

Abstract
We hypothesized that human variation in the activity of the ATP regenerating enzyme creatine kinase (CK) activity affects hypertension and cardiovascular disease risk. CK is tightly bound close to ATP-utilizing enzymes including Ca2+-ATPase, myosin ATPase, and Na+/K+-ATPase, where it rapidly regenerates ATP from ADP, H+, and phosphocreatine. Thus, relatively high CK was thought to enhance ATP-demanding processes including resistance artery contractility and sodium retention, and reduce ADP-dependent functions. In a series of studies of our group and others, CK was linked to hypertension and bleeding risk. Plasma CK after rest, used as a surrogate measure for tissue CK, was associated with high blood pressure and failure of antihypertensive therapy in case-control and population studies. Importantly, high tissue CK preceded hypertension in animal models and in humans, and human vascular tissue CK gene expression was strongly associated with clinical blood pressure. In line with this, CK inhibition substantially reduced the contractility of human resistance arteries ex vivo. We also presented evidence that plasma CK reduced ADP-dependent platelet aggregation. In subsequent intervention studies, the oral competitive CK inhibitor beta-guanidinopropionic acid (GPA) reduced blood pressure in spontaneously hypertensive rats (SHRs), and a 1-week trial of sub-therapeutic dose GPA in healthy men was uneventful. Thus, based on theoretical concepts, evidence was gathered in laboratory, case-control, and population studies that high CK is associated with hypertension and with bleeding risk, potentially leading to a new mode of cardiovascular risk reduction with CK inhibition.
Keywords: Creatine kinase (CK), hypertension, ADP, bleeding, ancestry groups
Introduction
Hypertension is an important worldwide public-health challenge (1). It is a common disease, with more than 25% of the adult people worldwide affected, totaling nearly a billion people. Because of its high frequency and concomitant risks of cardiovascular and kidney disease, hypertension has been called “the silent killer”. Hypertension is the main risk factor for premature mortality, and is ranked third as a cause of disability-adjusted life-years. Cardiovascular diseases are now responsible for 30% of all deaths worldwide (1).
Environmental as well as biological factors are thought to contribute to the occurrence of hypertension (1), but by modeling physiological systems controlling blood pressure, Guyton forwarded that sustained elevation of arterial pressure could be achieved through only two pathways: general vasoconstriction including the renal arteries, or excess renal sodium retention (2). The dominant role of the kidney was confirmed by the discovery that in all Mendelian forms of hypertension renal sodium reabsorption is affected (3). We explored these two pathways leading to hypertension in more detail, focusing on the energy demands of blood pressure generation.
Methods
In this paper, we summarize and synthesize the body of work of the past 20 years on the association between the activity of the ATP-regenerating enzyme creatine kinase (CK, EC 2.7.3.2) and cardiovascular function. A theoretical framework is presented detailing on the energetic demands of hypertension and how high CK activity may alter pressor responses on a molecular level. Furthermore, studies are described on tissue and plasma CK activity levels of different population subgroups and on the association of CK with blood pressure. Clinical and laboratory studies consider vascular contractility and salt retention in the context of high CK. We also address the response to antihypertensive drugs with high CK. Finally, important evidence is presented regarding the effect of CK inhibition on blood pressure, and we discuss related subjects such as obesity and bleeding risk with high CK, as well as future research directions. Data in parentheses are mean ± SE, and data in square brackets are 95% confidence intervals (CI) unless indicated otherwise.
Results
Theoretical modeling: the energy expenditure of blood pressure generation
Cardiovascular work
Mean arterial pressure depends on cardiac output and the total vascular peripheral resistance, expressed in the equation: MAP = CO × TPR; where MAP = mean arterial pressure, CO = cardiac output, and TPR = total peripheral resistance (4). The cardiovascular system is essentially a pump that propels fluid through a set of tubes of decreasing size. The heart beats around 100,000 times a day, pumping approximately 10 tons of blood through the body. This highly energy demanding function requires more ATP than any other organ, around 6 kg per day. To acquire this, the heart uses chemical energy stored in fatty acids and glucose to fuel actin-myosin interaction of myofibrils (5). The enzyme CK is central to cellular energy metabolism, as it rapidly regenerates ATP from phosphocreatine near subcellular locations of high energy demands. In established hypertension, cardiac output is normal or even low (4,6), and a raised vascular resistance is mostly responsible for the elevated pressure. Still, the heart has to generate the energy to create the cardiac output against this higher pressure.
The energy used by the heart to create and sustain pressure is partly “stored” through in the arteries that are actively stretched by the heart’s pumping action. This stored heart's energy is returned to the vascular system between heartbeats through passive elastic recoil. This recoil prevents the blood flow and pressure from falling to zero between heartbeats. Thus, there is a constant supply of blood to tissues. This keeps the efficiency of blood delivery high (7).
Total peripheral resistance of the small arteries is affected by blood viscosity, and by the diameter and total length of the vessels. The greatest contribution to peripheral resistance is made by the small (resistance) arteries and arterioles (8). Flow resistance (R) in a cylindrical tube, such as a blood vessel, can be calculated by Poiseuille’s formula: R= L × ŋ/ri4, where L= the length of vessel, ŋ= viscosity of fluid and ri = the inner radius of the vessel. Therefore, ri plays an important role, and even minute changes in ri will have marked effect on R (4). The natural baseline of ri is thought to be set by structurally determined dimensions and geometric arrangements. In hypertension, arteriolar and capillary rarefaction (obliteration of preexisting blood vessels) is the most commonly observed change that affects the structure of the microvascular network (4,9), while microvascular tonus is thought to be influenced by nervous and humoral factors. Vascular smooth muscle contraction is an active process, myosin light chains must be phosphorylated before shortening can occur. This phosphorylation is performed by myosin light chain kinase (MLCK) activated by the Calcium-Calmodulin complex. Tension maintenance in vascular smooth muscle is a graded phenomenon without twitch tension, and the vessels can generate and maintain tension along their entire length (7,10).
Smooth muscle is generally considered to have a slower metabolism and lower concentration of metabolites compared to striated muscle. However, metabolites and energetic processes are highly organized and structured. CK is present and active in mitochondria, contractile elements, membrane pumps, cytoplasm, where it is involved in distribution and regeneration of ATP (7,10,11).
Murphy et al. (11) discussed that smooth muscle has attached but dephosphorylated cross-bridges, which are slowly cycling cross-bridges that maintain tension with high economy of energy consumption. This state of the cross-bridges termed “latch”, has a 5-fold slower rate of detachment than phosphorylated cycling cross-bridges. Thus, tension is maintained at low Ca2+ with low ATP consumption. However, the energetic cost of generating tension is high (7,10,11). Oxidative and glycolytic utilization of glucose are increased in the aorta of hypertensive rats, and energy consumption is increased by 10–15% (12). This increase in energy consumption might be attributable to increased work (associated with increased tensions) and/or decreased energy efficiency (decrease in contribution of latch) (7,12).
Compared with heart muscle, vascular smooth muscle is extremely glycolytic (13). The flux of energy substrates in vascular smooth muscle is relatively slow compared to the heart in resting condition with ATP hydrolysis of respectively 1.2 vs. 4.3 µM/min/g ww). In stimulated tissue, the comparison changes to 4.3 and 32, respectively. During pathology such as hypertension, the vascular smooth muscle increases energy flux as reflected by significant increases in enzyme activities and net flux. Clark et al. (14) reported a 200% increase in citrate synthase in rat aorta during hypertension. This increase was resolved with adequate antihypertensive treatments.
Aside functional alterations there are structural changes to myocardium and arteries in response to high blood pressure. Muscle cells enlarge, with increased extracellular proteins. During the established phase of hypertension, total peripheral resistance remains increased, structural vascular changes cause decreased compliance, and changes in smooth muscle activity or sensitivity of the vessels to extracellular stimuli may increase peripheral resistance and work for the heart (4-6).
Renal work
Sodium retention in the kidney requires ATP, as Na+/K+-ATPase couples the energy of hydrolysis of ATP to the active exchange of three intracellular Na ions for two extracellular K. The filtration rate of human kidneys amounts to 180 L/day, which corresponds to 1.7 kg of NaCl. This is approximately 11 times our total extracellular space. The largest part of this filtered load must be conserved for the body and absorbed by renal tubules. The overall O2 consumption of both kidneys is approximately 20 L/day, which corresponds to 6 mol, or with a molecular weight of 573.1, about 3 kg of ATP/day (on the basis of 1 mol O2/6 mol ATP). This energy is invested in the absorption of 30 mol of Na+/day. In other words, 5 mol of Na+ are absorbed for each mol of ATP hydrolyzed to ADP (15,16).
Throughout the kidney, in the proximal nephron, in the thick ascending limb of the loop of Henle, as well as in the distal tubule, filtered Na+ is absorbed. Although the exact quantities are determined by many regulatory factors such as glomerulotubular balance, hormonal factors, sympathetic innervation, and acid base status, the essential component of absorption throughout the kidney is basolateral Na+/K+-ATPase. CK rapidly provides ATP to this ATPase (15,17). It can be concluded that both components of pressor responses, vasoconstriction and renal sodium retention, are highly energy demanding processes. To generate and sustain high blood pressure, the cellular energy system needs to regenerate ATP at a higher rate.
The enzyme CK and energy demands of hypertension
CK is the central regulatory enzyme of energy metabolism. The enzyme catalyzes the rapid and reversible transfer of the high-energy phosphate moiety (P) between creatine and ADP:
MgADP + PCreatine + H+ ↔ MgATP + Creatine.
Importantly, the rate of transfer of the phosphoryl group from phosphocreatine (Pcreatine) to ADP by CK is greater than the maximum rate of ATP generation by oxidative phosphorylation and glycolysis together (18,19). CK is specifically located at subcellular compartments where energy is produced such as in the mitochondrial intermembranous space and near glycolytic enzymes; as well as at compartments where energy is used, including near motor proteins involved in force generation at acto-myosin ATPase, at sarcoplasmatic reticulum-Ca2+-ATPase, and ATPases involved in ion transport, such as Na+/K+-ATPase and Ca2+-ATPase (18).
On the energy-producing site, dimeric/octameric mitochondrial CK facilitates the transphosphorylation of matrix-generated ATP into phosphocreatine and its subsequent export out of the mitochondrion. At subcellular compartments of energy consumption, ATP is directly regenerated by dimeric cytosolic CK via phosphocreatine, thus keeping local ATP/ADP ratios near ATPases very high, thereby increasing the thermodynamic efficiency of ATP hydrolysis. Thus, via phosphocreatine and creatine, the CK system buffers and transports energy produced by glycolysis and oxidative phosphorylation, to sites of energy production, such as myofibrils and membrane ion pumps, forming a spatial and temporal energy buffer (18-20). While phosphocreatine helps maintain the ATP concentration, creatine and ADP can signal the mitochondria to stimulate oxidative phosphorylation and ATP production.
Another important function of CK is proton buffering. Using ADP and H+ to synthesize ATP, CK prevents local and global acidification of the cell. Acidification hampers cell function and seriously diminishes the force of muscle contraction. Taking the metabolic enzymes and processes controlled by ADP, ATP, and pH into account, CK has a central role in intracellular homeostasis, and significantly higher activities of the enzyme could profoundly alter cellular metabolism and functions dependent on ADP, ATP, and pH (18-23).
CK and pressor responses
We proposed that relative high CK activity in cardiovascular muscle and other tissues with high energy demands increases cardiovascular contractile reserve, enhances trophic responses, and increases renal tubular ability to retain salt, facilitating the development of arterial hypertension (18).
Central to the effects of CK is the ability of the enzyme to lower ADP (18-23). Vascular smooth muscle contraction consists of a fast, force generating component at relatively high energy costs, and a slow tonic maintenance of tension (23-25), which is thought to depend on the ability to have attached but dephosphorylated cross bridges (latch) for which ADP is required (11,25-27). CK is thought to reduce ADP at the smooth muscle contractile proteins, leading to excessive shortening and contractile responses delaying or hindering latch formation (18). Even small increases in vascular contractility could profoundly increase blood pressure, as resistance to flow is strongly affected with small changes in ri. In addition, vascular CK activity is relatively low compared to striated muscle, with very rapid rates of myosin ATPase (7,23). Even small changes in vascular smooth muscle CK activity may then result in at least a local alteration in the levels of ATP, ADP, and H+ and profoundly enhance contractility (18,19) (Figure 1).
Figure 1.

Smooth muscle contraction with high creatine kinase activity. A model of the four-state contractile system of vascular smooth muscle adapted from Brewster (18), based on Murphy (25), and Hai and Murphy (26). Ca2+ initiates contraction with activation of MLCK. MLCK phosphorylates myosin light chains to activate myosin ATPase (MA) and the MyosinP-ADP interacts generates tension with actin through an Actin-MyosinP-ADP complex. Shortening occurs through cross bridge cycling. The MyosinP-ADP can be dephosphorylated to form the so-called “latch bridges”, which are slowly cycling cross bridges depending on ADP (27). Controlled exit from the latch bridge occurs by re-phosphorylation of myosin, while dephosphorylation of MyosinP and a decrease in Ca2+concentration inactivates the high-tension state. Smooth muscle creatine kinase (CK) activity is relatively low compared to striated muscle (23). With relatively high CK activity, lower ADP levels might hinder the ADP-dependent latch bridge formation and enhance myosin ATPase activity, leading to greater microvascular contractility (18). MLCK, myosin light chain kinase.
In addition, calcium-dependent, vasoconstrictive pathways as well as vasodilative RhoA/Rho kinase and nitric oxide (NO)-cGMP pathways, the main intracellular effectors of blood pressure regulating systems in vascular smooth muscle, converge on metabolic processes fueled by CK as a final common pathway to vascular contractility. Thus, CK is thought to have a strong modulatory effect on vascular contractility. Moreover, evidence indicates that high creatine demands with high CK might reduce the bioavailability of L-arginine and the rate of NO synthesis (19). Creatine and NO are both synthesized from L-arginine, but creatine synthesis demands more plasma L-arginine than NO synthesis, and the rate of NO synthesis is limited by the rate of endothelial L-arginine uptake (19).
Regarding cardiac function, higher tissue CK is understood to provide greater protection from myocardial ischemia, while promoting left ventricular hypertrophy (18). CK is closely involved in trophic responses of tissues. Growth promoting substances including growth hormone, sex steroids, vitamin D metabolites, and insulin-like growth factor-I increase the expression of cellular CK (28-30), and high tissue CK might enhance hyperplastic and hypertrophic reactions that sustain hypertension (18). Although the hypertrophic myocardium performs better under increased demands, naturally there is a limit to the metabolic effects of greater CK activity. With increasing workload, the ATP and H+ buffer function of CK will become increasingly inadequate and heart failure may occur (5,18).
The highest blood pressures are found in persons of West-African ancestry (AA) (31). As persons of West-AA were reported to have high plasma and skeletal muscle CK activity in anecdotal studies (18,32,33), questions arose as to whether blood pressure and CK activity are associated, whether vascular contractility and salt retention are enhanced with high CK, and whether CK inhibition reduces blood pressure.
Tissue and plasma CK
CK is expressed in a variety of cells and tissues with high and fluctuating energy demands, such as muscle, brain, and kidney (18). Nuclear genes on chromosomes 5, 15, 19, and 14 encode four CK subunits: mitochondrial sarcomeric (sMt), mitochondrial ubiquitous (uMt), cytoplasmatic muscle (M) and cytoplasmatic ubiquitous or brain (B) subunits respectively, with a preferential expression in skeletal muscle (sMt and M), cardiac muscle (sMt, M and B), and smooth muscle, brain, and other tissue (uMt and B) (18). In physiologic and pathological states, such as at rest, after exercise, or after tissue damage, CK is released from tissue and transported through lymphatic vessels to the circulation. Plasma CK activity is the resultant of tissue CK concentration, the release of CK from tissues, lymphatic flow, and CK clearance by the liver (19).
In a random population sample, we found that mean resting plasma CK activity is higher in men, and around 70% higher in healthy persons of AA as compared to persons of European ancestry (EA), (median value 149 vs. 88 IU/L) with persons of South Asian ancestry having intermediate values (34). The distribution of isoenzymes was similar (19,35,36). When subjected to electrophoresis, the plasma of healthy individuals contains predominately CK-MM isoenzyme, with less than 6% CK-MB and CK-BB (35,37). With frank tissue damage, large quantities of the enzyme enter the circulation through the lymphatic system, with the type of isoenzyme as an indication of the tissue of origin. CK has been used for the diagnosis of myocardial infarction and skeletal muscle disease (37,38). Although largely displaced by troponin (39,40), CK is still in use as a diagnostic enzyme to diagnose myopathy including during statin therapy for hypercholesterolemia, and more recently in CK-MB to estimate myocardial infarct size and clinical outcomes (41,42).
We hypothesized that the relatively high resting plasma CK activity levels found across West-AA populations are a physiological phenomenon mainly due to generalized higher tissue CK activity (18,34,43). Healthy persons of West-AA were found to have around twice the CK activity of persons of EA in skeletal muscle, with a predominance of fast, type II muscle fibers (33). We further assessed CK activity spectrophotometrically in multiple tissues with high and fluctuating energy demands including cerebrum, cerebellum, heart, renal artery, and skeletal muscle, obtained post-mortem in 17 men of EA and 10 of West-AA, mean age 62 years old (SE 4). Mean tissue CK activity was 76% higher with AA in all tissues studied [estimated marginal means 188.6 mU/mg (range, 148.8 to 228.4 mU/mg) protein, vs. 107.2 (95% CI, 76.7 to 137.7) with EA, P<0.01]. Our data implied a large variation in CK-dependent ATP buffer capacity across population’s ancestry subgroups, which may alter cellular function under high energy demands (44). The molecular basis of the coordinated expression of different isoenzymes at specific subcellular sites in specific tissues, and of these large differences in CK activity found between human subgroups are not known (18,19,33,43,44).
CK: a risk factor for cardiovascular disease
After it became clear that subgroups of the population had relatively high plasma and tissue CK, we conducted studies to refute the hypothesis that CK increases vascular contractility, renal sodium retention, blood pressure, and reduces ADP-dependent platelet aggregation. These studied are summarized and synthesized below.
Blood pressure
In a small cohort of 46 predominantly white men, aged 18 to 67 years, diagnosed with high CK activity without neuromuscular disease (i.e., idiopathic hyperCKemia) followed for up to 20 years, 48% of the subjects were hypertensive, as compared to 19% of random population controls (n=22,612, aged 20 to 65 years old), an odds ratio of 2.0 (range, 1.1 to 3.8) after adjustment for sex, age, and body mass index Importantly, we found that high CK had preceded the development of hypertension (45,46).
We further analyzed a random sample of the population of Amsterdam, of 1,444 citizens (503 white European, 292 South Asian, 580 West-African, and 69 of other ancestry), aged 34 to 60 years. Resting plasma CK activity was relatively low in women of EA, and was associated with an increase in systolic and diastolic pressure of respectively 8 mmHg (range, 3 to 13 mmHg) and 5 mmHg (range, 2 to 8 mmHg) per log CK increase after adjustment for age, sex, body mass index, and ethnicity (19) (Figure 2). Subclinical cardiovascular damage is unlikely to contribute to this association, as healthy hypertensive people with high resting plasma CK have normal isoenzyme distribution patterns (35,36,45).
Figure 2.
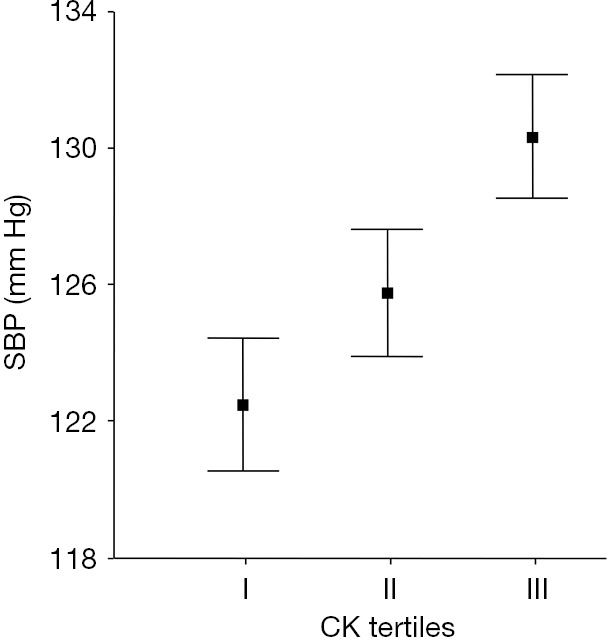
SBP by increasing plasma creatine kinase activity. Crude SBP according to resting plasma creatine kinase (CK) tertiles, adapted from Brewster et al. (19). I–III are the first through the third tertile of log plasma CK (IU/L), of a random population sample stratified by ethnicity, with respectively 447, 444 and 452 subjects in each tertile. Values are mean ± 2 (SEM). Similar values were found for DBP, and both SBP and DBP differed significantly among the CK tertiles (P<0.001; Kruskal-Wallis test). SBP, systolic blood pressure.
The association between CK and blood pressure was replicated in a population sample of the arctic city Tromsø (n=12,776, aged 30 to 87 years). The authors found a 3-mmHg increase in systolic blood pressure (SBP) per log plasma CK increase after adjustments including age, sex, BMI, and glucose (47). CK was obtained in non-resting conditions, potentially diluting this association (19,48). However, this carefully executed, large and independent replication study shows that even in subgroups with relatively low CK and in non-resting conditions, CK predicts blood pressure on a population level. Later studies in a large asymptomatic Asian cohort confirmed this finding (49).
Vascular contractility
We assessed contractility characteristics in isolated resistance arteries ex vivo from normotensive pregnant women of self-defined AA (n=11) and EA (n=20), mean age 34 years, who donated an omental fat pad during caesarean section from which resistance-sized arteries were isolated as previously described (50). We studied two main pathways of vasodilation after maximum contractility, inhibition of CK-dependent contractility and stimulation of the NO/cGMP pathway. First, the NO donor sodium nitroprusside (SNP) (10−9 to 10−4 mol/L) and bradykinin (10−10 to 10−7 mol/L)-induced vasodilation were studied, and lastly, the specific CK blocker dinitrofluorobenzene (DNFB) (10−7 to 10−6 mol/L) was added. All concentrations refer to final bath concentrations.
Morphology assessed with electron microscopy was similar in AA and EA women, but significantly greater maximum contractions to norepinephrine were seen in vessels from in AA women [14.0 mN (1.8 SE) compared with EA (8.9 mN (1.4 SE), P=0.02]. Furthermore, we found greater residual contractility after DNFB in AA, 55% (6 SE) vs. 28% (4 SE) in EA (P=0.001), and attenuated vasodilation after bradykinin in AA women, 103% (6 SE) vs. with white 84% (5 SE) in EA (P=0.023). Responses to SNP and amlodipine were similar. We concluded that compared with EA women, normotensive pregnant AA women displayed greater resistance artery contractility and evidence of higher vascular CK activity with attenuated nitric oxide synthesis (50).
However, since pregnancy is a strong CK inducer and we lacked data on human hypertension and CK, we further explored CK-dependency of vascular contractility in non-pregnant normotensive and hypertensive women. We included 19 consecutive non-pregnant women, mean age 42 years (SE 1.3), mean systolic/diastolic blood pressure respectively 142.6 (SE 5.9)/85.6 (3.4) mmHg (9 hypertensive), who donated an omental fat sample during abdominal surgery. We isolated resistance arteries and compared vasodilation after DNFB (10−6 mol/L) to SNP (10−6 mol/L) ex vivo. Additionally, we assessed predictors of vasoconstrictive force. DNFB reduced vascular contractility to 24.3% (SE 4.4), P<0.001, compared to baseline. SNP reduced contractility to 89.8% (SE 2.3). Maximum contractile force correlated with DNFB effect as a measure of CK (r=0.8), and with vessel diameter (r=0.7). The increase in contractile force was 16.5 (9.1 to 23.9) mN per unit DNFB effect in univariable, and 10.35 (2.10 to 18.60) mN in multivariable regression analysis. This study extended on our previous findings in pregnant normotensive women of CK-dependent microvascular contractility, indicating that CK contributes significantly to resistance artery contractility across human normotension and hypertension (51).
Finally, we assessed resistance artery CK isoenzyme mRNA using quantitative real-time polymerase chain reaction, and studied the association with blood pressure (52). Thirteen women were included, 6 normotensive and 7 hypertensive, mean age 42.9 years (SE, 1.6) and mean systolic/diastolic blood pressure, 144.8 (8.0)/86.5 (4.3) mmHg. Normalized CK B mRNA copy numbers, ranging from 5.2 to 24.4 (SE, 1.9), showed a near-perfect correlation with diastolic blood pressure (correlation coefficient, 0.9; 95% CI, 0.6–1.0) with similar results for SBP. Hypertensives displayed a 90% relative increase in resistance artery CK B mRNA compared with normotensives [normalized copy numbers respectively, 19.3 (SE, 2.0) vs. 10.1 (SE, 2.1), P<0.01]. Thus, CK was found to be a main predictor of blood pressure, and human resistance artery contractility was highly CK dependent, strengthening the evidence that this enzyme is involved in human hypertension. We theorized that hyperexpression of resistance artery CK serves to meet the increased metabolic demands of enhanced peripheral resistance in hypertension.
Sodium retention
As CK rapidly regenerates ATP for Na+/K+-ATPase driven sodium retention throughout the kidney (15,17,53), we assessed whether resting plasma CK is associated with sodium retention. Sixty healthy men (29 EA and 31 AA) with a mean age of 37.2 years (SE 1.2) were assigned to low sodium intake (<50 mmol/d) during 7 days, followed by 3 days of high sodium intake (>200 mmol/d). Sodium excretion (mmol/24-h) after high sodium was 260.4 (28.3) in the high CK tertile vs. 415.2 (26.3) mmol/24-h in the low CK tertile (P<0.001), with a decrease in urinary sodium excretion of 98.4 mmol/24-h for each increase in log CK, adjusted for age and AA (53). The findings did not reject the hypothesis of an association between CK and sodium retention, but more direct assessments of kidney CK are needed to establish whether variation in its activity is relevant for sodium sensitivity.
Failure of antihypertensive drug therapy
Since evidence indicates that CK enhances vascular contractility, salt retention, and pressor responses, we assessed whether CK was associated with therapy-resistance (54). We analyzed the Amsterdam cross-sectional, random multiethnic sample of the general population (n=1,444), aged 34–60 years. The primary outcome was the independent association between resting plasma CK and treated uncontrolled hypertension in the population, using multinomial logistic regression analysis. In line with our finding of an association with blood pressure, we found that hypertension prevalence was respectively, 26.8%; 30.8%; and 41.2% for the lowest (<88 IU/L) through the highest population CK tertile (>145 IU/L; P<0.001). Importantly, further analyses showed that CK was high in uncontrolled hypertension (Figure 3), and antihypertensive drug treatment failed in 72.9% of participants with high resting CK activity, vs. 46.7% with low CK (P=0.004). In logistic regression analysis, CK was the main predictor of treatment failure (adjusted OR 3.7; 95% CI, 1.2–10.9), independent of age, sex, BMI, fasting glucose, ethnicity, or education level. Thus, our hypothesis that high CK is associated with therapy resistance, was not refuted (54).
Figure 3.
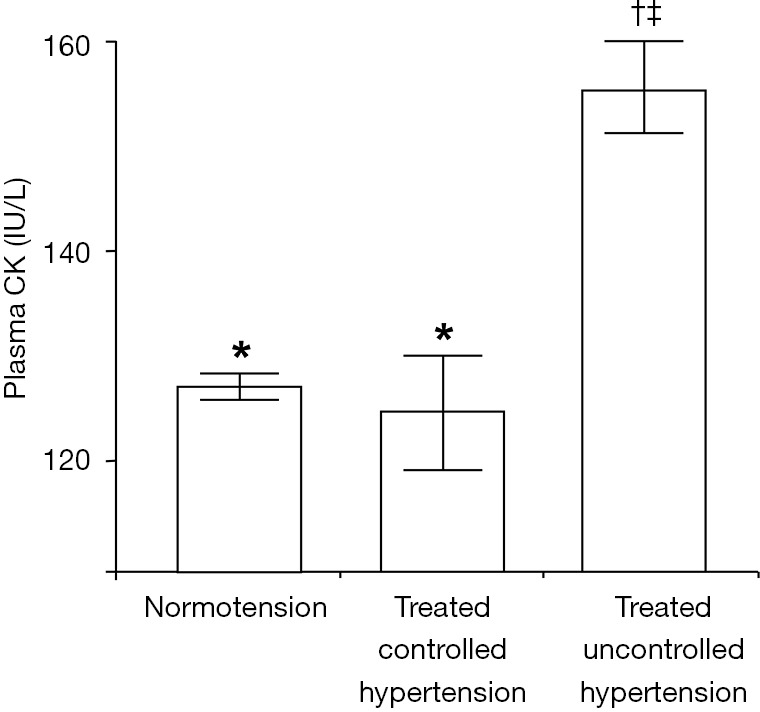
Plasma creatine kinase activity in normotension and treated hypertension. Values depict significant differences in mean plasma creatine kinase (CK) activity (SE) after rest in a random population sample as assessed with ANOVA (P<0.001) (54). *, in the post-test, CK levels between “normotension” and “controlled hypertension” were not significantly different. †‡, CK in “treated”, but “uncontrolled hypertension” was significantly higher than “normotension”, and “treated controlled hypertension” (P<0.001).
More specifically, we addressed the question how high CK might affect the response to different antihypertensive drugs (55-57). With AA as a proxy for high CK, we synthesized the evidence on the differential clinical efficacy of antihypertensive drugs. We retrieved 3,763 papers, and included 72 reports that mainly considered the 4 major classes of antihypertensive drugs, calcium blockers, diuretics, drugs that interfere with the renin-angiotensin system and β-adrenergic blockers. Pharmacokinetics, plasma renin and genetic polymorphisms did not well predict the response of patients of AA to antihypertensive drugs. We provided evidence indicating that low nitric oxide availability and high CK activity may explain individual responses to antihypertensive drugs (57).
The enhanced response to calcium blockers and diuretics, with lower efficacy of ACE inhibitors and beta-adrenergic blockers seen in AA patients is in accord with the enhanced ATP buffer capacity proposed with high tissue CK (Figures 4,5). CK fuels Ca2+-ATPase at the sarcoendoplasmic reticulum and thereby calcium uptake, as well the ATPases directly leading to vasoconstriction and renal sodium retention (17-24,50-53). Calcium antagonists and diuretics directly antagonize this effect. On the other hand, the reduced bioavailability of NO with high creatine demand may hamper the effect of ACE inhibitors as these drugs promote NO synthesis in the endothelium (58). Renin levels did not predict the efficacy of ACE inhibitors (57). We showed that high CK is associated with low vascular NO bioavailability in vitro (50,51), and L-Arginine was found to be low in persons of AA (59), with supplementation restoring NO bioavailability in vivo (60). Finally, beta-adrenergic blockers inhibit β2-mediated vasodilation, which is thought to induce greater peripheral vasoconstriction in the context of high vascular CK activity (55-57,61). The data indicate that resting plasma CK is a strong predictor of failure of antihypertensive therapy, stronger than glucose values, and that the specific pattern of response to antihypertensive drugs of a high CK subgroup is congruent with the molecular effects of CK on pressor responses.
Figure 4.

Molecular modulators of microvascular contractility. This schematic representation of vascular smooth muscle contraction is based on Brewster et al. (57). Creatine kinase (CK) is tightly bound near Ca2+ ATPase and myosin ATPase, and evidence suggests the enzyme is also located near myosin light chain (LC) kinase. Here, CK serves to rapidly regenerate ATP from phosphocreatine (Creatine ~ P) (18-27,57). Nitric oxide (NO) and creatine compete for bioavailable L-Arginine (19,57). Importantly, NO, RhoA/Rho kinase, and calcium-dependent pathways involved in the regulation of vascular contractility, converge on metabolic processes modulated by CK (57). Antihypertensive drugs and vasodilators may enhance NO-dependent pathways, such as in ACE inhibitor (ACE-i) induced NO synthesis, or inhibit CK-dependent pathways. Calcium blockers (CaB) directly antagonize CK dependent processes by reduction of cellular Ca2+ uptake and release from the sarcoendoplasmic reticulum (SER), while β-adrenergic agonists inhibit myosin LC kinase. High CK is thought to amplify contractile responses, including of β-adrenergic blockers and the high creatine demand with high CK is thought to compromise nitric oxide synthesis, further enhancing contractility (57). cGMP, guanosine cyclic 3',5'-(monophosphate); MLCP, myosin light chain phosphatase.
Figure 5.

Creatine kinase and sodium retention. Renal sodium retention is driven by basolateral Na+/K+ ATPase (15). Depicted is the kidney distal convoluted tubule. Creatine kinase (CK) is tightly bound near basolateral Na+/K+ ATPase to regenerate ATP for sodium retention (17,53). Thiazide diuretics may antagonize this effect indirectly as these drugs inhibit luminal Na+/Cl−-cotransport (53,57).
Obesity
The focus of our work has been on the enhanced cardiovascular contractility and renal sodium retention with high CK, but skeletal muscle characteristics have been reported to be relevant for hypertensive disease and obesity (62). CK activity is tightly coupled to glycolytic skeletal muscle type II fibers, lower oxidative capacity, and capillary rarefaction, which may contribute to the greater peripheral resistance and higher blood pressure levels seen in population subgroups with high CK (18,43,44,62,63). Furthermore, these fibers are fit for burst activity and typically display an attenuated mitochondrial fatty acid oxidation and glucose uptake. This promotes the storage of lipid as fat tissue resulting in insulin resistance and obesity, and resting plasma CK activity is independently associated with different markers of obesity in a multi-ethnic population (63).
Bleeding risk
We had proposed that high plasma CK might reduce plasma ADP and ADP-dependent platelet aggregation resulting in greater bleeding risk (64,65). CK is a main scavenger of ADP in the circulation (66). The enzyme can scavenge ADP as a binding protein, or convert ADP to ATP via its catalytic activity (64). As outlined above, high plasma CK occurs mainly in persons of AA ancestry, after exercise, and with damage of CK-rich tissue, including cardiac muscle, brain, and skeletal muscle (34,37,43,47-49). Taking the central role of ADP in platelet activation into account, we assessed whether high plasma CK inhibits ADP-induced platelet aggregation.
In a single-center study, we added CK MM isoenzyme in a clinically relevant activity range (500 to 4,000 IU/L), to low-CK plasma obtained from EA women. With increasing CK, ADP-induced platelet aggregation became increasingly attenuated and was completely abolished at CK 4,000 IU/L (64).
Secondly, we studied platelet aggregation in relation to endogenous CK activity in 9 healthy men, 4 AA and 5 EA and found that ADP-induced platelet aggregation became increasingly inhibited at higher levels of CK (64). Finally, we assessed ADP-induced platelet aggregation with high endogenous CK after exercise. One day after a bout of intensive exercise in a volunteer of West-AA, plasma CK was 4,664 IU/L, without clinical signs of rhabdomyolysis. At this CK level, ADP-induced platelet aggregation was completely abrogated. After 7 days of rest, CK normalized to 258 IU/L, with concomitant normalization of the ADP-induced platelet aggregation (64). To our knowledge, this was the first report suggesting that plasma CK in clinical relevant ranges may reduce platelet aggregation. High plasma CK levels may thus lead to less sticky platelets, and greater bleeding risk (64).
We subsequently analyzed the evidence on bleeding in subgroups of patients known to have high plasma CK. AA patients with ST-segment elevation myocardial infarction who underwent fibrinolysis were reported to have an unexplained higher risk of severe bleeding (adjusted OR 1.36; 95% CI, 1.14 to 1.62), with higher mortality (HR 2.83; 95% CI, 2.08 to 3.86), with similar findings after carotid endarterectomy (64,67,68). Furthermore, AA patients are at greater risk of microbleeds and bloody stroke after adjustment for blood pressure (OR 3.31; 95% CI, 1.14 to 9.57) (69). Importantly, a high unexplained bleeding risk was found in the AA subgroup of the CHARISMA study after the use of platelet ADP receptor inhibitor clopidogrel (adjusted HR 3.78; 95% CI, 1.35 to 10.60), despite lower plasma concentrations of the active drug (70,71). A higher bleeding risk was also observed in AA patients using dabigatran as compared to warfarin (65,72). Both dabigatran and warfarin interfere with the generation of the potent platelet agonist thrombin (73,74), but the direct thrombin inhibitor dabigatran has a stronger effect on thrombin-induced platelet aggregation (73). The effect of direct thrombin inhibitors on platelets is potentiated by inhibition of ADP receptors, as concurrent signaling through the ADP receptors is reported to be necessary for full platelet activation by thrombin (64,65,74). Therefore, we suggested that the risk of severe bleeding in AA patients using dabigatran might be due to synergy of the direct thrombin inhibitory effect with CK-induced reduction of ADP-dependent responses, to strongly inhibit platelet activation (65).
Other clinical conditions with highly elevated plasma CK might be relevant to study ADP-dependent platelet aggregation and bleeding risk, including the unexplained increased hemorrhagic stroke risk with statin use, especially in men (75); bleeding after myocardial infarction, in particular with the use of (multiple) antiplatelet therapy during peak CK activities around 24 hours after the event (37,64,76); as well as the extensive high CK tissue damage with highly elevated plasma CK in “trauma-induced coagulopathy” and “traumatic brain injury platelet dysfunction”, where an unexplained near-complete inhibition of ADP-dependent platelet aggregation and near uncontrollable hemorrhage with high mortality rates is observed (64).
Thus, highly elevated plasma CK might be a hitherto unrecognized factor that might greatly increase bleeding risk because of the resulting lower plasma ADP and ADP-dependent platelet aggregation (64,76), and subgroups of patients with high CK may be particularly sensitive to the use of platelet ADP receptor blockers. Further studies are needed to establish whether plasma CK induces a distinct coagulopathy, in particular with concomitant use of statins or antithrombotic drugs in persons with high plasma CK activity.
Intervention studies
CK Inhibition in the spontaneously hypertensive rat (SHR)
Because of the cardiovascular effects associated with high CK, we assessed whether CK inhibition with the creatine analogue and competitive inhibitor beta-guanidinopropionic acid (GPA) would safely reduce blood pressure. We studied the SHR, an animal model with high tissue CK activity that precedes the development of hypertension (77-79).
Creatine is either absorbed in the intestine from dietary sources or synthesized de novo. Kidney L-arginine:glycine amidinotransferase (AGAT, EC 2.1.4.1) catalyzes the first, rate-limiting step of L-arginine and glycine into guanidinoacetic acid, demanding a major part of bioavailable L-arginine (80,81). Subsequently, liver S-adenosyl-L-methionine:N-guanidinoacetate methyltransferase (GAMT, EC 2.1.1.2), methylates guanidinoacetic acid to creatine, which is utilized by peripheral tissues after cellular uptake through a membrane bound creatine transporter.
AGAT also synthesizes GPA, from L-arginine and beta-alanine. The estimated GPA plasma concentration in healthy rats is 0.06 (SD 0.02) µmol/L, and clearance is probably renal (80,82). The physiological function is unknown, but when given to animals GPA reduces the flux through the cytoplasmic CK reaction, by competitive inhibition of creatine, in the gut, at AGAT, or at the cellular creatine transporter. Reduced intracellular creatine attenuates the non-enzymatic conversion to creatinine (80,81). GPA can also be phosphorylated by cytoplasmic, but not mitochondrial CK, to PGPA. This further reduces CK-dependent ATP buffer capacity, as GPA and PGPA are “inefficient substrates” for the CK reaction (80).
In previous studies, the reduced flux through the CK reaction with GPA induced greater resistance against brain ischemia. Furthermore, a shift from high CK, predominantly glycolytic type II skeletal muscle fibers, to low CK, predominantly oxidative type I fibers was reported, resulting in greater endurance capacity, enhanced glucose tolerance, and weight loss. Left ventricular systolic pressure, cardiac output, and rate of tension development were unchanged in the unstressed heart or during high workload in the intact animal, but mortality after acutely induced myocardial infarction was increased (80), an effect that was not replicated in myocardial infarction induced in creatine-depleted knock-out mice (83). Since blood pressure, vascular contractility, kidney function, or lipid spectrum were not studied, we randomly assigned 16 male 16-week-old SHR to GPA (3%) in chow vs. standard chow with blood pressure as the main outcome (79).
In line with previous studies on creatine depletion (80,81,83), animals appeared healthy and displayed normal physical activity after GPA in our study, without apparent adverse effects. GPA reduced blood pressure with 42.7 (5.5) systolic and 35.6 (5.0) mmHg diastolic (P<0.001) vs. controls. Heart rate did not significantly change. Mean NO-dependent vasodilation at 10−5 mmol/L methacholine in mesenteric artery was 24% higher with GPA (52.0, vs. 41.7% in controls). In line with a diuretic effect (84) we found higher renin mRNA in the kidney, normalized copy number 1.7 (0.2) with GPA vs. 1.1 (0.2) in controls (P=0.03). Furthermore, we found evidence of a shift to low CK type I skeletal muscle fibers. Since type I fibers have more oxidative metabolism, with a higher capillary density and NO generation (62,63), this shift might have contributed to blood pressure lowering and weight loss with GPA. Although body weight was lower in the GPA group throughout the trial, this was not significant at day 28 [GPA 338 g (5 g) vs. control 356 g (9 g), P=0.06]. In addition, mean heart weight and mean heart weight/body weight ratio were not significantly different between GPA and control groups as previously reported (79). Plasma creatinine concentration was lower with GPA as expected (11.6 mg/L with GPA vs. 24.1 mg/L in controls; P<0.001).
Finally, plasma HDL-cholesterol unexpectedly increased with 46% after GPA. The mode of action is unclear. Rats are naturally deficient in cholesteryl ester transfer protein, with relatively high HDL fractions (85). Theoretically, GPA might have increased HDL through augmentation of the concentration of Apo A-I, mimicking of the functionality of Apo A-I, enhanced reverse cholesterol transport, or inhibition of endothelial lipase (86). In line with the mitochondrial dependency of ABC transporter proteins (87), we speculate that the stimulatory effect of GPA on mitochondrial function promotes reverse cholesterol transport, but further study is needed.
To our knowledge, this was the first report on the blood pressure lowering effect of reversible competitive CK inhibition. This experimental evidence adds to the data indicating that the enzyme is involved in the pathophysiology of hypertension. In addition, the presented data also indicate that creatine is not indispensable, and suggests that CK is potentially a new systemic arterial hypertension treatment target.
First-in-human tolerance study with CK inhibition
After collecting evidence indicating that CK is intimately involved in hypertension and that the CK inhibitor GPA reduces blood pressure in a high-CK-hypertensive animal model, we assessed the tolerability of a sub-therapeutic GPA dose in healthy men (88).
In this active and placebo-controlled, triple-blind, single-center trial, we recruited 24 healthy men (18–50 years old, BMI 18.5–29.9 kg/m2) in the Netherlands. Participants were randomized (1:1:1) to one week daily oral GPA 100 mg, creatine 5 g, or matching placebo. Creatine, which has an identical molecular formula to GPA, was chosen to assess the effect of the synergist. The average daily rate of creatine synthesis in healthy omnivorous males is estimated to be 1.3 g. We used 5 g as recommended in studies on creatine supplementation (88). The GPA dose, 100 mg, was calculated according to the Food and Drug Administration (FDA) ‘Guidance on Estimating the Maximum Safe Starting Dose in Initial Clinical Trials in Adult Healthy Volunteers’ (89), to ensure the safety of the human volunteers. The primary outcome of the trial was the tolerability of GPA in humans, in an intent-to-treat analysis.
All 24 randomized participants received the allocated intervention, and 23 completed the study. One participant in the placebo arm dropped out for personal reasons. This participant experienced no side effects (including during a re-challenge with the assigned drug). GPA was well tolerated, without serious or severe adverse events. No changes were found in blood pressure, weight, ADP-dependent platelet aggregation, heart rate, cardiac contractility, cardiac output and total peripheral resistance monitored noninvasively. Furthermore, no abnormalities were reported in clinical safety parameters, including physical examination, laboratory studies, and a 12-Lead ECG. At day 8, mean plasma GPA was 213.88 (SE 0.07) in the GPA arm vs. 32.75 (SE 0.00) nmol/L with placebo arm (P<0.01).
This first-in-human study raised no safety or tolerability concerns, including no adverse effects reported and no significant differences detected compared to baseline or placebo in physical examination, biochemistry or cardiovascular function, including blood pressure, liver enzymes, cardiac contractility, and QT interval.
Conclusions
Blood pressure reduction may be challenging despite the availability of several classes of antihypertensive drugs, and a substantial proportion of treated hypertensive patients does not achieve blood pressure control despite adequate treatment (54-57,90,91).
Risk factors for poor blood pressure control include obesity, age, AA, the presence of diabetes or end organ damage; but non-adherence of the patient, the white-coat effect, therapeutic inertia of the physician, dietary factors, or the concomitant use of blood pressure increasing drugs may also contribute. However, a subgroup of patients with uncomplicated, primary hypertension remains uncontrolled despite adequate use of antihypertensive drugs, and antihypertensive acting via new mechanisms might aid in achieving better control in these patients (90,91).
We presented a new theoretical model on the creation of high blood pressure and therapy resistance, based on the energy needed to create high blood pressure. We conducted laboratory, clinical, and population studies on CK and hypertension, and developed a novel modus of blood pressure lowering in CK inhibition. We hypothesized that hypertension is a highly energy demanding condition, and that therefore, individuals with high activity of the energy buffering enzyme CK might create higher blood pressures. We detailed on the main energy buffer and ATP-regenerating function of CK, with the enzyme tightly bound near motor proteins involved in force generation at acto-myosin ATPase, and near ion channels at Na+/K+-ATPase and Ca2+-ATPase, where it rapidly buffers ADP to regenerate ATP from phosphocreatine, enhancing the function of these ATPases (18).
Thus, the aggregated evidence indicates that tissue CK increases hypertension risk and that circulating plasma CK increases bleeding risk, both through enhanced ADP utilization. CK is a main independent determinant of blood pressure, resistance artery contractility, sodium excretion, obesity, and of failure of antihypertensive therapy (19,34,43-47,50-57,62-65). Our study in a high CK hypertension animal model indicated that CK inhibition with oral GPA lowers blood pressure (79), and our first in human tolerance study with GPA was uneventful (88). Future studies should focus on safe reduction of tissue CK and associated cardiovascular disease risk, and address bleeding risk and the safe use of statins and antithrombotic therapy in patients with high plasma CK.
Acknowledgements
This paper was funded by the Creatine Kinase Foundation.
Footnotes
Conflicts of Interest: Lizzy M. Brewster is an inventor on NL patent WO/2012/138226 (filed).
References
- 1.Poulter NR, Prabhakaran D, Caulfield M. Hypertension. Lancet 2015;386:801-12. 10.1016/S0140-6736(14)61468-9 [DOI] [PubMed] [Google Scholar]
- 2.Guyton AC. Arterial Pressure and Hypertension. Philadelphia, PA: W.B. Saunders, 1980. [Google Scholar]
- 3.Lifton RP, Gharavi AG, Geller D. Molecular mechanisms of human hypertension. Cell 2001;104:545-56. 10.1016/S0092-8674(01)00241-0 [DOI] [PubMed] [Google Scholar]
- 4.Folkow B, Hansson L Sivertson R. Structural vascular factors in the pathogeneisis of hypertension. In: Birkenhager WH, Reid JL, eds. Handbook of hypertension. Volume 1 Clinical aspects of essential hypertension. Amsterdam: Elsevier, 1983. [Google Scholar]
- 5.Neubauer S. The failing heart. N Engl J Med 2007;356:1140-51. 10.1056/NEJMra063052 [DOI] [PubMed] [Google Scholar]
- 6.Johansen PL. The heamodynamics of essential hypertension. In, Birkenhager WH, Reid JL, eds. Handbook of hypertension. Volume 1 Clinical aspects of essential hypertension. Amsterdam: Elsevier, 1983. [Google Scholar]
- 7.Clark JF, Pyne-Geithman G. Vascular smooth muscle function: The physiology and pathology of vasoconstriction. Pathophysiology 2005;12:35-45. 10.1016/j.pathophys.2005.02.007 [DOI] [PubMed] [Google Scholar]
- 8.Burton A. Physiology and Biophysics of the Circulation. An Introductory Text. Chicago: Year Book Medical, 1965. [Google Scholar]
- 9.Noon JP, Walker BR, Webb DJ, et al. Impaired microvascular dilatation and capillary rarefaction in young adults with a predisposition to high blood pressure. J Clin Invest 1997;99:1873-79. 10.1172/JCI119354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakayama S, Clark JF. Smooth muscle and NMR review: an overview of smooth muscle metabolism. Mol Cell Biochem 2003;244:17-30. 10.1023/A:1022431101158 [DOI] [PubMed] [Google Scholar]
- 11.Murphy RA, Rembold CM, Hai CM. Contraction in smooth muscle: what is latch? Prog Clin Biol Res 1990;327:39-50. [PubMed] [Google Scholar]
- 12.Daly MM, Gambetta-Gurpide E. The respiration and cytochrome oxidase activity of rat aorta in experimental hypertension. J Exp Med 1959;109:187-95. 10.1084/jem.109.2.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishida Y, Takagi-Ohta K. Lactate production of mammalian intestinal and vascular smooth muscles under aerobic and hypoxic conditions. J Smooth Muscle Res 1996;32:61-7. 10.1540/jsmr.32.61 [DOI] [PubMed] [Google Scholar]
- 14.Clark JF, Radda GK, Boehm EA. The effects of anti-hypertensive therapy on the structural, mechanical and metabolic properties of the rat aorta. J Muscle Res Cell Motil 2000;21:255-67. 10.1023/A:1005646614308 [DOI] [PubMed] [Google Scholar]
- 15.Greger R. Physiology of renal sodium transport. Am J Med Sci 2000;319:51-62. 10.1097/00000441-200001000-00005 [DOI] [PubMed] [Google Scholar]
- 16.Deetjen P, Kramer K. Die Abhängigkeit des O2-Verbrauchs der Niere von der Na-Rückresorption. Pfluegers Arch Eur J Physiol 1961;273:636-42. 10.1007/BF00361632 [DOI] [PubMed] [Google Scholar]
- 17.Ikeda K. Localization of brain type creatine kinase in kidney epithelial cell subpopulations in rat. Experientia 1988;44:734-5. 10.1007/BF01959143 [DOI] [PubMed] [Google Scholar]
- 18.Brewster LM, Clark JF, van Montfrans GA. Is greater tissue activity of creatine kinase the genetic factor increasing hypertension risk in black people of sub-Saharan African descent? J Hypertens 2000;18:1537-44. 10.1097/00004872-200018110-00002 [DOI] [PubMed] [Google Scholar]
- 19.Brewster LM, Mairuhu G, Bindraban NR, et al. Creatine kinase activity is associated with blood pressure. Circulation 2006;114:2034-9. 10.1161/CIRCULATIONAHA.105.584490 [DOI] [PubMed] [Google Scholar]
- 20.Wallimann T, Wyss M, Brdiczka D, Nicolay K, et al. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the 'phosphocreatine circuit' for cellular energy homeostasis. Biochem J 1992;281:21-40. 10.1042/bj2810021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hardin CD, Raeymaekers L, Paul RJ. Comparison of endogenous and exogenous sources of ATP in fueling Ca2+ uptake in smooth muscle plasma membrane vesicles. J Gen Physiol 1992;99:21-40. 10.1085/jgp.99.1.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spitzer E, Grosse R, Kuprijanov V, et al. Demonstration of a digitalis-sensitive sarcolemmal Ca2+-pump functionally coupled with a membrane associated creatine phosphokinase. Acta Biol Med Ger 1981;40:1111-22. [PubMed] [Google Scholar]
- 23.Clark JF. The creatine kinase system in smooth muscle. Mol Cell Biochem 1994;133-134:221-32. 10.1007/BF01267956 [DOI] [PubMed] [Google Scholar]
- 24.Somlyo AP, Wu X, Walker LA, et al. Pharmacomechanical coupling: the role of calcium, G-proteins, kinases and phosphatases. Rev Physiol Biochem Pharmacol 1999;134:201-34. [DOI] [PubMed] [Google Scholar]
- 25.Murphy RA. What is special about smooth muscle? The significance of covalent crossbridge regulation. FASEB J 1994;8:311-8. 10.1096/fasebj.8.3.8143937 [DOI] [PubMed] [Google Scholar]
- 26.Hai CM, Murphy RA. Cross-bridge phosphorylation and regulation of latch state in smooth muscle. Am J Physiol 1988;254:C99-106. 10.1152/ajpcell.1988.254.1.C99 [DOI] [PubMed] [Google Scholar]
- 27.Khromov A, Somlyo AV, Somlyo AP. MgADP promotes a catch-like state developed through force-calcium hysteresis in tonic smooth muscle. Biophys J 1998;75:1926-34. 10.1016/S0006-3495(98)77633-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iyengar MR, Iyengar CW, Chen HY, et al. Expression of creatine kinase isoenzyme during oogenesis and embryogenesis in the mouse. Dev Biol 1983;96:263-8. 10.1016/0012-1606(83)90327-5 [DOI] [PubMed] [Google Scholar]
- 29.Wallimann T, Hemmer W. Creatine kinase in non-muscle tissues and cells. Mol Cell Biochem 1994;133-134:193-220. 10.1007/BF01267955 [DOI] [PubMed] [Google Scholar]
- 30.Somjen D, Knoll E, Kohen F, et al. Effects of phytoestrogens on DNA synthesis and creatine kinase activity in vascular cells. Am J Hypertens 2001;14:1256-62. 10.1016/S0895-7061(01)02214-2 [DOI] [PubMed] [Google Scholar]
- 31.Nwankwo T, Yoon SS, Burt V, et al. Hypertension among adults in the United States: National Health and Nutrition Examination Survey, 2011-2012. NCHS Data Brief 2013;133:1-8. [PubMed] [Google Scholar]
- 32.Meltzer HY. Factors affecting serum creatine phosphokinase levels in the general population: the role of race, activity and age. Clin Chim Acta 1971;33:165-72. 10.1016/0009-8981(71)90264-6 [DOI] [PubMed] [Google Scholar]
- 33.Ama PFM, Simoneau JA, Boulay MR, et al. Skeletal muscle characteristics in sedentary black and caucasian males. J Appl Physiol 1986;61:1758-61. 10.1152/jappl.1986.61.5.1758 [DOI] [PubMed] [Google Scholar]
- 34.Brewster LM, Mairuhu G, Sturk A, et al. Distribution of creatine kinase in the general population: implications for statin therapy. Am Heart J 2007;154:655-61. 10.1016/j.ahj.2007.06.008 [DOI] [PubMed] [Google Scholar]
- 35.Edelman B, Koch TR, Raab C. Evaluation of the new Beckman electrophoretic method for CK-MB: comparison with ion exchange chromatography and investigation of a reference outpatient population. Clin Biochem 1981;14:39-44. 10.1016/0009-9120(81)90180-6 [DOI] [PubMed] [Google Scholar]
- 36.Van Steirteghem AC, Robertson EA, Zweig MH. Distribution of serum concentrations of creatine kinase MM and BB isoenzymes measured by radioimmunoassay. Clin Chim Acta 1979;93:25-8. 10.1016/0009-8981(79)90240-7 [DOI] [PubMed] [Google Scholar]
- 37.Cabaniss CD. Creatine Kinase. In: Walker HK, Hall WD, Hurst JW, editors. Clinical Methods: The History, Physical, and Laboratory Examinations. 3rd edition. Boston: Butterworths, 1990. [PubMed] [Google Scholar]
- 38.Karmen A, Wroblewski F, La Due JS. Transaminase activity in human blood. J Clin Invest 1955;34:126-31. 10.1172/JCI103055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ibanez B, James S, Agewall S, et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J 2018;39:119-77. 10.1093/eurheartj/ehx393 [DOI] [PubMed] [Google Scholar]
- 40.American College of Emergency Physicians ; Society for Cardiovascular Angiography and Interventions, O’Gara PT, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013;61:e78-140. 10.1016/j.jacc.2012.11.019 [DOI] [PubMed] [Google Scholar]
- 41.Dohi T, Maehara A, Brener SJ, et al. Utility of peak creatine kinase-MB measurements in predicting myocardial infarct size, left ventricular dysfunction, and outcome after first anterior wall acute myocardial infarction (from the INFUSE-AMI trial). Am J Cardiol 2015;115:563-70. 10.1016/j.amjcard.2014.12.008 [DOI] [PubMed] [Google Scholar]
- 42.Hartman MHT, Eppinga RN, Vlaar PJJ, et al. The contemporary value of peak creatine kinase-MB after ST-segment elevation myocardial infarction above other clinical and angiographic characteristics in predicting infarct size, left ventricular ejection fraction, and mortality. Clin Cardiol 2017;40:322-8. 10.1002/clc.22663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brewster LM. On the association between creatine kinase and blood pressure. The Netherlands, Amsterdam: University of Amsterdam, 2007. [Google Scholar]
- 44.Brewster LM, Coronel CM, Sluiter W, et al. Ethnic differences in tissue creatine kinase activity: an observational study. PLoS One 2012;7:e32471. 10.1371/journal.pone.0032471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brewster LM, van Bree S, Reijneveld JC, et al. Hypertension risk in idiopathic hyperCKemia. J Neurol 2008;255:11-5. 10.1007/s00415-008-0651-y [DOI] [PubMed] [Google Scholar]
- 46.Brewster LM, de Visser M. Persistent hyperCKemia: fourteen patients studied in retrospect. Acta Neurol Scand 1988;77:60-3. 10.1111/j.1600-0404.1988.tb06975.x [DOI] [PubMed] [Google Scholar]
- 47.Johnsen SH, Lilleng H, Wilsgaard T, et al. Creatine kinase activity and blood pressure in a normal population: the Tromsø study. J Hypertens 2011;29:36-42. 10.1097/HJH.0b013e32834068e0 [DOI] [PubMed] [Google Scholar]
- 48.Nicholson GA, McLeod JG, Morgan G, et al. Variable distributions of serum creatine kinase reference values. Relationship to exercise activity. J Neurol Sci 1985;71:233-45. 10.1016/0022-510X(85)90062-0 [DOI] [PubMed] [Google Scholar]
- 49.Yen CH, Wang KT, Lee PY, et al. Gender-differences in the associations between circulating creatine kinase, blood pressure, body mass and non-alcoholic fatty liver disease in asymptomatic Asians. PLoS One 2017;12:e0179898. 10.1371/journal.pone.0179898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brewster LM, Taherzadeh Z, Volger S, C, et al. Ethnic differences in resistance artery contractility of normotensive pregnant women. Am J Physiol Heart Circ Physiol 2010;299:H431-6. 10.1152/ajpheart.00919.2009 [DOI] [PubMed] [Google Scholar]
- 51.Taherzadeh Z, Karamat FA, Ankum WM, et al. The effect of creatine kinase inhibition on contractile properties of human resistance arteries. Am J Hypertens 2016;29:170-7. 10.1093/ajh/hpv078 [DOI] [PubMed] [Google Scholar]
- 52.Karamat FA, Oudman I, Ris-Stalpers C, et al. Resistance artery creatine kinase mRNA and blood pressure in humans. Hypertension 2014;63:68-73. 10.1161/HYPERTENSIONAHA.113.01352 [DOI] [PubMed] [Google Scholar]
- 53.Brewster LM, Oudman I, Nannan Panday RV, et al. Creatine kinase and renal sodium excretion in African and European men on a high sodium diet. J Clin Hypertens (Greenwich) 2018;20:334-41. 10.1111/jch.13182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oudman I, Kewalbansing P, Valkengoed I, et al. Creatine kinase is associated with failure of hypertension treatment. J Hypertens 2013;31:1025-31. 10.1097/HJH.0b013e32835f5c29 [DOI] [PubMed] [Google Scholar]
- 55.Brewster LM, van Montfrans GA, Kleijnen J. Systematic review: antihypertensive drug therapy in black patients. Ann Intern Med 2004;141:614-27. 10.7326/0003-4819-141-8-200410190-00009 [DOI] [PubMed] [Google Scholar]
- 56.Brewster LM, van Montfrans GA, Oehlers GP, et al. Systematic review: antihypertensive drug therapy in patients of African and South Asian ethnicity. Intern Emerg Med 2016;11:355-74. 10.1007/s11739-016-1422-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brewster LM, Seedat YK. Why do hypertensive patients of African ancestry respond better to calcium blockers and diuretics than to ACE inhibitors and β-adrenergic blockers? A systematic review. BMC Med 2013;11:141-7. 10.1186/1741-7015-11-141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Desta B, Vanhoutte PM, Boulanger CM. Inhibition of the angiotensin converting enzyme by perindoprilat and release of nitric oxide. Am J Hypertens 1995;8:1S-6S. 10.1016/0895-7061(95)00026-L [DOI] [PubMed] [Google Scholar]
- 59.Glyn MC, Anderssohn M, Lüneburg N, et al. Ethnicity-specific differences in L-arginine status in South African men. J Hum Hypertens 2012;26:737-43. 10.1038/jhh.2011.103 [DOI] [PubMed] [Google Scholar]
- 60.Houghton JL, Philbin EF, Strogatz DS, et al. The presence of African American race predicts improvement in coronary endothelial function after supplementary L-arginine. J Am Coll Cardiol 2002;39:1314-22. 10.1016/S0735-1097(02)01781-3 [DOI] [PubMed] [Google Scholar]
- 61.Veterans Administration Cooperative Study Group on Antihypertensive Agents Comparison of propranolol and hydrochlorothiazide for the initial treatment of hypertension. I. Results of short-term titration with emphasis on racial differences in response. JAMA 1982;248:1996-2003. 10.1001/jama.1982.03330160044023 [DOI] [PubMed] [Google Scholar]
- 62.Brewster LM, van Montfrans GA. Creatine kinase and hypertension. J Clin Hypertens (Greenwich) 2008;10:506. 10.1111/j.1751-7176.2008.tb00001.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haan YC, Oudman I, Diemer FS, et al. Creatine kinase as a marker of obesity in a multi-ethnic population. Mol Cell Endocrinol 2017;442:24-31. 10.1016/j.mce.2016.11.022 [DOI] [PubMed] [Google Scholar]
- 64.Horjus DL, Nieuwland R, Boateng KB, et al. Creatine kinase inhibits ADP-induced platelet aggregation. Sci Rep 2014:9;4:6551. [DOI] [PMC free article] [PubMed]
- 65.Horjus DL, van Montfrans GA, Brewster LM. Bleeding in Patients of African Ancestry Using Dabigatran: A Potential Effect of Creatine Kinase. JAMA Intern Med 2015;175:1243. 10.1001/jamainternmed.2015.1274 [DOI] [PubMed] [Google Scholar]
- 66.Tschopp TB, Baumgartner HR. Enzymatic removal of ADP from plasma: unaltered platelet adhesion but reduced aggregation on subendothelium and collagen fibrils. Thromb Haemost 1976;35:334-41. 10.1055/s-0038-1647926 [DOI] [PubMed] [Google Scholar]
- 67.Mehta RH, Stebbins A, Lopes RD, et al. Race, Bleeding, and Outcomes in STEMI Patients Treated with Fibrinolytic Therapy. Am J Med 2011;124:48-57. 10.1016/j.amjmed.2010.07.028 [DOI] [PubMed] [Google Scholar]
- 68.Brown HA, Sullivan MC, Gusberg RG, et al. Race as a predictor of morbidity, mortality, and neurologic events after carotid endarterectomy. J Vasc Surg 2013;57:1325-30. 10.1016/j.jvs.2012.10.131 [DOI] [PubMed] [Google Scholar]
- 69.Copenhaver BR, Hsia AW, Merino JG, et al. Racial differences in microbleed prevalence in primary intracerebral hemorrhage. Neurology 2008;71:1176-82. 10.1212/01.wnl.0000327524.16575.ca [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mak KH, Bhatt DL, Shao M, et al. Ethnic variation in adverse cardiovascular outcomes and bleeding complications in the Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) study. Am Heart J 2009;157:658-65. 10.1016/j.ahj.2008.08.031 [DOI] [PubMed] [Google Scholar]
- 71.Desta Z, Zhao X, Shin JG., et al. Clinical significance of the cytochrome P450 2C19 genetic polymorphism. Clin Pharmacokinet 2002;41:913-58. 10.2165/00003088-200241120-00002 [DOI] [PubMed] [Google Scholar]
- 72.Hernandez I, Baik SH, Piñera A, et al. Risk of bleeding with dabigatran in atrial fibrillation JAMA Intern Med 2015;175:18-24. 10.1001/jamainternmed.2014.5398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Renda G, Malatesta G, Lanuti P, et al. Effects of the direct thrombin inhibitor dabigatran etexilate vs warfarin on platelet function in patients with atrial fibrillatio. Eur Heart J 2013;34:1430 10.1093/eurheartj/eht308.P1430 [DOI] [Google Scholar]
- 74.Nylander S, Mattsson C, Ramström S, et al. Synergistic action between inhibition of P2Y12/P2Y1 and P2Y12/ thrombin in ADP- and thrombin-induced human platelet activation. Br J Pharmacol 2004;142:1325-31. 10.1038/sj.bjp.0705885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goldstein LB, Amarenco P, Szarek M, et al. SPARCL Investigators Hemorrhagic stroke in the Stroke Prevention by Aggressive Reduction in Cholesterol Levels study. Neurology 2008;70:2364-70. 10.1212/01.wnl.0000296277.63350.77 [DOI] [PubMed] [Google Scholar]
- 76.Tantry US, Bonello L, Aradi D, et al. Consensus and update on the definition of on-treatment platelet reactivity to adenosine diphosphate associated with ischemia and bleeding. J Am Coll Cardiol 2013;62:2261-73. 10.1016/j.jacc.2013.07.101 [DOI] [PubMed] [Google Scholar]
- 77.Jin X, Xia L, Wang LS, et al. Differential protein expression in hypertrophic heart with and without hypertension in spontaneously hypertensive rats. Proteomics 2006;6:1948-56. 10.1002/pmic.200500337 [DOI] [PubMed] [Google Scholar]
- 78.Seccia TM, Atlante A, Vulpis V, et al. Mitochondrial energy metabolism in the left ventricular tissue of spontaneously hypertensive rats: abnormalities in both adeninenucleotide and phosphate translocators and enzyme adenylate-kinase and creatine-phosphokinase activities. Clin Exp Hypertens 1998;20:345-58. 10.3109/10641969809052126 [DOI] [PubMed] [Google Scholar]
- 79.Karamat FA, Oudman I, Haan YC, et al. Creatine kinase inhibition lowers systemic arterial blood pressure in spontaneously hypertensive rats: a randomized controlled trial. J Hypertens 2016;34:2418-26. 10.1097/HJH.0000000000001090 [DOI] [PubMed] [Google Scholar]
- 80.Oudman I, Clark JF, Brewster LM. The effect of the creatine analogue beta-guanidinopropionic acid on energy metabolism: a systematic review. PLoS One 2013;8:e52879. 10.1371/journal.pone.0052879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wyss M, Kaddurah-Daouk R. Creatine and creatinine metabolism. Physiol Rev 2000;80:1107-213. 10.1152/physrev.2000.80.3.1107 [DOI] [PubMed] [Google Scholar]
- 82.Marescau B, Deshmukh DR, Kockx M, et al. Guanidino compounds in serum, urine, liver, kidney, and brain of man and some ureotelic animals. Metabolism 1992;41:526-32. 10.1016/0026-0495(92)90213-T [DOI] [PubMed] [Google Scholar]
- 83.Lygate CA, Aksentijevic D, Dawson D, et al. Living without creatine: unchanged exercise capacity and response to chronic myocardial infarction in creatine-deficient mice. Circ Res 2013;112:945-55. 10.1161/CIRCRESAHA.112.300725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kitami Y, Hiwada K, Kokubu T. Kidney renin gene expression in spontaneously hypertensive rats. J Hypertens 1989;7:727-31. 10.1097/00004872-198909000-00006 [DOI] [PubMed] [Google Scholar]
- 85.Oschry Y, Eisenberg S. Rat plasma lipoproteins: re-evaluation of a lipoprotein system in an animal devoid of cholesteryl ester transfer activity. J Lipid Res 1982;23:1099-106. [PubMed] [Google Scholar]
- 86.Degoma EM, Rader DJ. Novel HDL-directed pharmacotherapeutic strategies. Nat Rev Cardiol 2011;8:266-77. 10.1038/nrcardio.2010.200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Graham A. Mitochondrial regulation of macrophage cholesterol homeostasis. Free Radic Biol Med 2015;89:982-92. 10.1016/j.freeradbiomed.2015.08.010 [DOI] [PubMed] [Google Scholar]
- 88.Karamat FA, Horjus DL, Haan YC, et al. The acute effect of beta-guanidinopropionic acid vs. creatine or placebo in healthy men (ABC-Trial): a randomized controlled first-in-human trial. Br J Clin Pharmacol 2017;83:2626-35. 10.1111/bcp.13390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.US Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research. Guidance for industry. Estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. Available online: https://www.fda.gov/downloads/drugs/guidances/ucm078932.pdf
- 90.Laurent S, Schlaich M, Esler M. New drugs, procedures, and devices for hypertension. Lancet 2012;380:591-600. 10.1016/S0140-6736(12)60825-3 [DOI] [PubMed] [Google Scholar]
- 91.Pimenta E, Calhoun DA. Drug Development for Hypertension: Do We Need Another Antihypertensive Agent for Resistant Hypertension? Curr Hypertens Rep 2016;18:25. 10.1007/s11906-016-0634-9 [DOI] [PubMed] [Google Scholar]