetic information into a risk prediction model for prioritization of treatment strategies, as well as using multi‐omic analyses to discover novel drug targets with companion diagnostics. Further research is also required to ensure the appropriate use of this information to empower individuals and healthcare professionals to make personalized decisions for achieving the optimal outcome.
Keywords: Genomics, Pharmacogenetics, Precision medicine
Introduction
Diabetes mellitus is a group of chronic diseases characterized by elevated blood glucose levels due to different causes with abnormal β‐cell biology playing a pivotal role. Over recent decades, the population affected by diabetes has been increasing in both developed and developing countries. The International Diabetes Federation (IDF) recently estimated that 425 million people are affected by diabetes worldwide1. Amongst them, 80% live in low‐ and middle‐income nations2, and a large proportion reside in the Asia–Pacific region2. In a Chinese nationwide survey carried out in 2013, 11.6% of adults aged >18 years had diabetes; that is, one in nine Chinese adults were affected3, 4.
Diabetes increases the risk of premature mortality and morbidity as a result of multisystem complications during the lifelong disease course5. These include atherosclerotic cardiovascular diseases, renal failure, visual loss, foot ulcer and amputation, all of which are highly preventable and treatable. It has been estimated that 68% of adults with diabetes aged >65 years died of some form of coronary heart disease (CHD), whereas 16% died of stroke6. Additionally, diabetes is associated with morbidities, such as cancer, depression, and physical and mental disabilities7, 8. The chronic and complex nature of diabetes denotes that affected patients are lifelong users of healthcare services once diagnosed with diabetes. If uncontrolled, diabetes can lead to escalating burden and costs to society and families. In this context, the cost of diabetes management was estimated to be approximately $US850 billion in 2017, and is expected to increase to $US958 billion by 2045, the majority of which will be used to treat complications1.
Until recently, most management algorithms proposed stepwise treatment escalation upon failure with oral blood glucose‐lowering drugs leading to insulin treatment9, 10. In the Hong Kong Diabetes Register, during a median follow‐up duration of 3 years, 45.6 and 57.9% of patients treated with metformin and sulfonylurea (SU) monotherapy, respectively, required escalation of treatment10. In randomized clinical trials (RCTs) where treatment is usually closely monitored, patients treated with monotherapy or combination treatment using metformin and SU tend to progressively deteriorate in their glycemic control. That said, optimal glycemic control with glycated hemoglobin (HbA1c) <6.5% and certain drugs, such as thiazolidinediones (TZD), have been shown to improve the sustainability of glycemic control11. There are also reports suggesting that early intensive glycemic control without undue risk of hypoglycemia could lead to diabetes remission12. Given the plethora of medications, the challenge lies in matching the right drug with the right patient at the right time to obtain the best clinical outcome.
The pathophysiology of diabetes is complex with multiple causes, phenotypes, trajectories and consequences13, 14. Theoretically, there can be many diabetes subphenotypes characterized by different combinations of molecular features, pathophysiological processes, risk factors, complications and comorbidities. These phenotypes can be altered by self‐management, quality of care and drug treatments, all of which can influence clinical outcomes. To this end, both genetic and environmental factors can interact to alter pathophysiological processes including β‐cell regeneration and survival, as well as insulin secretion and action5. Given these vast numbers of causal and mediating factors and their near‐infinite combinations, most clinical guidelines have now replaced the traditional one‐size‐fits‐all with a more individualized and holistic approach in diabetes management15. It is against this diversity of clinical observations that have led to the advocacy of personalized healthcare delivery16. With increasing understanding of the molecular mechanisms of diabetes and the documentation of different subphenotypes, there is a case to include this new knowledge to improve the precision of diagnosis and classification in order to optimize care, maximize treatment efficacy and reduce undesirable side‐effects17, 18, 19, 20.
Insights from Genetic Studies
The completion of the Human Genome Project has provided new insights into the genomic architecture consisting of >30 billion nucleotide base pairs punctuated by regions that show marked interindividual variations. These variations can take the form of single‐nucleotide polymorphism (SNP) or structural variations with copy number variations, deletions or insertions. Traditionally, variants with <1% in the population are referred as mutations21, whereas variants with >5% are known as common variants22. In the past decade, the proliferation of genome‐wide association studies (GWAS) has led to huge progress in genetic research of common diseases and traits. Until then, the candidate gene approach was used to discover the genetic association of diseases based on prior knowledge of the pathogenic significance of a gene and its gene product (i.e., protein). With the development of microarray technology, since the early 2000s, genome‐wide screening has been used to discover associations between diseases of interest and genomic variations without prior assumptions. This approach has led to the discovery of novel pathogenic pathways in an unbiased manner and proven to be highly efficient in discovering common genetic variants17. Although the effect size of these common variants is considerably weaker than that of rare variants, the additive effects through gene–gene interactions can be modest in common diseases, such as diabetes. In contrast, carriers of rare variants that are often associated with amino acid and thus protein changes are more likely to have significant structural or functional impairments. Because of their early disease manifestation, many affected individuals could have reduced reproductive fitness and survival. As such, during human evolution, these unfavorable genetic variants might be reduced to low frequencies through natural selection forces23.
After nearly a decade of intensive research, it is now evident that the GWAS‐identified genetic loci might merely represent a tag of the causal genes in a distant site24. In the human genome, linkage disequilibrium is a prominent architectural feature whereby gene sequences, SNPs within a gene and/or a genomic region containing a large number of variants of multiple genes are co‐inherited as a block. In most GWAS, microarrays containing millions of SNPs discovered in the Human Genome Project scattered throughout the genome are used to interrogate disease association, usually in case–control cohorts. After adjusting for multiple comparisons, variants associated with a disease or trait might simply represent a “signpost” of another genetic variant nearby, which is the true source of association. In rare diseases, the associated uncommon variants are usually causal and located in coding exonic regions with protein changes. By contrast, in common diseases or complex traits, multiple genetic variants that lie within a pathophysiological pathway/network are often implicated. These genetic variants are usually located in non‐coding regions (e.g., intergenic, promoter, intronic), many of which are regulatory sites through epigenetic mechanisms (e.g., binding with transcription factors, activators or repressors, deoxyribonucleic acid methylation, chromatin modification, non‐coding ribonucleic acid). Sequence variations and/or SNPs in these regions can lead to subtle changes in a gene network to explain the interindividual variations of multiple traits25.
Using both candidate gene and GWAS approaches, hundreds of genes and genetic loci associated with type 1 diabetes, type 2 diabetes and diabetic complications have been discovered. Many of these type 2 diabetes loci are implicated in islet development, notably β‐cell biology. These include β‐cell development26, 27, 28, 29 and survival28, insulin secretion and action28, 29, and glucose processing30. Some of these type 2 diabetes loci also overlap with genetic variants implicated in aging28, adiposity28, autoimmune diseases31, cancer32 and CVD33. Despite these discoveries, the functional significance of many of these putative loci has not been characterized28, 34, 35, 36, 37. In the present review article, we have summarized the current state of knowledge on the genetics of the different subtypes of diabetes and its complications.
Type 1 diabetes
Type 1 diabetes results from autoimmune destruction of the pancreatic islet, due to interaction between genetic susceptibility, perturbed immunology and environmental factors. A number of studies have confirmed the associations of type 1 diabetes with allelic variants at the human leukocyte antigen (HLA) class I and class II gene regions, non‐HLA susceptibility genes, and innate immunity genes38, 39. In the type 1 diabetes Genetics Consortium consisting of European populations, mutations in INS (insulin), PTPN22 (protein tyrosine phosphatase, non‐receptor type 22), IL2RA (interleukin‐2 receptor subunit alpha), IFIH1 (interferon induced with helicase C domain 1), CTLA4 (cytotoxic T‐lymphocyte associated protein 4) loci, and TCF7‐P19T (transcription factor 7‐P19T) have been reported using candidate gene approaches40. In addition, >40 distinct locations have been discovered in GWAS, some of which are potential treatment targets41, 42. In a large prospective study of patients with type 1 diabetes in the USA, researchers have reported three SNPs associated with increased risk of type 1 diabetes (rs10517086_A, rs1534422_G and rs2327832_G in tumor necrosis factor, alpha‐induced protein 3 [TNFAIP3]) and one with decreased risk (rs1004446_A in INS)43. Most of these putative genes were associated with autoimmune activation, overexpression of inflammatory or intrinsic immune response genes, modulation of intracellular signaling, or destruction of β‐cells44, 45. Some genes were associated with inflammatory responses to infection with enteroviruses, which have been implicated in the onset of type 1 diabetes46.
Type 2 diabetes
Type 2 diabetes and its complications are classical examples of polygenic and complex diseases as a result of interactions between multiple genetic and environmental factors16. To date, >128 distinct signals at 113 loci have been associated with type 2 diabetes47, 48, 49. The majority of these SNPs are associated with islet development and glucose sensing, as well as insulin synthesis, secretion, signaling or resistance; whereas others are associated with metabolic traits, such as obesity, which frequently coexists with diabetes50. Abnormal β‐cell biology is a pathway specific to diabetes, which is often unmasked by other factors, such as obesity, low‐grade inflammation and aging. Thus, variants associated with insulin secretion located in, or near, the following 18 genes are of particular interest. These include SLC2A2 (solute carrier family 2 member2), GCK (glucokinase), GCKR (glucokinase receptor), SUR1 (sulfonylurea receptors type 1), Kir6.2 (potassium voltage‐gated channel subfamily J member 11), CACNA1E (calcium voltage‐gated channel subunit alpha1 E), CAPN10 (calpain 10), HHEX (hematopoietically expressed homeobox), IDE (insulin degrading enzyme), PPARGC1A (PPAR gamma coactivator 1 alpha), SLC30A8 (solute carrier family 30 member 8), WFS1 (wolframin ER transmembrane glycoprotein), UCP2 (uncoupling protein 2), PTGS2 (prostaglandin‐endoperoxide synthase 2), CDKN2A/B (cyclin‐dependent kinase inhibitor 2A/B), CDKAL1 (CDK5 regulatory subunit associated protein 1 like 1), SLC2A1 (solute carrier family 2 member 1) and MTNR1B (melatonin receptor 1B), which have been shown to influence insulin secretion pathways or β‐cell function. That said, a gene mutation of TBC1D4 (TBC1 domain family member 4) associated with muscle insulin resistance has also been associated with type 2 diabetes51. Of note, several of these genetic variants were associated with postprandial glucose elevation, which highlights the importance of using the 75‐g oral glucose tolerance to detect carriers of these mutations or variants52.
Although many of these variants have an effect size expressed as an odds ratio (OR) ranging from 1.01 to 1.71, because of their common nature (5–95%, depending on the nomenclature in denoting the risk‐conferring variant), many individuals might carry multiple genetic variants. These gene–gene interactions can substantially increase their risk of having diabetes, especially in the presence of risk factors, such as obesity, resulting in early age of onset. Using Hong Kong Chinese individuals as an example, our group first reported the additive effects of genetic variants of seven genes associated with type 2 diabetes discovered in the first wave of GWAS, where individuals in the top tertile of the genetic risk score (GRS) had a two‐ to threefold higher risk of type 2 diabetes than those with lower GRS (Figure 1). In the same analysis, we first reported the interethnic differences in minor allele frequency (MAF) and effect sizes of these SNPs, which were first discovered in Caucasians followed by validation in Asian populations53. In line with the interethnic differences in the linkage disequilibrium54, we subsequently reported the association of SNPs with higher MAF within the same loci associated with type 2 diabetes in a Chinese population. Although the majority of these loci are shared by Caucasian and Asian populations, there are loci, such as paired box 4 (PAX4) that are uniquely found in Asians but not Caucasians55, 56. These interethnic differences in effect size and MAF can give rise to different population‐attributable risk; that is, the explained variance of type 2 diabetes prevalence within a population. Depending on the background risk for type 2 diabetes in a particular population (e.g., lifestyle, culture and other environmental factors), the impact of these genetic variants on the population and individual risk can vary substantially depending on the combinations of these genetic, environmental and lifestyle factors. Thus, in the most unfavorable scenario, a person with high GRS, suboptimal perinatal nurturing, obesity and health adverse lifestyles (e.g., unhealthy diet, physical inactivity, poor sleep, psychosocial stress, smoking, excess use of alcohol) in later life and delayed intervention might develop diabetes at a substantially younger age with prolonged exposure to hyperglycemia, and high risk of premature complications and death. By contrast, a person who has low GRS, optimal upbringing, healthy lifestyle and early intervention might expect to have good health and longevity. Between these two extremes, there can be huge variations with considerable opportunities for changing the trajectories depending on self‐management, quality of care and access to care (Figure 2)57.
Figure 1.
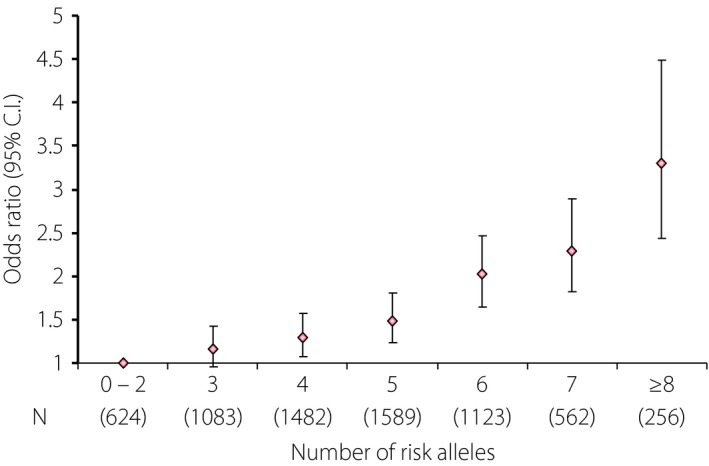
Effect of an increasing number of risk alleles and type 2 diabetes risk in the combined Chinese and Korean samples (adapted with permission from supplementary text of Ng et al.53).
Figure 2.
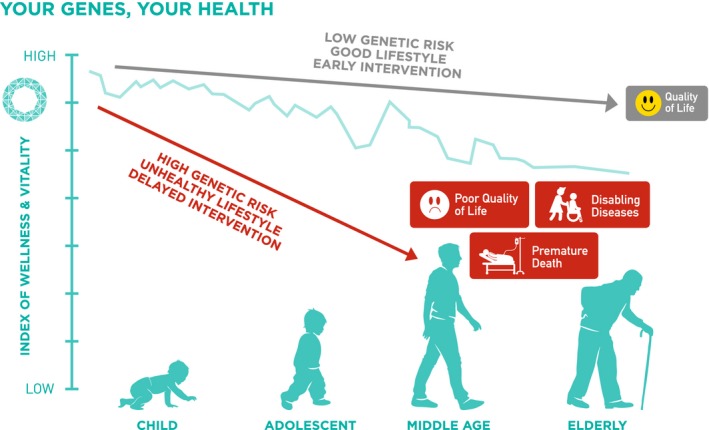
The basic concept of personalized intervention and substantial interaction of genetic and modifiable risk factors. Figure reproduced with permission from GemVCare.
Gestational diabetes mellitus
Gestational diabetes mellitus (GDM) is defined as hyperglycemia first manifest in pregnancy. The latter is a diabetogenic condition characterized by multiple hormonal changes with increased insulin resistance. Thus, in women with a genetic predisposition, GDM might be the first manifestation for their high lifetime risk for diabetes, and represent a high‐risk group for monitoring and early intervention. Not dissimilar to type 2 diabetes, multiple genetic studies have reported loci associated with GDM or glucose traits during pregnancy58, 59, with some loci overlapping with that for type 2 diabetes58, 59. In a study carried out in Poland, researchers reported better performance of using type 2 diabetes‐ or obesity‐associated genetic variants with ORs ranging from 1.67 to 2.88 for each SNP to predict GDM than using age, history of previous births and pre‐pregnancy body mass index60. Given the young age of women with GDM, multi‐ethnic meta‐analyses are currently ongoing to discover additional loci associated with GDM, which might provide new insights into the genetic basis of not just GDM, but also young‐onset diabetes where etiologies remain to be elucidated61.
Maturity‐onset diabetes of the young
Maturity‐onset diabetes of the young (MODY) is an uncommon form of monogenetic diabetes due to a single gene alteration and accounts for 1–2% of all cases of diabetes. It is a group of autosomal dominant disorders characterized by non‐ketotic and/or non‐acute presentation, typical of type 2 diabetes, but occurring at a younger age, usually before the age of 25 years62. Because of their rapid failure with oral drugs and/or young onset of presentation, MODY patients can be misdiagnosed as type 1 diabetes. Alternatively, because of their low risk of ketosis, they might simply be classified as having type 2 diabetes63, 64. To date, most of the evidence points to multiple mutations/variants implicated in pathways predominantly linked to β‐cell biology. Up to now, there are at least 14 genetic subtypes of MODY each with distinct clinical characteristics65, 66 and responsible genes (Table 1). In these young individuals with familial early‐onset diabetes with or without typical features, genetic testing is required to increase the precision of diagnosis, which has implications on treatment selection and family screening17, 67.
Table 1.
Summary of genetic mutations associated with maturity‐onset diabetes of the young
| Subtype | Gene | Location | Etiology | Features |
|---|---|---|---|---|
| MODY 1(82) | HNF4α | 20q13.12 | Insulin secretion defect | Progressive hyperglycemia |
| MODY 2(75) | Glucokinase | 7p13 | Glucose sensing and insulin secretion defect | Early onset; mild hyperglycemia, minor microvascular disease |
| MODY 3(83) | HNF1α | 12q24.31 | Insulin secretion defect | Progressive hyperglycemia, sensitive to SU |
| MODY 4(84) | PDX1/IPF1 | 13q12.2 | Insulin secretion defect | Early onset |
| MODY 5(85) | HNF1β | 17q12 | Insulin secretion defect | Variable age at onset, range infancy to adult; progressive hyperglycemia, renal cysts; renal failure, require insulin treatment |
| MODY 6(86) | NeuroD1 | 2q31.3 | Insulin secretion defect | Early onset |
| MODY 7(87) | KLF11 | 2p25.1 | Insulin secretion defect | Very rare |
| MODY 8(88) | CEL | 9q34.13 | β‐cell defect | Endocrine and exocrine pancreatic insufficiency |
| MODY 9(89) | PAX4 | 7q32.1 | Little data | Very rare |
| MODY 10(90) | INS | 11p15.5 | Insulin secretion defect | Diagnosed in patients aged in their 20s to 30s. Can cause neonatal diabetes, antibody negative type 1 diabetes, and MODY |
| MODY 11(91) | BLK | 8p23.1 | Defect in insulin synthesis and secretion | Onset often before age 25 years; some patients require insulin for treatment |
| MODY 12(92) | ABCC8 | 11p15.1 | Little data | Frequent cause of neonatal diabetes, but can rarely cause MODY |
| MODY 13(93) | KCNJ11 | 11p15.1 | Insulin secretion defect | Sulfonylurea therapy effective |
| MODY 14(94) | APPL1 | 3p14.3 | Defect in insulin signaling pathway | With elevated FBG and HbA1C and onset between 30s and 50s |
This table was adapted from Anik et al.80 ABCC8, adenosine triphosphate‐binding cassette, subfamily C (CFTR/MRP), member 8; APPL1, the adaptor protein, phosphotyrosine interaction, PH domain, and leucine zipper containing 1; ATP, adenosine triphosphate; BLK, B‐lymphocyte kinase; CEL, carboxyl ester lipase; GCK, glucokinase; HNF4A, hepatocyte nuclear factor 4α; INS, insulin; IPF1, insulin promoter factor 1; KCNJ11, potassium channel, inwardly rectifying subfamily J, member 11; KLF11, Kruppel‐like factor 11; MODY, maturity‐onset diabetes of the young; NEUROD1, neurogenic differentiation 1; OAD, oral antidiabetic agents; PAX4, paired‐box‐containing gene 4; PDX1, pancreatic and duodenal homeobox 1; PNDM, permanent neonatal diabetes.
The commonest types of MODY include MODY 2, with mutations in the glucokinase (GCK) gene; MODY 3 and MODY 1, with mutations in hepatocyte nuclear factor (HNF)1α and HNF‐4α genes, respectively. These mutations were discovered in atypical families of young‐onset diabetes where mapping of chromosomal regions was linked to diabetes or related traits in affected family members followed by gene sequencing in these linked genomic regions. In Caucasian populations, using a strict definition of MODY characterized by multiple generations of diabetes with age of diagnosis <25 years, defects in these three genes (GCK, HNF1α and HNF4α) account for approximately 80% of MODY series67.
Using linkage analysis, researchers first discovered segregation of various mutations in GCK genes with affected family members. Different variants might be found in different families and thus are often referred to as private mutations, possibly as a result of marriages amongst closely related individuals. These private mutations can only be detected by re‐sequencing the whole gene in the proband. Often, the nature of these mutations might not have been characterized and their causative nature can only be validated by comparing their co‐segregation in affected family members68, 69. Carriers of these GCK mutations typically have impaired glucose sensing and hence higher set points of plasma glucose for stimulation of insulin release than non‐carriers. These individuals usually have mild fasting hyperglycemia with HbA1c of 6–7%. They also tend to have little response to oral blood glucose‐lowering agents or insulin. Despite this chronic fasting hyperglycemia, cross‐sectional analysis of patients with GCK mutations suggested a low risk of microvascular (prevalence 1%, 95% confidence interval [CI] 0–5%) and macrovascular complications (prevalence 4%, 95% CI 1–10%), at least in Caucasians70. Thus, treatment with blood glucose‐lowering drugs is often not necessary in patients with GCK mutations, although the co‐occurrence of other common genetic variants and lifestyle factors might impair β‐cell function and increase the risk of long‐term complications. In Chinese patients with young‐onset diabetes diagnosed before the age of 40 years, nearly 60% had a positive family history, with some of them harboring autoimmune markers as well as common and uncommon genetic variants71. Indeed, there is now increasing evidence showing the aggressive clinical course of these young patients who have 1.5‐fold higher risk of cardiovascular–renal disease and premature death than their peers with late‐onset diabetes72, 73.
Long‐term prognosis aside, during pregnancy, the interactions amongst the pattern of inheritance of MODY genetic variants to the offspring, maternal genetic background and intrauterine hyperglycemic environment can have complex effects on fetal outcome. In the case of GCK, offspring who are carriers of mutations inherited from either the father and/or mother tend to have lower birthweight by 520 g compared with non‐carriers74. Here, intensive glycemic control in the non‐affected mother with GDM of an affected offspring can lead to serious dysplasia in MODY 2 offspring. By contrast, in a mother who is a carrier of MODY 2 mutations, chronic hyperglycemia might cause fetal hyperinsulinemia, and insulin treatment will be required to prevent macrosomia and islet hyperplasia in the non‐carrier fetus75. So, theoretically, it might be informative to test for MODY 2 mutations in young women with GDM or young‐onset diabetes and their partners, as well as the fetus using fetal deoxyribonucleic acid, although a study will be required to inform practice.
Amongst these MODY subtypes, MODY 3 is the commonest and has been reported in most populations. Both MODY 1 and MODY 3 mutations influence the development and survival of pancreatic β‐cells with common features of early‐onset of diabetes (usually before the age of 25 years) and progressive deterioration of glycemic control. Affected individuals often require treatment escalation and develop microvascular complications76, 77. Yet, screening family members identified through a proband can lead to early diagnosis and treatment with few complications as compared with affected family members who are diagnosed late, resulting in poor glycemic control and complications78.
Diagnosis of MODY 1 and MODY 3 also have therapeutic implications, as these carriers show hypersensitivity to SU. As the primary genetic defect in MODY 1 and MODY 3 relates to transcriptional efficiency of insulin, the use of SU can bypass this defect by acting directly on the adenosine triphosphate (ATP)‐sensitive potassium (KATP) channel to promote insulin secretion, often at doses one‐quarter that of used in patients with common forms of type 2 diabetes79.
Other rare forms of MODY have also been reported. Affected patients with mutations in HNF1‐β (MODY 5) could have pancreas agenesis, renal dysfunction, genital tract malformations and liver abnormalities80, and often require insulin treatment. MODY 12 and MODY 13 are associated with mutations in ATP‐binding cassette, subfamily, member 8 (ABCC8) and potassium channel, inwardly rectifying subfamily J, member 11 (KCNJ11), which encode subunits of the KATP channel. These individuals show better response to SU than insulin therapy65, 81. Other MODY types are very rare, and their characteristics are summarized in Table 1.
Neonatal diabetes
Several national registers show that the incidence of neonatal diabetes, diagnosed before the age of 6 months, is approximately one in 100,000 live births82. The possibility of monogenic diabetes should always be considered in patients with a history of neonatal diabetes or familial young‐onset diabetes irrespective of whether they have type 1 diabetes or type 2 diabetes presentation. In the case of neonatal diabetes, 50–60% of patients have a transient form of the disease with spontaneous remission within a few months‐of‐age, whereas the remaining have permanent neonatal diabetes. Amongst the reported mutations associated with neonatal diabetes (Table 2), nearly half were as a result of mutations in the genes encoding the KATP (ABCC8 and KCNJ11)65 with good response to SU65, 83.
Table 2.
Summary of genetic mutations associated with neonatal diabetes
| Type of diabetes | Gene | Location | Affected protein | Features | Usual age of onset |
|---|---|---|---|---|---|
| PNDM | KCNJ11 | 11p15.1 | Kir6.2 | Most common type of PNDM | Within 3–6 months |
| PNDM | ABCC8 | 11p15.1 | SUR1‐sulfonylurea receptor 1 | Rare | 1–3 months |
| PNDM | GCK | 7p13 | Glucokinase | Rare | 1 week |
| PNDM | INS | 11p15.5 | Insulin | Rare | Birth to 6 months |
| PNDM | IPF1; also known as PDX1 | 13q12.2 | Insulin promoter factor 1 | Rare | 1 week |
| PNDM | PTF1A | 10p12.2 | Pancreas transcription factor 1A | Rare. Associated with cerebellar agenesis and severe neurological dysfunction. | At birth |
| PNDM | FOXP3, | Xp11.23 | Forkhead box P3 | Rare. Immune dysregulation, polyendocrinopathy, and enteropathy, X‐linked (IPEX) syndrome | Sometimes present at birth |
| PNDM | EIF2AK3, | 2p11.2 | Eukaryotic translation initiation factor 2‐alpha kinase 3 | Rare. Wolcott–Rallison syndrome | 3 months |
| PNDM | GATA4 | 8p23.1 | A zinc finger transcription factor | Rare. Variable diabetes phenotypes. | Birth to 3 months |
| PNDM | GATA6 | 18q11C.2 | A zinc finger transcription factor | Rare. Associated with structural heart defects, biliary tract and gut anomalies, and other endocrine abnormalities. | Birth to 3 months |
| PNDM | GLIS3 | 9p24.2 | Zinc finger protein GLIS3 (GLI similar protein 3) | Rare. With congenital hypothyroidism | Birth to 3 months |
| PNDM | RFX6 | 6q22.1 | RFX6, a transcription factor | Rare. Mitchell–Riley syndrome. With pancreatic hypoplasia, intestinal atresia and gall bladder hypoplasia | Birth to 6 months |
| PNDM | NEUROD1 | 2q31.3 | Neurogenic differentiation factor 1 | Normal pancreatic exocrine function. With cerebellar hypoplasia, sensorineural deafness and visual impairment. | Birth to 6 months |
| PNDM | NEUROG3 | 10q22.1 | Neurogenin‐3 | Severe insulin deficiency and pancreatic exocrine insufficiency due to hypoplastic pancreas | Birth to childhood |
| PNDM | HNF1B, TCF2 | 17q12 | Hepatocyte nuclear factor‐1‐beta | A syndrome of neonatal diabetes mellitus and renal abnormalities | Birth to 6 months |
| PNDM | PAX6 | 11p13 | PAX6, a transcription factor | With brain malformations, microcephaly, and microphthalmia | Birth to 6 months |
| PNDM | SLC19A2 | 1q24.2 | A thiamine transporter | Also known as Rogers syndrome | Birth to 6 months |
| PNDM | MNX1 | 7q36.3 | Motor neuron and pancreas homeobox 1 | With developmental delays, sacral agenesis and imperforated anus | Birth to 6 months |
| PNDM | NKX2‐2 | 20p11.22 | NK2 homeobox 2 | With developmental delays, hypotonia, short stature and deafness | Birth to 6 months |
| PNDM | IER3IP1 | 18q21.1 | Immediate early response 3 Interacting protein 1 | With microcephaly, lissencephaly, and epileptic encephalopathy | Birth to 6 months |
| PNDM | MHC | 6p21.3 | Major histocompatibility complex | With methylmalonic acidemia and agenesis of pancreatic beta cells | Birth to 6 months |
| TNDM | PLAGL1/HYMAI | 6q24.2 | PLAG1: pleomorphic adenoma gene‐like 1 or; HYMAI: hydatiform mole‐associated and imprinted transcript | Most common form of NDM | Birth to 3 months |
| TNDM1 | ZFP57 | 6P22.1 | KRAB zinc finger proteins | Rare | Birth to 6 months |
| TNDM2 | ABCC8 | 11p15.1 | SUR1‐sulfonylurea receptor 1 | Rare | Birth to 6 months |
| TNDM3 | KCNJ11 | 11p15.1 | Kir6.2 | Uncommon cause of TNDM, but most common cause of PNDM | Birth to 6 months |
| TNDM | HNF1β | 17q12 | Hepatocyte nuclear factor 1B | Rare | Birth to 6 months |
Data were obtained from Online Mendelian Inheritance in Man (OMIM) (accessed on 20171001). NDM, neonatal diabetes mellitus; PNDM, permanent diabetes mellitus; TNDM, transient diabetes mellitus.
Using Genomic Markers to Predict Diabetes and its Complications
Type 1 diabetes
With better understanding of the genetic architecture of diabetes, there is increasing interest in using allelic variants in susceptibility genes to predict diabetes risk. In type 1 diabetes, anti‐islet autoantibodies might precede the onset of diabetes, albeit not invariably84. In contrast, increased risks of type 1 diabetes have been associated with genetic variations in HLA class I gene, HLA class II gene, non‐HLA susceptibility genes and innate immunity genes39. In a population‐based survey involving 4,574 cases and 1,207 controls in Germany, researchers have developed a GRS consisting of >40 SNPs of HLA genes and non‐HLA genes to predict the risk of type 1 diabetes85. In the testing set, the AUC (area under the curve) of the receiver operating characteristic curve was 0.87 and 0.84 in the validation set85. In another study involving 257 children from Denmark (84% Caucasians), the cumulative GRS of GWAS type 1 diabetes loci had predictive utility with a positive relationship between HbA1c levels and the number of risk alleles86. The loci included IFIH1 (interferon‐induced helicase C domain‐containing protein 1), CTLA4 (cytotoxic T‐lymphocyte protein 4), PTPN22 (tyrosine‐protein phosphatase non‐receptor type 22), IL18RAP (interleukin‐18 receptor accessory protein), SH2B3 (SH2B adapter protein 3), KIAA0350 (a gene predicted to code a sugar‐binding, C‐type lectin), COBL (protein cordon‐bleu) and ERBB3 (receptor tyrosine‐protein kinase erbB‐3), which could effectively stratify individuals into different risk groups for future development of islet autoantibodies and progression to type 1 diabetes87. The median AUC for receiver operating characteristic curves using diabetes as the outcome was 0.588 (P = 0.001) in the whole cohort and 0.656 (P < 0.001) in the HLA‐risk children. Using auto‐antibodies as outcomes, the respective median AUC was 0.521 (P = 0.945) for the whole cohort and 0.573 (P = 0.02) in the HLA‐risk children. These intermediate AUCs highlight the importance of other external or host factors yet to be discovered.
Here, it is important to point out that in developing a screening program, aside from predictive and analytical utility of the biomarker, clinical utility is a major consideration. For screening to be cost‐effective, the disease should be relatively common and preferably with a lag phase where effective preventive strategies can be implemented. As there are no effective preventive measures for type 1 diabetes, the use of genetic markers to identify at‐risk individuals remains debatable as compared with type 2 diabetes, which is common and potentially preventable.
Type 2 diabetes
Because of the devastating nature of type 2 diabetes, which affects one in 11 adults, and its potentially preventable nature through lifestyle modification and early use of medications (e.g., metformin and acarbose)88, 89, there are global efforts to discover type 2 diabetes‐associated genetic variants. The use of these variants can potentially identify high‐risk individuals for intensive lifestyle modification or early treatment, depending on genetic loading. These personalized approaches should complement public health awareness and promotion measures (e.g., healthy city, healthy lifestyle, anti‐smoking, sugar tax, food labeling etc.,) in order to curb the rising trend of type 2 diabetes. Amongst the 100 genetic loci associated with type 2 diabetes, genetic variants of transcription factor 7‐like 2 (TCF7L2) have been found to confer susceptibility to type 2 diabetes in multiple ethnic groups. However, there are interethnic differences in terms of location, MAF and effect size of genetic variants within this locus90, 91, 92, 93. In a large‐scale genome‐wide replication study in the French population, the researchers combined the risk alleles of 15 loci, which effectively discriminated type 2 diabetes susceptibility with an AUC of 0.86 on receiver operating characteristic curve analysis94. In the Framingham Offspring Study, genetic and metabolic biomarkers showed complementary predictive effects for the risk of type 2 diabetes. The AUC for the combined model (GRS and metabolic traits) were 0.820 vs 0.641 for GRS alone (P < 0.0001) or 0.803 for metabolic traits alone (P = 0.01)95. In a prospective study carried out in the UK, a GRS based on 65 risk variants detected 19.9%, the phenotypes‐based Framingham risk model 30.7% and a combined model 37.3% of incident type 2 diabetes. The respective AUCs were 0.60 (95% CI 0.58–0.62), 0.75 (95% CI 0.73–0.77), and 0.76 (95% CI 0.75–0.78)96. In another Japanese study, the use of 11 validated variants predicted the risk of type 2 diabetes with an AUC of 0.7297.
These reports showed that incorporation of genetic risks only marginally increased the discriminative power of clinical or metabolic biomarkers. However, with the discovery of more risk loci associated with diabetes‐related traits (e.g., glucose or insulin levels), GRS predictive of phenotypes had better performance in predicting diabetes. In a large‐scale study, a GWAS‐derived doubly‐weighted GRS that included the effect of 1,000 SNPs was reported to predict incident type 2 diabetes with a hazard ratio of 3.45 (95% CI 2.31–5.17) amongst carriers of top the quintile of GRS vs the lowest quintile98.
Although there are meta‐analysis data89 confirming that lifestyle and drug treatment can prevent or delay the onset of type 2 diabetes, which might reduce complications and death in the long term99, there are ongoing debates on the most appropriate and cost‐effective implementation strategy in real‐world practice. By using GRS to select high‐risk individuals for detailed evaluation of modifiable lifestyle and cardio‐metabolic risk factors, we might increase the precision of subject selection and thus, the cost‐effectiveness of these diabetes prevention programs, although definitive studies or modeling are required to confirm such a proposition.
Diabetic complications
The burden of diabetes lies in its multisystem complications. The familial clustering of diabetic complications, notably diabetic kidney disease, has provided the premise for genetic association studies for diabetic complications100. In a large‐scale case–control analysis of Caucasians including 2,563 type 1 diabetes patients with chronic kidney disease (CKD) and 2,593 type 1 diabetes non‐CKD controls, three variants (rs1989248 near CNTNAP2 [contactin associated protein like 2], rs61277444 in PTPN13 [protein tyrosine phosphatase, non‐receptor type 13] and rs7562121 in AFF3 [AF4/FMR2 family member 3]) were associated with CKD in type 1 diabetes101. Using genome‐wide genotyping data, the heritability for CKD phenotypes in type 1 diabetes varied from 0.35 to 0.59, with a heritability of 0.47 for end‐stage renal disease. These estimates were comparable with earlier reports from family‐based cohorts102. In line with the epidemiological findings of risk association of metabolic syndrome with CKD103, in a recent meta‐analysis, genetic variants for obesity and body mass index were associated with CKD, raising the possibility of using these variants to identify type 1 diabetes or type 2 diabetes patients at risk of developing CKD101, 104. In Europeans, the carnosinase gene (CNDP1) polymorphism predicted the risk of progression to diabetic nephropathy (defined as persistent albuminuria ≥300 mg/24 h) and end‐stage renal disease (defined as the need to start chronic dialysis or kidney transplantation) in patients with type 1 diabetes105. In Chinese patients with type 2 diabetes, genetic polymorphisms implicated in CVD in Caucasians were found to predict complications. In the case of CVD, SNPs of IL5RA (interleukin‐5 alpha subunit), LPL (lipoprotein lipase), ITGA2 (integrin subunit alpha2) and NOS3 (endothelial nitric oxide synthase) genes predicted ischemic stroke106, whereas that of PON1 (paraoxonase 1), PON2 (paraoxonase 2), CETP (cholesteryl ester transfer protein), ITGA2 (intergrin‐α2β1) and LTA (lymphotoxin‐α) predicted CKD, independent of conventional risk factors and treatment107. These findings suggested that patients with these genetic predispositions might need to be treated more intensively or that target‐specific therapies might need to be developed to reduce the risk of these complications.
Heritability for CHD in type 2 diabetes based on family‐clustering analysis was estimated to be approximately 50%108. This is comparable with the estimated heritability of 0.41 for carotid intima‐medial thickness, an indicator of subclinical atherosclerosis109. A large number of GWAS‐associated loci for CHD have been discovered in general population. In a recent analysis that examined their significance in patients with type 2 diabetes, researchers reported positive associations with five SNPs (rs4977574 [CDKN2A/2B, cyclin‐dependent kinase inhibitor 2A/2B], rs12526453 (PHACTR1, phosphatase and actin regulator 1), rs646776 (CELSR2‐PSRC1‐SORT1, cadherin EGF LAG seven‐pass G‐type receptor 2‐proline and serine rich coiled‐coil 1‐sortilin 1), rs2259816 (HNF‐1A, HNF1 homeobox A), and rs11206510 (PCSK9, proprotein convertase subtilisin/kexin type 9) with direction and combined ORs of 1.17–1.25 similar to the general populations110. In a subsequent GWAS, a novel genetic variant (rs1091102) near the glutamate‐ammonia ligase (GLUL) gene was found to be associated with CHD in type 2 diabetes111, although the functional significance of many of these loci and their interactions remain to be clarified.
Genetic analysis reported 18–27% heritability for any diabetic retinopathy and 25–52% for proliferative diabetic retinopathy in type 1 diabetes and type 2 diabetes patients102. In epidemiological analysis and RCTs, blood pressure and blood glucose were the main determinants for diabetic retinopathy, whereas risk associations with dyslipidemia were less consistent. In a recent study utilizing Mendelian randomization, researchers failed to detect any causal association between genetically determined lipid fractions and the risk of diabetic retinopathy112.
Compared with the large number of loci associated with type 1 diabetes and type 2 diabetes, there are only a few GWAS‐confirmed and replicated genetic variants for diabetic complications102, 113, 114. Amongst these complications, retinopathy and nephropathy are particularly relevant to diabetes because of the long‐term effects of glucotoxocity on microvasculature, as supported by the confirmed benefits of blood glucose lowering in RCT. Given the challenges in optimizing glycemic control, discovering causal pathways for these complications might lead to new biology and new diagnostic and treatment strategies. In contrast, the considerable phenotypic heterogeneity, large number of confounders (e.g., age, sex, disease duration, smoking, obesity, control of blood lipids, blood glucose and blood pressure), multiple treatment effects (e.g., renin–angiotensin system inhibitors and lipid‐lowering drugs) and other non‐diabetes‐related factors (e.g., glomerulonephritis, renal stones and drug toxicity) can confound the genetic analysis of diabetic complications. In addition, there is a need to compare the utility and performance of genetic vs traditional clinical markers in predicting complications. To this end, a large sample size with well‐defined phenotypes and prospective follow up will be pivotal in our search for causal variants for diabetic complications independent of these risk factors100, 102.
Using Genomic Markers to Guide Diabetes Prevention and Treatment
Lifestyle changes including diet and exercise accompanied by weight reduction have been shown to prevent diabetes or improve metabolic control in individuals with diabetes. Recently, individuals’ genotypes have been shown to be associated with variable responses to lifestyle interventions in individuals with prediabetes115, 116, 117 and women at risk of GDM118. In the USA and Japan, researchers examined whether genetic counseling for individuals at risk of having diabetes would motivate behavioral change, but failed to show significant improvement, at least in the short term119, 120, 121. Prospective analysis of the Diabetes Prevention Study also showed that intensive lifestyle modification was effective regardless of genetic risk122, and was effective even in individuals with the highest genetic risk123. Other researchers have evaluated the attitudes to genetic testing of patients or those at risk. In the USA, surveys amongst individuals who are at high‐risk for developing type 2 diabetes (e.g., with positive family history, obesity) showed that many of them acknowledged the important role of genetic factors in the development of type 2 diabetes, and that lifestyle modification was more effective than drug treatment124. These findings suggested that their belief in genetic causality did not result in thoughts of futility and suboptimal self‐reported health behaviors124. To this end, there are many factors that can determine one's behavior, notably parental and peer influence. Thus, it is unlikely that genetic counseling alone can lead to behavioral change. Rather, awareness of one's genetic predisposition might activate behavioral changes, while education, support and feedback are used to sustain behavioral change.
There is general consensus that population measures will be required to control the diabetes epidemic, while a detection and prevention program targeted at high‐risk individuals with linkage to the healthcare system will be complementary. From a personal perspective, at risk individuals might be interested to know their own risk in order to make decisions depending on their value system and priorities, which might change over time. These are important questions in the post‐GWAS era, which will need to be addressed in order to translate this information into clinical utility for prevention purposes.
By contrast, the use of genetic testing to diagnose, classify and treat people with monogenic diabetes is widely accepted. Some experts recommended genetic testing for monogenic diabetes in patients with atypical features of type 1 diabetes or type 2 diabetes, or features suggestive of subtypes of monogenic diabetes125. Although autoimmune type 1 diabetes is still the predominant form of diabetes in young children and adults, especially in Caucasian populations, diabetes diagnosed below the age of 6 months or familial young‐onset diabetes (e.g., diagnosed below the age of 40 years) should be considered for potential diagnosis of monogenic diabetes. Leaving costs aside, there are now technologies, including microarray and sequencing, that can be used to confirm the molecular diagnosis of neonatal diabetes and MODY. These diagnoses carry prognostic and treatment implications including drug choices, planning for pregnancy, management during pregnancy and family screening, as discussed in previous section. As children and young adults with diabetes have to face long disease duration, precise diagnosis and personalized treatment are important for optimizing care in order to preserve quality of life and reduce long‐term complications and treatment costs.
In an international cohort study, early referral of patients with neonatal diabetes for comprehensive genetic testing has led to identification of causal variants in >80% of cases. These precise diagnoses have led to more personalized diabetes treatment and fewer complications than usual care126. In a retrospective study in the USA, patients with neonatal diabetes who carried KCNJ11 mutations had good response to early treatment with SU treatment, supporting the use of genetic diagnosis to individualize therapy127.
Caucasian patients with mutations in GCK (MODY 2) confirmed by genetic testing could successfully discontinue their antidiabetic medications and be treated with diet alone66, 70, 128. Although classical teaching shows that these patients have a low risk of developing microvascular complications, given the increasing understanding of the genotypic heterogeneity, interethnic differences in the patterns of young‐onset diabetes, and the effects of disease duration and other risk factors (e.g., obesity), more detailed phenotyping and comprehensive genotyping, as well as long‐term follow up, will be required to monitor treatment responses and disease progression in these young individuals.
Patients with the HNF1α (MODY 3) or HNF4α gene (MODY 1) mutations are often misdiagnosed as type 1 diabetes and are treated with insulin at presentation. However, on correct diagnosis with molecular testing, treatment can be successfully switched from insulin to SU with improved glycemic control, even in patients who have been treated with insulin for a long time79, 129. That said, with increasing disease duration, some patients with MODY 3 will eventually require additional treatment, such as dipeptidyl peptidase‐4 inhibitors on top of SU to optimize glycemic control130, 131. In contrast, patients with MODY 5 are often insensitive to SU treatment, and require early commencement of insulin therapy132. In another proof‐of‐concept study, the risk genotype of α2A‐adrenergic receptor was found to be associated with impaired insulin secretion in type 2 diabetes patients, which could be corrected by an α2A‐adrenergic receptor antagonist, illustrating the potential of personalized genotype‐specific treatment of type 2 diabetes133.
Due to these variable treatment responses, nearly two decades ago, molecular genetic testing was proposed to improve the precision of diagnosis, disease classification and treatment selection76. With better understanding of the genomic architecture and increasing affordability of technologies, molecular testing is expected to be more popular, especially in young patients with atypical presentation for personalized therapy. Although RCT remains the gold standard in evaluating treatment efficacy, given the uncommon and heterogeneous nature of monogenic diabetes, practicing physicians will need to take on a more active role in establishing registers of MODY and young‐onset diabetes with detailed documentation of phenotypes, treatment responses and clinical outcomes in order to fill the knowledge gap and inform clinical practice and healthcare planning.
Pharmacogenomics in Diabetes
Pharmacogenomics is the study of how genes affect a person's response to drugs, which might help practitioners to select treatment with maximal efficacy and minimal side effects134. In the USA, the Mayo Clinic has launched a study to evaluate the impact of precision medicine by integrating preemptive sequencing of pharmacogenomics data and electronic medical record with clinical decision support135. In a preliminary analysis, approximately 99% of participants carry variants of at least one of five well‐characterized pharmacogenomics genes (CYP2D6 [cytochrome P450 family 2 subfamily D member 6], CYP2C19 [cytochrome P450 family 2 subfamily C member 19], SLCO1B1 [solute carrier organic anion transporter family member 1B1], CYP2C9 [cytochrome P450 family 2 subfamily C member 9] and VKORC1 [vitamin K epoxide reductase complex subunit 1]) that might cause adverse drug‐related events (ADE)136. That study highlighted the potential value of pharmacogenomic testing in optimizing treatment, and reducing ADE and healthcare costs.
In the field of diabetes, apart from heterogeneity in etiology, patients also show marked variations in their responses to various blood glucose‐lowering drugs137. Discoveries of genetic polymorphisms associated with different degrees of efficacy and risk of ADE for different therapeutic agents have provided new insights into the genetic determinants for treatment responses in diabetes138. Metformin is often regarded as the first‐line treatment in type 2 diabetes. Its primary actions include increased glucose uptake by muscles, liver and adipose tissues, and reduced hepatic glucose output with improved insulin resistance. Metformin might also act on the gut to release incretin, enhance insulin secretion and improve glucose homeostasis139. In a GWAS analysis, the heritability of glucose response to metformin was estimated to be up to 0.34 in patients with type 2 diabetes9. Although heterogeneity in disease etiology and behavioral factors might contribute to such variation, genetic differences in treatment responses can be important139. Metformin is transported across cellular membranes through organic cation transporters (OCTs) of different isoforms. This is followed by excretion through bile and urine by multidrug and toxin extrusion (MATE) transporters. Here, OCT1 is coded by the solute carrier family 22 member 1 (SLC22A1), which is primarily involved in hepatic uptake, whereas OCT2 (coded by SLC22A2) is involved in tubular secretion140, 141. Genetic variants in these molecules, namely, OCT1, OCT2, MATE1 and MATE2, are associated with both metformin efficacy and intolerance142. For example, SNP rs6622342 AA and rs594709 in the SLC22A1 gene (OCT1) were associated with increased response to metformin amongst South Indian143 and Chinese patients with type 2 diabetes144. In another study of Chinese patients with type 2 diabetes, SLC22A2 808G>T (rs316019) was associated with greater HbA1c reduction, along with reduced renal clearance145. Multiple variants in the SLC22A2 gene (OCT2), for instance, c.596C>T (rs201919874), c.602C>T and c.808G>T (rs316019), were associated with reduced renal clearance of metformin146. In the analysis of the Diabetes Prevention Program, the minor allele of SLC47A1 (solute carrier family 47 member 1, encoding MATE1) variant rs8065082 (C>T) was associated with a lower incidence of diabetes in individuals treated with metformin compared with the major allele, with a hazard ratio of 0.78 (P = 0.02)147. In Chinese patients with type 2 diabetes, increased response to metformin amongst carriers of SLC47A1 rs2289669 (G>A) have also been reported148. In contrast, the SLC47A2 (solute carrier family 47 member 2, encoding MATE2) variant rs12943590 was associated with poor treatment response to metformin in a USA population149.
Along with metformin, SU is commonly prescribed in type 2 diabetes, and works by binding to the SU receptor 1 to stimulate insulin secretion. Genetic polymorphisms of ABCC8 (SU receptor 1), KCNJ11 (Kir6.2), CYP2C9, TCF7L2 and NOS1AP (nitric oxide synthase 1 adaptor protein) genes were associated with altered responses to SU134. In Chinese patients with type 2 diabetes, a variant in ABCC8 (S1369A) was consistently found to be associated with a favorable response to SU150. Other comparative studies have shown that carriers of KCNJ11 variants had a better response to SU than insulin151, 152. It is noteworthy that some of these genes were implicated in MODY and neonatal diabetes, and thus their sensitivity to SU therapy might be due to their primary defect rather than changes in drug metabolism.
In contrast, cytochrome P450 2C9 (CYP2C9) is the main enzyme involved in the metabolism of SU. Carriers of CYP2C9 variants (rs1057910, rs1799853, CYP2C9*3) have reduced SU clearance153 and increased sensitivity to SU therapy. In these individuals, a lower dose is advised to reduce the risk of hypoglycemia142. In contrast, carriers of variants of TCF7L2 (T/T genotype at rs12255372), a well‐documented gene for type 2 diabetes, have been found to be associated with reduced responsiveness to SUs154. Apart from pharmacogenetic responses, these diverse genotype‐treatment responses might reflect the degree of β‐cell dysfunction as a result of these genetic variants, which might influence their response to SU.
TZDs bind to and activate peroxisome proliferator‐activated receptor gamma (PPAR‐γ), a member of the nuclear receptor family regulating carbohydrates and lipid metabolism. The PPAR γ2 Pro12Ala variant is associated with a reduced risk of type 2 diabetes155 and improved insulin sensitivity156, 157. Patients with type 2 diabetes who carried the risk‐conferring PPAR γ2 Pro12Ala variant had a significant response to TZD158, 159. In a recent study, carriers of variants in CYP2C8 and SLCO1B1 also had improved responses to rosiglitazone, highlighting the importance of transporters and metabolizing genes in pharmacogenetics160.
For newer blood glucose‐lowering drugs, such as dipeptidyl peptidase‐4 inhibitors and sodium–glucose cotransporter 2 (SGLT‐2) inhibitors, there are few pharmacogenetic studies of treatment efficacy so far. In an association study, researchers reported an association between rs7202877 near CTRB1/2 (chymotrypsinogen B1/2) and lower HbA1c response to dipeptidyl peptidase‐4 inhibitors161. Although pharmacogenetic variants might provide new perspectives on precision medicine, given the large number of factors that can influence insulin secretion, insulin action and drug metabolism, the availability of large cohorts or datasets with well‐defined phenotypes and genomic data is essential to detect patterns of treatment responses (safety, tolerability and efficacy) in different patients groups, as well as to explore repositioning of drug indications based on genomic discoveries. As an illustration, a minor missense variant (Ala316Thr; rs10305492) in the glucagon‐like peptide‐1 receptor gene was associated with reduced fasting plasma glucose, type 2 diabetes risk and cardiovascular risk162. These genetic associations are in line with the continuum of hyperglycemia, type 2 diabetes and CVD. Recently, several cardiovascular outcome studies have confirmed the beneficial effects of glucagon‐like peptide‐1 receptor agonist in reducing CVD163, raising the possibility of using genetic information to validate therapeutic targets and guide drug development162.
Conclusion
Great advances have been made in our exploration of the genetic architecture of diabetes and its complications, which have provided the foundations for the development of genomic medicine in diabetes. Genetic analysis can help discriminate subgroups of patients with specific molecular defects who otherwise are classified under the broad umbrella of type 1 diabetes or type 2 diabetes, and provide the basis for optimal preventive or therapeutic interventions23. Genetic testing is considered to be effective in increasing the precision of diagnosis and treatment selection in individuals with known monogenic diabetes including MODY and neonatal diabetes. Comprehensive testing using sequencing can now identify causal mutations in >80% of patients with monogenic diabetes126, and serves as a promising framework for precision medicine in diabetes126. In contrast, advances in pharmacogenomics have the potential to predict individual response and ADE. For example, patients with genetic variants associated with increased responsiveness to metformin and SU as a result of reduced clearance might also be at risk of side‐effects with deteriorating renal function, whereas patients with variants associated with increased responsiveness to TZD might benefit from the drug, despite the general decline in the use of this drug class (Figure 3).
Figure 3.
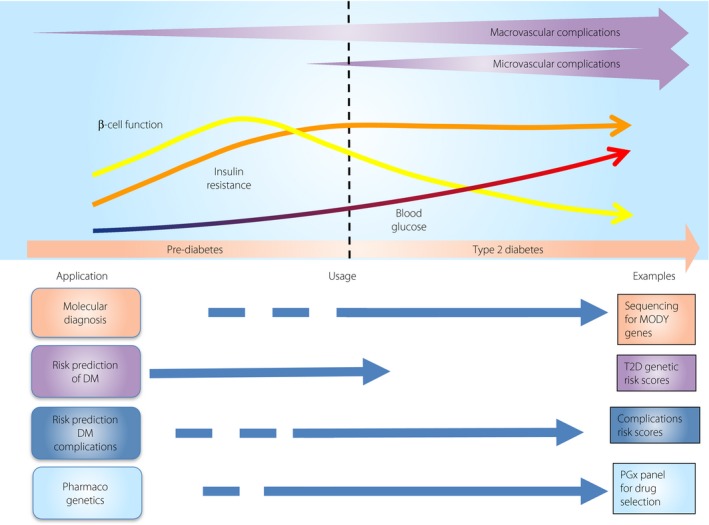
Potential application of precision medicine in diabetes at different stages of diabetes. DM, diabetes mellitus; MODY, maturity‐onset diabetes of the young; T2D, type 2 diabetes.
Continuing advances in genotyping and genome sequencing technologies, together with increasing sample sizes, will lead to the discovery of an expanding list of risk variants for diabetes and its complications, as well as drug responses. Questions remain regarding the additional information required to define the clinical utility of these data, and how best to incorporate them into routine diabetes care. Future areas of research include defining the functional impact of these identified variants on gene regulation or expression, further exploration of the extent to which genetic factors contribute to the observed phenotype, gene–environment interactions and gene–treatment responses. In addition to advances in “genomics,” there is also a need to integrate other types of “omics” (e.g., epigenomics, proteomics, metabolomics, lipidomics, transcriptomics, immunomics) to provide a full landscape of the genomic structure/function and their correlations with disease pathways, phenotypes and treatment response. To this end, incorporation of such “omics” markers into clinical care would require an optimized framework to integrate genomic data into clinic records for evaluation of the validity, efficacy and cost‐effectiveness of clinical testing. Most importantly, these initiatives must be accompanied by ongoing training and education of clinicians on how to use these data appropriately164. In addition, more research efforts are required to build the clinical evidence and roadmap for achieving consensus and developing guidelines in genomic medicine165 in our pursuit of precision medicine for making the right diagnosis and giving the right drug at the right time for the right outcome.
Disclosure
JCNC serves on advisory boards and/or steering committees of research and education programs sponsored by Amgen, Astra Zeneca, Bayer, Boehringer Ingelheim, Lilly, Merck Sharpe Dohme, Pfizer and Sanofi. Her affiliated institutions, the Chinese University of Hong Kong and Asia Diabetes Foundation, had received research and educational grants from these companies. RCWM received research funding from AstraZeneca, Bayer, Boehringer Ingelheim, Merck Sharp & Dohme, Pfizer and Takeda for carrying out clinical trials, speaker honorarium or consultancy in advisory boards. All proceeds have been donated to the Chinese University of Hong Kong to support diabetes research. JCNC and RCWM are co‐founders of GemVCare, a diabetes genetic testing laboratory, which was established through support from the Technology Start‐up Support Scheme for Universities (TSSSU) from the Hong Kong Government Innovation and Technology Commission (ITC).
Acknowledgments
RCWM and JCNC acknowledge support from the Research Grants Council Theme‐based Research Scheme (T12‐402/13N), the Health and Medical Research Fund from the Food and Health Bureau of the Government of the Hong Kong Special Administrative Region (13140761), the Focused Innovation Scheme, the Vice‐Chancellor One‐off Discretionary Fund of the Chinese University of Hong Kong, and the Chinese University of Hong Kong‐Shanghai Jiao Tong University Joint Research Collaboration Fund.
J Diabetes Investig 2018; 9: 998–1015
References
- 1. International Diabetes Federation . IDF Diabetes Atlas, 8th edn Available from: http://wwwdiabetesatlasorg/ 2017 Accessed December 19th, 2017. [Google Scholar]
- 2. Nanditha A, Ma RC, Ramachandran A, et al Diabetes in Asia and the Pacific: implications for the global epidemic. Diabetes Care 2016; 39: 472–485. [DOI] [PubMed] [Google Scholar]
- 3. Chan JC, Zhang Y, Ning G. Diabetes in China: a societal solution for a personal challenge. Lancet Diabetes Endocrinol 2014; 2: 969–979. [DOI] [PubMed] [Google Scholar]
- 4. Xu Y, Wang L, He J, et al Prevalence and control of diabetes in Chinese adults. JAMA 2013; 310: 948–959. [DOI] [PubMed] [Google Scholar]
- 5. Ingelfinger JR, Rosen CJ. Cardiac and renovascular complications in type 2 diabetes–is there hope? N Engl J Med 2016; 375: 380–382. [DOI] [PubMed] [Google Scholar]
- 6. Worel JN, Hayman LL. Preventing cardiovascular disease in patients with diabetes: new evidence informs changes in standards of care. J Cardiovasc Nurs 2016; 31: 198–200. [DOI] [PubMed] [Google Scholar]
- 7. Gregg EW, Sattar N, Ali MK. The changing face of diabetes complications. Lancet Diabetes Endocrinol 2016; 4: 537–547. [DOI] [PubMed] [Google Scholar]
- 8. Cummings DM, Kirian K, Howard G, et al Consequences of comorbidity of elevated stress and/or depressive symptoms and incident cardiovascular outcomes in diabetes: results from the reasons for geographic and racial differences in stroke (REGARDS) study. Diabetes Care 2016; 39: 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou K, Donnelly L, Yang J, et al Heritability of variation in glycaemic response to metformin: a genome‐wide complex trait analysis. Lancet Diabetes Endocrinol 2014; 2: 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiang GZ, Luk AO, Yang XL, et al Progression to treatment failure among Chinese patients with type 2 diabetes initiated on metformin versus sulphonylurea monotherapy‐The Hong Kong Diabetes Registry. Diabetes Res Clin Pract 2016; 112: 57–64. [DOI] [PubMed] [Google Scholar]
- 11. Kahn SE, Haffner SM, Heise MA, et al Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med 2006; 355: 2427–2443. [DOI] [PubMed] [Google Scholar]
- 12. Weng J, Li Y, Xu W, et al Effect of intensive insulin therapy on beta‐cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: a multicentre randomised parallel‐group trial. Lancet 2008; 371: 1753–1760. [DOI] [PubMed] [Google Scholar]
- 13. Franks PW, Pearson E, Florez JC. Gene‐environment and gene‐treatment interactions in type 2 diabetes: progress, pitfalls, and prospects. Diabetes Care 2013; 36: 1413–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pearson E. RD Lawrence lecture 2013. Stratified approaches to the management of diabetes. Diabet Med 2014; 31: 393–398. [DOI] [PubMed] [Google Scholar]
- 15. Pearson ER. Personalized medicine in diabetes: the role of ‘omics’ and biomarkers. Diabet Med 2016; 33: 712–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Malandrino N, Smith RJ. Personalized medicine in diabetes. Clin Chem 2011; 57: 231–240. [DOI] [PubMed] [Google Scholar]
- 17. Smith RJ, Nathan DM, Arslanian SA, et al Individualizing therapies in type 2 diabetes mellitus based on patient characteristics: what we know and what we need to know. J Clin Endocrinol Metab 2010; 95: 1566–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thomsen SK, Gloyn AL. Human genetics as a model for target validation: finding new therapies for diabetes. Diabetologia 2017; 60: 960–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hattersley AT, Patel KA. Precision diabetes: learning from monogenic diabetes. Diabetologia 2017; 60: 769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McCarthy MI. Painting a new picture of personalised medicine for diabetes. Diabetologia 2017; 60: 793–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karki R, Pandya D, Elston RC, et al Defining “mutation” and “polymorphism” in the era of personal genomics. BMC Med Genomics 2015; 8: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hoffmann TJ, Witte JS. Strategies for imputing and analyzing rare variants in association studies. Trends Genet 2015; 31: 556–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Florez JC. Precision medicine in diabetes: is it time? Diabetes Care 2016; 39: 1085–1088. [DOI] [PubMed] [Google Scholar]
- 24. Frayling TM. Genome‐wide association studies provide new insights into type 2 diabetes aetiology. Nat Rev Genet 2007; 8: 657–662. [DOI] [PubMed] [Google Scholar]
- 25. Fan B, Luk AOY, Chan JCN, et al MicroRNA and diabetic complications: a clinical perspective. Antioxid Redox Signal 2017. 10.1089/ars.2017.7318 [DOI] [PubMed] [Google Scholar]
- 26. Zeggini E. Replication of genome‐wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 2007; 317: 1035–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sladek R, Rocheleau G, Rung J, et al A genome‐wide association study identifies novel risk loci for type 2 diabetes. Nature 2007; 445: 881–885. [DOI] [PubMed] [Google Scholar]
- 28. Scott LJ, Mohlke KL, Bonnycastle LL, et al A genome‐wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 2007; 316: 1341–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cho AH, Killeya‐Jones LA, O'Daniel JM, et al Effect of genetic testing for risk of type 2 diabetes mellitus on health behaviors and outcomes: study rationale, development and design. BMC Health Serv Res 2012; 12: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Saxena R, Voight BF, Lyssenko V, et al Genome‐wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007; 316: 1331–1336. [DOI] [PubMed] [Google Scholar]
- 31. Todd JA, Walker NM, Cooper JD, et al Robust associations of four new chromosome regions from genome‐wide analyses of type 1 diabetes. Nat Genet 2007; 39: 857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Frayling TM, Colhoun H, Florez JC. A genetic link between type 2 diabetes and prostate cancer. Diabetologia 2008; 51: 1757–1760. [DOI] [PubMed] [Google Scholar]
- 33. Menzaghi C, Trischitta V, Doria A. Genetic influences of adiponectin on insulin resistance, type 2 diabetes, and cardiovascular disease. Diabetes 2007; 56: 1198–1209. [DOI] [PubMed] [Google Scholar]
- 34. Yamauchi T, Hara K, Maeda S, et al A genome‐wide association study in the Japanese population identifies susceptibility loci for type 2 diabetes at UBE2E2 and C2CD4A‐C2CD4B. Nat Genet 2010; 42: 864–868. [DOI] [PubMed] [Google Scholar]
- 35. Li HX, Gan W, Lu L, et al A genome‐wide association study identifies GRK5 and RASGRP1 as type 2 diabetes loci in Chinese Hans. Diabetes 2013; 62: 291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hara K, Fujita H, Johnson TA, et al Genome‐wide association study identifies three novel loci for type 2 diabetes. Hum Mol Genet 2014; 23: 239–246. [DOI] [PubMed] [Google Scholar]
- 37. Hakonarson H, Grant SFA, Bradfield JP, et al A genome‐wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature 2007; 448: 591–597. [DOI] [PubMed] [Google Scholar]
- 38. Nejentsev S, Howson JMM, Walker NM, et al Localization of type 1 diabetes.susceptibility to the MHC class I genes HLA‐B and HLA‐A. Nature 2007; 450: 887–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Park Y. Prediction of the risk of type 1 diabetes from polymorphisms in candidate genes. Diabetes Res Clin Pract 2004; 66(Suppl 1): S19–S25. [DOI] [PubMed] [Google Scholar]
- 40. Julier C, Akolkar B, Concannon P, et al The Type I Diabetes Genetics Consortium ‘Rapid Response’ family‐based candidate gene study: strategy, genes selection, and main outcome. Genes Immun 2009; 10: S121–S127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ram R, Mehta M, Nguyen QT, et al Systematic evaluation of genes and genetic variants associated with type 1 diabetes susceptibility. J Immunol 2016; 196: 3043–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jia X, Yu H, Zhang H, et al Integrated analysis of different microarray studies to identify candidate genes in type 1 diabetes. J Diabetes 2017; 9: 149–157. [DOI] [PubMed] [Google Scholar]
- 43. Krischer JP, Liu X, Lernmark A, et al The influence of type 1 diabetes genetic susceptibility regions, age, sex, and family history on the progression from multiple autoantibodies to type 1 diabetes: a TEDDY study report. Diabetes 2017; 66: 3122–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Planas R, Pujol‐Borrell R, Vives‐Pi M. Global gene expression changes in type 1 diabetes: insights into autoimmune response in the target organ and in the periphery. Immunol Lett 2010; 133: 55–61. [DOI] [PubMed] [Google Scholar]
- 45. Sharp RC, Abdulrahim M, Naser ES, et al Genetic variations of PTPN2 and PTPN22: role in the pathogenesis of type 1 diabetes and Crohn's disease. Front Cell Infect Microbiol 2015; 5: 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hober D, Alidjinou EK. Enteroviral pathogenesis of type 1 diabetes: queries and answers. Curr Opin Infect Dis 2013; 26: 263–269. [DOI] [PubMed] [Google Scholar]
- 47. Kaul N, Ali S. Genes, genetics, and environment in type 2 diabetes: implication in personalized medicine. DNA Cell Biol 2016; 35: 1–12. [DOI] [PubMed] [Google Scholar]
- 48. Fuchsberger C, Flannick J, Teslovich TM, et al The genetic architecture of type 2 diabetes. Nature 2016; 536: 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Scott RA, Scott LJ, Magi R, et al An expanded genome‐wide association study of type 2 diabetes in Europeans. Diabetes 2017; 66: 2888–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Karaderi T, Drong AW, Lindgren CM. Insights into the genetic susceptibility to type 2 diabetes from genome‐wide association studies of obesity‐related traits. Curr Diab Rep 2015; 15: 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Moltke I, Grarup N, Jørgensen ME, et al A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature 2014; 512: 190–193. [DOI] [PubMed] [Google Scholar]
- 52. Manousaki D, Kent JW, Haack K, et al Toward precision medicine: TBC1D4 disruption is common among the Inuit and leads to underdiagnosis of type 2 diabetes. Diabetes Care 2016; 39: 1889–1895. [DOI] [PubMed] [Google Scholar]
- 53. Ng MC, Park KS, Oh B, et al Implication of genetic variants near TCF7L2, SLC30A8, HHEX, CDKAL1, CDKN2A/B, IGF2BP2, and FTO in type 2 diabetes and obesity in 6,719 Asians. Diabetes 2008; 57: 2226–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ng MC, Wang Y, So WY, et al Ethnic differences in the linkage disequilibrium and distribution of single‐nucleotide polymorphisms in 35 candidate genes for cardiovascular diseases. Genomics 2004; 83: 559–565. [DOI] [PubMed] [Google Scholar]
- 55. Ng MC, Tam CH, Lam VK, et al Replication and identification of novel variants at TCF7L2 associated with type 2 diabetes in Hong Kong Chinese. J Clin Endocrinol Metab 2007; 92: 3733–3737. [DOI] [PubMed] [Google Scholar]
- 56. Ma RC, Chan JC. Type 2 diabetes in East Asians: similarities and differences with populations in Europe and the United States. Ann N Y Acad Sci 2013; 1281: 64–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Park KS. Prevention of type 2 diabetes mellitus from the viewpoint of genetics. Diabetes Res Clin Pract 2004; 66(Suppl 1): S33–S35. [DOI] [PubMed] [Google Scholar]
- 58. Kwak SH, Kim S‐H, Cho YM, et al A genome‐wide association study of gestational diabetes mellitus in Korean women. Diabetes 2012; 61: 531–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wu L, Cui L, Tam WH, et al Genetic variants associated with gestational diabetes mellitus: a meta‐analysis and subgroup analysis. Sci Rep 2016; 6: 30539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hryniewicka J, Zbucka‐Kretowska M, Wawrusiewicz‐Kurylonek N, et al Genetic variants associated with type 2 diabetes and obesity better predict gestational diabetes than traditional risk factors. Diabetologia 2014; 57: S448. [Google Scholar]
- 61. Chan JCN, Yeung R, Luk A. The Asian diabetes phenotypes: challenges and opportunities. Diabetes Res Clin Pract 2014; 105: 135–139. [DOI] [PubMed] [Google Scholar]
- 62. Bonfig W, Hermanns S, Warncke K, et al GCK‐MODY (MODY 2) caused by a Novel p.Phe330Ser mutation. ISRN Pediatr 2011; 2011: 676549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Doria A, Patti ME, Kahn CR. The emerging genetic architecture of type 2 diabetes. Cell Metab 2008; 8: 186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ledermann H. Maturity‐onset diabetes of the young (MODY) at least ten times more common in Europe than previously assumed? Diabetologia 1995; 38: 1482. [DOI] [PubMed] [Google Scholar]
- 65. Pearson ER, Flechtner I, Njolstad PR, et al Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med 2006; 355: 467–477. [DOI] [PubMed] [Google Scholar]
- 66. Vaxillaire M, Froguel P. Monogenic diabetes: implementation of translational genomic research towards precision medicine. J Diabetes 2016; 8: 782–795. [DOI] [PubMed] [Google Scholar]
- 67. McCarthy MI, Hattersley AT. Molecular diagnostics in monogenic and multifactorial forms of type 2 diabetes. Expert Rev Mol Diagn 2001; 1: 403–412. [DOI] [PubMed] [Google Scholar]
- 68. Froguel P, Vaxillaire M, Sun F, et al Close linkage of glucokinase locus on chromosome 7p to early‐onset non‐insulin‐dependent diabetes mellitus. Nature 1992; 356: 162–164. [DOI] [PubMed] [Google Scholar]
- 69. Hattersley AT, Turner RC, Permutt MA, et al Linkage of type 2 diabetes to the glucokinase gene. Lancet 1992; 339: 1307–1310. [DOI] [PubMed] [Google Scholar]
- 70. Steele AM, Shields BM, Wensley KJ, et al Prevalence of vascular complications among patients with glucokinase mutations and prolonged, mild hyperglycemia. JAMA 2014; 311: 279–286. [DOI] [PubMed] [Google Scholar]
- 71. Ng MC, Lee SC, Ko GT, et al Familial early‐onset type 2 diabetes in Chinese patients: obesity and genetics have more significant roles than autoimmunity. Diabetes Care 2001; 24: 663–671. [DOI] [PubMed] [Google Scholar]
- 72. Chan JC, Lau ES, Luk AO, et al Premature mortality and comorbidities in young‐onset diabetes: a 7‐year prospective analysis. Am J Med 2014; 127: 616–624. [DOI] [PubMed] [Google Scholar]
- 73. Luk AO, Lau ES, So WY, et al Prospective study on the incidences of cardiovascular‐renal complications in Chinese patients with young‐onset type 1 and type 2 diabetes. Diabetes Care 2014; 37: 149–157. [DOI] [PubMed] [Google Scholar]
- 74. Hattersley AT, Beards F, Ballantyne E, et al Mutations in the glucokinase gene of the fetus result in reduced birth weight. Nat Genet 1998; 19: 268–270. [DOI] [PubMed] [Google Scholar]
- 75. Spyer G, Macleod KM, Shepherd M, et al Pregnancy outcome in patients with raised blood glucose due to a heterozygous glucokinase gene mutation. Diabet Med 2009; 26: 14–18. [DOI] [PubMed] [Google Scholar]
- 76. Hattersley AT. Maturity‐onset diabetes of the young: clinical heterogeneity explained by genetic heterogeneity. Diabet Med 1998; 15: 15–24. [DOI] [PubMed] [Google Scholar]
- 77. Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity‐onset diabetes of the young. N Engl J Med 2001; 345: 971–980. [DOI] [PubMed] [Google Scholar]
- 78. Ng MC, Li JK, So WY, et al Nature or nurture: an insightful illustration from a Chinese family with hepatocyte nuclear factor‐1 alpha diabetes (MODY3). Diabetologia 2000; 43: 816–818. [DOI] [PubMed] [Google Scholar]
- 79. Pearson ER, Starkey BJ, Powell RJ, et al Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet 2003; 362: 1275–1281. [DOI] [PubMed] [Google Scholar]
- 80. Anik A, Catli G, Abaci A, et al Maturity‐onset diabetes of the young (MODY): an update. J Pediatr Endocrinol Metab 2015; 28: 251–263. [DOI] [PubMed] [Google Scholar]
- 81. Zung A, Glaser B, Nimri R, et al Glibenclamide treatment in permanent neonatal diabetes mellitus due to an activating mutation in Kir6.2. J Clin Endocrinol Metab 2004; 89: 5504–5507. [DOI] [PubMed] [Google Scholar]
- 82. Grulich‐Henn J, Wagner V, Thon A, et al Entities and frequency of neonatal diabetes: data from the diabetes documentation and quality management system (DPV). Diabet Med 2010; 27: 709–712. [DOI] [PubMed] [Google Scholar]
- 83. Sagen JV, Raeder H, Hathout E, et al Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: patient characteristics and initial response to sulfonylurea therapy. Diabetes 2004; 53: 2713–2718. [DOI] [PubMed] [Google Scholar]
- 84. Knip M, Korhonen S, Kulmala P, et al Prediction of type 1 diabetes in the general population. Diabetes Care 2010; 33: 1206–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Winkler C, Krumsiek J, Buettner F, et al Feature ranking of type 1 diabetes susceptibility genes improves prediction of type 1 diabetes. Diabetologia 2014; 57: 2521–2529. [DOI] [PubMed] [Google Scholar]
- 86. Brorsson CA, Nielsen LB, Andersen ML, et al Genetic risk score modelling for disease progression in new‐onset type 1 diabetes patients: increased genetic load of islet‐expressed and cytokine‐regulated candidate genes predicts poorer glycemic control. J Diabetes Res 2016; 2016: 9570424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Winkler C, Krumsiek J, Lempainen J, et al A strategy for combining minor genetic susceptibility genes to improve prediction of disease in type 1 diabetes. Genes Immun 2012; 13: 549–555. [DOI] [PubMed] [Google Scholar]
- 88. Padwal R, Majumdar SR, Johnson JA, et al A systematic review of drug therapy to delay or prevent type 2 diabetes. Diabetes Care 2005; 28: 736–744. [DOI] [PubMed] [Google Scholar]
- 89. Haw JS, Galaviz KI, Straus AN, et al Long‐term sustainability of diabetes prevention approaches: a systematic review and meta‐analysis of randomized clinical trials. JAMA Intern Med 2017; 177: 1808–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Miyake K, Horikawa Y, Hara K, et al Association of TCF7L2 polymorphisms with susceptibility to type 2 diabetes in 4,087 Japanese subjects. J Hum Genet 2008; 53: 174–180. [DOI] [PubMed] [Google Scholar]
- 91. Cropano C, Santoro N, Groop L, et al The rs7903146 variant in the TCF7L2 gene increases the risk of prediabetes/type 2 diabetes in obese adolescents by impairing beta‐cell function and hepatic insulin sensitivity. Diabetes Care 2017; 40: 1082–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wang J, Kuusisto J, Vanttinen M, et al Variants of transcription factor 7‐like 2 (TCF7L2) gene predict conversion to type 2 diabetes in the Finnish Diabetes Prevention Study and are associated with impaired glucose regulation and impaired insulin secretion. Diabetologia 2007; 50: 1192–1200. [DOI] [PubMed] [Google Scholar]
- 93. Zhang C, Qi L, Hunter DJ, et al Variant of transcription factor 7‐like 2 (TCF7L2) gene and the risk of type 2 diabetes in large cohorts of U.S. women and men. Diabetes 2006; 55: 2645–2648. [DOI] [PubMed] [Google Scholar]
- 94. Cauchi S, Meyre D, Durand E, et al Post genome‐wide association studies of novel genes associated with type 2 diabetes show gene–gene interaction and high predictive value. PLoS ONE 2008; 3: e2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Walford GA, Porneala BC, Dauriz M, et al Metabolite traits and genetic risk provide complementary information for the prediction of future type 2 diabetes. Diabetes Care 2014; 37: 2508–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Talmud PJ, Cooper JA, Morris RW, et al Sixty‐five common genetic variants and prediction of type 2 diabetes. Diabetes 2015; 64: 1830–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Miyake K, Yang W, Hara K, et al Construction of a prediction model for type 2 diabetes mellitus in the Japanese population based on 11 genes with strong evidence of the association. J Hum Genet 2009; 54: 236–241. [DOI] [PubMed] [Google Scholar]
- 98. Lall K, Magi R, Morris A, et al Personalized risk prediction for type 2 diabetes: the potential of genetic risk scores. Genet Med 2017; 19: 322–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Li G, Zhang P, Wang J, et al Cardiovascular mortality, all‐cause mortality, and diabetes incidence after lifestyle intervention for people with impaired glucose tolerance in the Da Qing Diabetes Prevention Study: a 23‐year follow‐up study. Lancet Diabetes Endocrinol 2014; 2: 474–480. [DOI] [PubMed] [Google Scholar]
- 100. Ma R, Cooper ME. Genetics of diabetic kidney disease‐from the worst of nightmares to the light of dawn? J Am Soc Nephrol 2017; 28: 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Sandholm N, Van Zuydam N, Ahlqvist E, et al The genetic landscape of renal complications in type 1 diabetes. J Am Soc Nephrol 2017; 28: 557–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ma RC. Genetics of cardiovascular and renal complications in diabetes. J Diabetes Investig 2016; 7: 139–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Thomas G, Sehgal AR, Kashyap SR, et al Metabolic syndrome and kidney disease: a systematic review and meta‐analysis. Clin J Am Soc Nephrol 2011; 6: 2364–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev 2013; 93: 137–188. [DOI] [PubMed] [Google Scholar]
- 105. Alkhalaf A, Bakker S, Bilo H, et al A polymorphism in the gene encoding carnosinase (CNDP1) as a predictor of mortality and progression from nephropathy to end‐stage renal disease in type 1 diabetes mellitus. Diabetologia 2010; 53: 2562–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Luk AO, Wang Y, Ma RC, et al Predictive role of polymorphisms in interleukin‐5 receptor alpha‐subunit, lipoprotein lipase, integrin A2 and nitric oxide synthase genes on ischemic stroke in type 2 diabetes–an 8‐year prospective cohort analysis of 1327 Chinese patients. Atherosclerosis 2011; 215: 130–135. [DOI] [PubMed] [Google Scholar]
- 107. Wang Y, Luk AO, Ma RC, et al Predictive role of multilocus genetic polymorphisms in cardiovascular disease and inflammation‐related genes on chronic kidney disease in Type 2 diabetes–an 8‐year prospective cohort analysis of 1163 patients. Nephrol Dial Transplant 2012; 27: 190–196. [DOI] [PubMed] [Google Scholar]
- 108. Barakat K, Hitman G. Genetic susceptibility to macrovascular complications of type 2 diabetes mellitus. Best Pract Res Clin Endocrinol Metab 2001; 15: 359–370. [DOI] [PubMed] [Google Scholar]
- 109. Lange LA, Bowden DW, Langefeld CD, et al Heritability of carotid artery intima‐medial thickness in type 2 diabetes. Stroke 2002; 33: 1876–1881. [DOI] [PubMed] [Google Scholar]
- 110. Qi L, Parast L, Cai T, et al Genetic susceptibility to coronary heart disease in type 2 diabetes: 3 independent studies. J Am Coll Cardiol 2011; 58: 2675–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Qi L, Qi Q, Prudente S, et al Association between a genetic variant related to glutamic acid metabolism and coronary heart disease in individuals with type 2 diabetes. JAMA 2013; 310: 821–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Sobrin L, Chong YH, Fan Q, et al Genetically determined plasma lipid levels and risk of diabetic retinopathy: a Mendelian randomization study. Diabetes 2017; 66: 3130–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Dahlstrom E, Sandholm N. Progress in defining the genetic basis of diabetic complications. Curr Diab Rep 2017; 17: 80. [DOI] [PubMed] [Google Scholar]
- 114. Kwak SH, Park KS. Genetic studies on diabetic microvascular complications: focusing on genome‐wide association studies. Endocrinol Metab 2015; 30: 147–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Pan Q, Delahanty LM, Jablonski KA, et al Variation at the melanocortin 4 receptor gene and response to weight‐loss interventions in the diabetes prevention program. Obesity 2013; 21: E520–E526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Uusitupa M. Gene–diet interaction in relation to the prevention of obesity and type 2 diabetes: evidence from the Finnish Diabetes Prevention Study. Nutr Metab Cardiovasc Dis 2005; 15: 225–233. [DOI] [PubMed] [Google Scholar]
- 117. Laukkanen O, Pihlajamaki J, Lindstrom J, et al Polymorphisms of the SUR1 (ABCC8) and Kir6.2 (KCNJ11) genes predict the conversion from impaired glucose tolerance to type 2 diabetes. The Finnish Diabetes Prevention Study. J Clin Endocrinol Metab 2004; 89: 6286–6290. [DOI] [PubMed] [Google Scholar]
- 118. Grotenfelt NE, Wasenius NS, Rono K, et al Interaction between rs10830963 polymorphism in MTNR1B and lifestyle intervention on occurrence of gestational diabetes. Diabetologia 2016; 59: 1655–1658. [DOI] [PubMed] [Google Scholar]
- 119. Grant RW, Meigs JB, Florez JC, et al Design of a randomized trial of diabetes genetic risk testing to motivate behavior change: the Genetic Counseling/lifestyle Change (GC/LC) Study for Diabetes Prevention. Clin Trials 2011; 8: 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Waxler JL, O'Brien KE, Delahanty LM, et al Genetic counseling as a tool for type 2 diabetes prevention: a genetic counseling framework for common polygenetic disorders. J Genet Couns 2012; 21: 684–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Nishigaki M, Tokunaga‐Nakawatase Y, Nishida J, et al The effect of genetic counseling for adult offspring of patients with type 2 diabetes on attitudes toward diabetes and its heredity: a randomized controlled trial. J Genet Couns 2014; 23: 762–769. [DOI] [PubMed] [Google Scholar]
- 122. Hivert MF, Christophi CA, Franks PW, et al Lifestyle and metformin ameliorate insulin sensitivity independently of the genetic burden of established insulin resistance variants in diabetes prevention program participants. Diabetes 2016; 65: 520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Hivert MF, Jablonski KA, Perreault L, et al Updated genetic score based on 34 confirmed type 2 diabetes Loci is associated with diabetes incidence and regression to normoglycemia in the diabetes prevention program. Diabetes 2011; 60: 1340–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Gallagher P, King HA, Haga SB, et al Patient beliefs and behaviors about genomic risk for type 2 diabetes: implications for prevention. J Health Commun 2015; 20: 728–735. [DOI] [PubMed] [Google Scholar]
- 125. Slingerland AS. Monogenic diabetes in children and young adults: challenges for researcher, clinician and patient. Rev Endocr Metab Disord 2006; 7: 171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. De Franco E, Flanagan SE, Houghton JA, et al The effect of early, comprehensive genomic testing on clinical care in neonatal diabetes: an international cohort study. Lancet 2015; 386: 957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Thurber BW, Carmody D, Tadie EC, et al Age at the time of sulfonylurea initiation influences treatment outcomes in KCNJ11‐related neonatal diabetes. Diabetologia 2015; 58: 1430–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. McCarthy MI, Zeggini E, Jafarmohammad B, et al Analysis of overlap between type 2 diabetes signals identified through genome‐wide linkage and association approaches. Diabetes 2008; 57: A321–A322. [Google Scholar]
- 129. Shepherd M, Shields B, Ellard S, et al A genetic diagnosis of HNF1A diabetes alters treatment and improves glycaemic control in the majority of insulin‐treated patients. Diabet Med 2009; 26: 437–441. [DOI] [PubMed] [Google Scholar]
- 130. Katra B, Klupa T, Skupien J, et al Dipeptidyl peptidase‐IV inhibitors are efficient adjunct therapy in HNF1A maturity‐onset diabetes of the young patients–report of two cases. Diabetes Technol Ther 2010; 12: 313–316. [DOI] [PubMed] [Google Scholar]
- 131. Lumb AN, Gallen IW. Treatment of HNF1‐alpha MODY with the DPP‐4 inhibitor Sitagliptin. Diabet Med 2009; 26: 189–190. [DOI] [PubMed] [Google Scholar]
- 132. Thanabalasingham G, Owen KR. Diagnosis and management of maturity onset diabetes of the young (MODY). BMJ 2011; 343: d6044. [DOI] [PubMed] [Google Scholar]
- 133. Tang Y, Axelsson AS, Spegel P, et al Genotype‐based treatment of type 2 diabetes with an alpha2A‐adrenergic receptor antagonist. Sci Transl Med 2014; 6: 257ra139. [DOI] [PubMed] [Google Scholar]
- 134. Loganadan NK, Huri HZ, Vethakkan SR, et al Genetic markers predicting sulphonylurea treatment outcomes in type 2 diabetes patients: current evidence and challenges for clinical implementation. Pharmacogenomics J 2016; 16: 209–219. [DOI] [PubMed] [Google Scholar]
- 135. Bielinski SJ, Olson JE, Pathak J, et al Preemptive genotyping for personalized medicine: design of the right drug, right dose, right time‐using genomic data to individualize treatment protocol. Mayo Clin Proc 2014; 89: 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ji Y, Skierka JM, Blommel JH, et al Preemptive pharmacogenomic testing for precision medicine: a comprehensive analysis of five actionable pharmacogenomic genes using next‐generation DNA sequencing and a customized CYP2D6 genotyping cascade. J Mol Diagn 2016; 18: 438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Maruthur NM, Gribble MO, Bennett WL, et al The pharmacogenetics of type 2 diabetes: a systematic review. Diabetes Care 2014; 37: 876–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Pearson E. Pharmacogenetics in diabetes. Curr Diab Rep 2009; 9: 172–181. [DOI] [PubMed] [Google Scholar]
- 139. Zhou K, Pedersen HK, Dawed AY, et al Pharmacogenomics in diabetes mellitus: insights into drug action and drug discovery. Nat Rev Endocrinol 2016; 12: 337–346. [DOI] [PubMed] [Google Scholar]
- 140. Wang D‐S, Jonker JW, Kato Y, et al Involvement of organic cation transporter 1 in hepatic and intestinal distribution of metformin. J Pharmacol Exp Ther 2002; 302: 510–515. [DOI] [PubMed] [Google Scholar]
- 141. Kimura N, Masuda S, Tanihara Y, et al Metformin is a superior substrate for renal organic cation transporter OCT2 rather than hepatic OCT1. Drug Metab Pharmacokinet 2005; 20: 379–386. [DOI] [PubMed] [Google Scholar]
- 142. Singh S, Usman K, Banerjee M. Pharmacogenetic studies update in type 2 diabetes mellitus. World J Diabetes 2016; 7: 302–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Umamaheswaran G, Praveen RG, Damodaran SE, et al Influence of SLC22A1 rs622342 genetic polymorphism on metformin response in South Indian type 2 diabetes mellitus patients. Clin Exp Med 2015; 15: 511–517. [DOI] [PubMed] [Google Scholar]
- 144. Xiao D, Guo Y, Li X, et al The impacts of SLC22A1 rs594709 and SLC47A1 rs2289669 polymorphisms on metformin therapeutic efficacy in Chinese type 2 diabetes patients. Int J Endocrinol 2016; 2016: 4350712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Hou W, Zhang D, Lu W, et al Polymorphism of organic cation transporter 2 improves glucose‐lowering effect of metformin via influencing its pharmacokinetics in Chinese type 2 diabetic patients. Mol Diagn Ther 2015; 19: 25–33. [DOI] [PubMed] [Google Scholar]
- 146. Song IS, Shin HJ, Shim EJ, et al Genetic variants of the organic cation transporter 2 influence the disposition of metformin. Clin Pharmacol Ther 2008; 84: 559–562. [DOI] [PubMed] [Google Scholar]
- 147. Jablonski KA, McAteer JB, de Bakker PI, et al Common variants in 40 genes assessed for diabetes incidence and response to metformin and lifestyle intervention in the diabetes prevention program. Diabetes 2010; 59: 2672–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. He R, Zhang D, Lu W, et al SLC47A1 gene rs2289669 G>A variants enhance the glucose‐lowering effect of metformin via delaying its excretion in Chinese type 2 diabetes patients. Diabetes Res Clin Pract 2015; 109: 57–63. [DOI] [PubMed] [Google Scholar]
- 149. Choi JH, Yee SW, Ramirez AH, et al A common 5′‐UTR variant in MATE2‐K is associated with poor response to metformin. Clin Pharmacol Ther 2011; 90: 674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Feng Y, Mao GY, Ren XW, et al Ser1369Ala variant in sulfonylurea receptor gene ABCC8 is associated with antidiabetic efficacy of gliclazide in Chinese type 2 diabetic patients. Diabetes Care 2008; 31: 1939–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Hamming KS, Soliman D, Matemisz LC, et al Coexpression of the type 2 diabetes susceptibility gene variants KCNJ11 E23K and ABCC8 S1369A alter the ATP and sulfonylurea sensitivities of the ATP‐sensitive K(+) channel. Diabetes 2009; 58: 2419–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Li Q, Chen M, Zhang R, et al KCNJ11 E23K variant is associated with the therapeutic effect of sulphonylureas in Chinese type 2 diabetic patients. Clin Exp Pharmacol Physiol 2014; 41: 748–754. [DOI] [PubMed] [Google Scholar]
- 153. Kirchheiner J, Brockmöller J, Meineke I, et al Impact of CYP2C9 amino acid polymorphisms on glyburide kinetics and on the insulin and glucose response in healthy volunteers. Clin Pharmacol Ther 2002; 71: 286–296. [DOI] [PubMed] [Google Scholar]
- 154. Pearson ER, Donnelly LA, Kimber C, et al Variation in TCF7L2 influences therapeutic response to sulfonylureas: a GoDARTs study. Diabetes 2007; 56: 2178–2182. [DOI] [PubMed] [Google Scholar]
- 155. Altshuler D, Hirschhorn JN, Klannemark M, et al The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet 2000; 26: 76–80. [DOI] [PubMed] [Google Scholar]
- 156. Koch M, Rett K, Maerker E, et al The PPARgamma2 amino acid polymorphism Pro 12 Ala is prevalent in offspring of Type II diabetic patients and is associated to increased insulin sensitivity in a subgroup of obese subjects. Diabetologia 1999; 42: 758–762. [DOI] [PubMed] [Google Scholar]
- 157. Stumvoll M, Wahl HG, Loblein K, et al Pro12Ala polymorphism in the peroxisome proliferator‐activated receptor‐gamma2 gene is associated with increased antilipolytic insulin sensitivity. Diabetes 2001; 50: 876–881. [DOI] [PubMed] [Google Scholar]
- 158. Hsieh MC, Lin KD, Tien KJ, et al Common polymorphisms of the peroxisome proliferator‐activated receptor‐gamma (Pro12Ala) and peroxisome proliferator‐activated receptor‐gamma coactivator‐1 (Gly482Ser) and the response to pioglitazone in Chinese patients with type 2 diabetes mellitus. Metabolism 2010; 59: 1139–1144. [DOI] [PubMed] [Google Scholar]
- 159. Kang ES, Park SY, Kim HJ, et al Effects of Pro12Ala polymorphism of peroxisome proliferator‐activated receptor gamma2 gene on rosiglitazone response in type 2 diabetes. Clin Pharmacol Ther 2005; 78: 202–208. [DOI] [PubMed] [Google Scholar]
- 160. Dawed AY, Donnelly L, Tavendale R, et al CYP2C8 and SLCO1B1 variants and therapeutic response to thiazolidinediones in patients with type 2 diabetes. Diabetes Care 2016; 39: 1902–1908. [DOI] [PubMed] [Google Scholar]
- 161. t Hart LM, Fritsche A, Nijpels G, et al The CTRB1/2 locus affects diabetes susceptibility and treatment via the incretin pathway. Diabetes 2013; 62: 3275–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Scott RA, Freitag DF, Li L, et al A genomic approach to therapeutic target validation identifies a glucose‐lowering GLP1R variant protective for coronary heart disease. Sci Transl Med 2016; 8: 341ra76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Bethel MA, Patel RA, Merrill P, et al Cardiovascular outcomes with glucagon‐like peptide‐1 receptor agonists in patients with type 2 diabetes: a meta‐analysis. Lancet Diabetes Endocrinol 2018; 6: 105–113. [DOI] [PubMed] [Google Scholar]
- 164. Floyd JS, Psaty BM. The application of genomics in diabetes: barriers to discovery and implementation. Diabetes Care 2016; 39: 1858–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Schully SD, Lam TK, Dotson WD, et al Evidence synthesis and guideline development in genomic medicine: current status and future prospects. Genet Med 2015; 17: 63–67. [DOI] [PMC free article] [PubMed] [Google Scholar]