

Abstract
Dimethyl sulfoxide (DMSO) is used to treat many diseases/symptoms. The molecular basis of the pharmacological actions of DMSO has been unclear. We hypothesized that DMSO exerts some of these actions by enhancing TGF-β activity. Here we show that DMSO enhances TGF-β activity by ~3–4-fold in Mv1Lu and NMuMG cells expressing Smad-dependent luciferase reporters. In Mv1Lu cells, DMSO enhances TGF-β-stimulated expression of P-Smad2 and PAI-1. It increases cell-surface expression of TGF-β receptors (TβR-I and/or TβR-II) by ~3–4-fold without altering their cellular levels as determined by 125I-labeled TGF-β-cross-linking/Western blot analysis, suggesting the presence of large intracellular pools in these cells. Sucrose density gradient ultracentrifugation/Western blot analysis reveals that DMSO induces recruitment of TβR-II (but not TβR-I) from its intracellular pool to plasma-membrane microdomains. It induces more recruitment of TβR-II to non-lipid raft microdomains than to lipid rafts/caveolae. Mv1Lu cells transiently transfected with TβR-II-HA plasmid were treated with DMSO and analyzed by indirect immunofluoresence staining using anti-HA antibody. In these cells, TβR-II-HA is present as a vesicle-like network in the cytoplasm as well as in the plasma membrane. DMSO causes depletion of TβR-II-HA-containing vesicles from the cytoplasm and co-localization of TβR-II-HA and cveolin-1 at the plasma membrane. These results suggest that DMSO, a fusogenic substance, enhances TGF-β activity presumably by inducing fusion of cytoplasmic vesicles (containing TβR-II) and the plasma membrane, resulting in increased localization of TβR-II to non-lipid raft microdomains where canonical signaling occurs. Fusogenic activity of DMSO may play a pivotal role in its pharmacological actions involving membrane proteins with large cytoplasmic pools.
Keywords: TGF-β ENHANCER, TβR-II, CYTOPLASMIC VESICLES, NON-LIPID RAFT MICRODOMAINS, CANONICAL SIGNALING
Dimethyl sulfoxide (DMSO) is a colorless liquid substance. It dissolves both polar and nonpolar compounds and is miscible in a variety of organic solvents as well as water. It is widely used as a cryoprotectant and a solvent vehicle for agents used in biochemical, cell biological, and animal experiments [David, 1972; Santos et al., 2003; Capriotti and Capriotti, 2012]. A known salient pharmacological action of DMSO is its ability to scavenge free radicals [David, 1972]. In many countries, DMSO is prescribed for a variety of diseases (e.g., interstitial cystitis and arthritis) and symptoms (e.g., pain, inflammation, and elevated intercranial pressure) [David, 1972; Fowler, 1981; Santos et al., 2003; Capriotti and Capriotti, 2012]. In the United States, DMSO is approved by the FDA for treatment of interstitial cystitis [Swanson, 1985]. DMSO is one of the most studied but least understood pharmaceutical agents. The molecular basis of the pharmacological actions of DMSO remains largely unknown. DMSO has been shown to exhibit anti-inflammatory, anti-atherosclerotic, and anti-tumor activities in relevant animal models [Gorog and Kovacs, 1968; Debons et al., 1987; See and Xia, 1992]. These are similar to those activities (anti-inflammation, anti-atherosclerosis, and tumor suppressor) of TGF-β that are known to be mainly mediated by Smad-dependent canonical signaling [Grainger and Metcalfe, 1995; Kulkarni et al., 1995; Derynck et al., 2001].
We hypothesized that DMSO, which alone does not exhibit inherent TGF-β activity, is an enhancer of TGF-β activity mediated by canonical signaling [Heldin et al., 1997; Massagué, 1998; Huang and Huang, 2005; Akhurst, 2012]. DMSO is effective in treating certain diseases and symptoms in which canonical TGF-β signaling is involved. The TGF-β enhancer activity of DMSO may be responsible for its effects on these diseases and symptoms. To test this hypothesis, we determined the effects of DMSO on TGF-β-stimulated luciferase activity and transcriptional activation in normal mink lung epithelial (Mv1Lu) cells and normal mouse mammary epithelial (NMuMG) cells expressing Smad-dependent luciferase reporters. To determine the mechanisms of the DMSO effects, we examined the cell-surface expression and plasma-membrane microdomain localization of the type I TGF-β receptor (TβR-I) and type II TGF-β receptor (TβR-II) in Mv1Lu cells treated with DMSO. In this communication, we show that DMSO is a potent enhancer ofTGF-β activity. It enhances TGF-β activity by inducing recruitment of TβR-II, and TβR-I and TβR-II from cytoplasmic vesicles and lipid rafts/caveolae, respectively, to non-lipid raft microdomains where canonical signaling occurs. These results suggest that the activity of DMSO, a cell fusogenic substance [Geron and Meiri, 1985; Faisst and Paweletz, 1987], to induce fusion of intracellular (cytoplasmic) vesicles and plasma membranes is responsible for recruiting TβR-II, and TβR-I and TβR-II from cytoplasmic vesicles and lipid rafts/caveolae, respectively, to nonlipid raft microdomains.
MATERIALS AND METHODS
MATERIALS
Mv1Lu and NMuMG cells were obtained from ATCC (Manassas, VA). Na [125I] (17 Ci/mg) was obtained from ICN Biochemicals (Irvine, CA). DMEM, high molecular mass protein standards (myosin, 205 kDa; β-galactosidase, 116 kDa; phosphorylase, 97 kDa; bovine serum albumin, 66 kDa), chloramine-T, disuccinimidyl suberate (DSS) and other biochemical reagents were obtained from Sigma (St Louis, MO). TGF-β (TGF-β1) was purchased from Austral Biologicals (San Ramon, CA). Rabbit polyclonal antibodies to caveolin-1 (N-20), early endosome antigen 1 (EEA1), hemagglutinin (HA) epitope [Tsukazaki et al., 1998], P-Smad2, Smad2, P-Erk1/2, Erk1/2, α-actin, P-JNK, JNK, P-p38, p38, α-tubulin, TβR-I (ALK-5) and TβR-II, and SB-505124 [DaCosta Byfield et al., 2004] were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). TGF-β peptide antagonist [Huang et al., 1997; Huang and Huang, 2005; Singer et al., 2009] was synthesized by C S Bio Co. (Menlo Park, CA). The luciferase assay system was obtained from Promega (Madison, WI). The TβR-II-HA plasmid was purchased from Addgene (Cambridge, MA). The COL1A2-luc plasmid was constructed as described [Poncelet et al., 1999]. The SBE4-luciferase reporter plasmid was constructed as described [Jonk et al., 1998].
PAI-1-LUCIFERASE, COL1A2-LUCIFERASE, AND SBE4-LUCIFERASE ACTIVITY ASSAYS IN MLE, Mv1Lu, AND NMuMG CELLS, RESPECTIVELY
MLE cells-Clone 32 are Mv1Lu cells stably expressing the PAI-1-luciferase reporter plasmid (PAI-1-luc) driven by the PAI-1 promoter [Abe et al., 1994; Chen et al., 2009]. MLE cells were grown to near-confluence on 12-well dishes and then treated with 50 pM or several concentrations of TGF-β in the presence of DMSO at the concentrations as indicated ±30 μM SB-505124 [DaCosta Byfield et al., 2004] or TGF-β peptide antagonist [Huang et al., 1997; Huang and Huang, 2005; Singer et al., 2009] at 37°C for 5 h. Treated cells were then lysed in 100 μl of lysis buffer (Promega). The PAI-1-luciferase activity of cell lysates (~20 μg protein) was then assayed using the luciferase kit from Promega.
Mv1Lu or NMuMG cells were transiently transfected with collagen promoter luciferase plasmid (COL1A2-luc) or SBE4-luciferase (SBE4-luc) plasmid using electroporation [Poncelet et al., 1999]. The COL1A2-luc or SBE4-luc was constructed by linking the human COL1A2 promoter [Poncelet et al., 1999] or mouse JunB promoter [Jonk et al., 1998] with the firefly luciferase gene. In electroporation, Mv1Lu or NMuMG cell suspension was mixed with 14 μg/ml plasmid DNA, transferred into an electroporation cuvette (0.4 cm gap, Bio-Rad) and pulsed (950 μF, 250 V, Gene Pulser II, Bio-Rad). After electroporation, cells were grown on 12-well cluster plates for 24 h, pretreated with different concentrations of DMSO at 37°C for 1 h, incubated with 50 or 100 pM TGF-β at 37°C for 4 h and lysed in 100 μl of lyses buffer (Promega). COL1A2-luciferase or SBE4-luciferase activity of the cell lysates (~20 μg protein) was assayed as described above.
QUANTITATIVE ANALYSIS OF PAI-1 mRNA (RELATIVE TO β-ACTIN mRNA) BY REAL-TIME RT-PCR
PAI-1 mRNA was quantified using real-time RT-PCR as described previously [Chen et al., 2009]. Mv1Lu cells were treated with 50 pM TGF-β and several concentrations of DMSO in serum-free DMEM. After stimulation of the cells with TGF-β and DMSO at 37°C for 2 h, RNAs from treated and untreated cells were isolated using the Trizol B (Teltext, TX) according to the manufacturer’s instructions. cDNAs were made from the isolated RNAs using MuLV reverse transcriptase (Applied Biosystems) and 1 μg RNA. The reverse transcription reaction was performed under the following conditions: 42°C for 15 min, 99°C for 5 min, and 4°C for 5 min. The SYBR green master mix was used with 200 nM of each primer. The real-time RT-PCR was performed at 2 min at 94°C for one cycle followed by 1 min at 94°C, 0.45 min at 60°C, and 1 min at 72°C for 35 cycles using a Bio-Rad Chrom 4 Thermocycler. The values of each experimental condition were normalized to the level of β-actin in a parallel sample. The primer sequences used were as follows: PAI-1 forward: 5′-GCCCTACTTCTTCAGGCTGTTC-3′; PAI-1 reverse: 5′-GAACAGCCTGAAGAAGTAGGGC-3′; β-actin forward: 5′-AGCCATGTACGTAGCCATCCAGGCTC-3′; and β-actin reverse: 5-TGGGTACATGGTGGTACCACCAGACA-3′.
QUANTITATIVE WESTERN BLOT ANALYSIS
Mv1Lu cells grown to near confluence on 12-well culture dishes were treated with 50 pM TGF-β in the presence of DMSO at the concentration indicated in serum-free DMEM (0.5 ml/well) at 37°C for 1 h. Treated cells were lysed by SDS sample buffer. Cell lysates with equal amounts of protein (200 μg) were analyzed by 7.5% SDS-PAGE followed by Western blot analysis using anti-Smad2, anti-P-Smad2, anti-P-Erk1/2, and anti-Erk1/2, anti-TβR-I, anti-TβR-II, anti-β-actin, and anti-caveolin-1 antibodies [Chen et al., 2007]. The relative levels of Smad2/P-Smad2, Erk1/2/P-Erk1/2, TβR-I/β-actin, TβR-II/β-actin, TβR-I/caveolin-1, TβR-II/caveolin-1 were determined by densitometry.
CELL-SURFACE 125I-TGF-β-CROSS-LINKING
Cell-surface 125I-TGF-β)-cross-linking was performed at 0°C using the cross-linking agent DSS according the published procedures [Huang and Huang, 2005; Chen et al., 2006, 2007, 2008, 2009], 125I-TGF-β-cross-linked cell lysates were analyzed by 7.5% SDS-polyacrylamide gel electrophoresis followed by autoradiography or quantification using a Phosphoimager. Mv1Lu cells grown to near-confluence on 6-well culture dishes were treated with several concentrations of DMSO at 37°C. Alter 1.5 h, treated cells were washed with cold binding buffer and incubated with 100 pM 125I-TGF-β in the presence or absence of 100-fold excess of unlabeled TGF-β in binding buffer containing 0.2% bovine serum albumin (BSA) at 0°C for 2.5 h. One hundred-fold excess of unlabeled TGF-β completely blocked 125I-TGF-β-cross-linking of TGF-β receptors. 125I-TGF-β-cross linked cells were then washed with cold binding buffer in the absence of bovine serum albumin and then incubated with 30 μM DSS at 0°C for 15 min. 125I-TGF-β-cross-linked cell lysates were analyzed by 7.5% SDS polyacrylamide gel electrophoresis (SDS-PAGE) and autoradiography. 125I-TGF-β-cross-linked TβR-I (TβR-I*) and 125I-TGF-β-cross-linked TβR-II (TβR-II*) on the SDS-PAGE gel (dried) were quantified using a PhosphoImager.
SEPARATION OF LIPID RAFTS/CAVEOLAE AND NON-LIPID RAFT MICRODOMAINS OF PLASMA MEMBRANES BY SUCROSE DENSITY GRADIENT ULTRACENTRIFUGATION
Mv1Lu were grown on 100 mm dishes (5 × 106 cells per dish). Cells were then incubated with 1.5% (v/v) DMSO or without DMSO (control) at 37°C for 1.5 h [Chen et al., 2007]. After two washes with ice cold phosphate-buffered saline, cells were scraped into 0.85 ml of 500 mM sodium carbonate, pH 11.0. Homogenization was carried out with 10 strokes of a tightfitting Dounce homogenizer followed by three 20-second bursts of an ultrasonic disintegrator (Soniprep 150; Fisher Scientific) to disrupt cell membranes, as described previously [Chen et al., 2007]. The homogenates were adjusted to 45% sucrose by addition of 0.85 ml of 90% sucrose in 25 mM 2-(N-morpholino) ethanesulfonic acid, pH 6.5, 0.15 M NaCl (MBS), and placed at the bottom of an ultracentrifuge tube. A discontinuous sucrose gradient was generated by overlaying 1.7 ml of 35% sucrose and 1.7 ml of 5% sucrose in MBS on the top of the 45% sucrose solution and it was then centrifuged at 200,000g for 16–20 h in an SW55 TI rotor. Ten 0.5-ml fractions were collected from the top of the tube, and a portion of each fraction was analyzed by SDS-PAGE followed by Western blot analysis using antibodies to TβR-I and TβR-II. The relative amounts of TβR-I and TβR-II on the blot were quantified by densitometry. Fractions 4–6, and fractions 8 and 9 contained caveolin-1 and EEA-1, respectively [Zhang et al., 2005; Chen et al., 2007].
INDIRECT IMMUNOFLUORESCENCE STAINING OF TβR-II-HA AND CAVEOLIN-1 IN Mv1Lu CELLS TRANSIENTLY TRANSFECTED WITH TβR-II-HA PLASMID
Mv1Lu cells transiently transfected with TβR-II-HA plasmid [Tsukazaki et al., 1998] were grown to 50% confluence on coverslips overnight. Transfected cells were then pretreated with 1.5% (v/v) DMSO at 37°C for 1.5 h and stimulated with and without 100 pM TGF-β for 30 min. After TGF-β stimulation, cells were fixed in methanol at −20°C for 15 min, washed with PBS and treated with 0.2% gelatin in PBS for 1 h. Fixed cells were then incubated overnight with a mouse antibody against hemagglutinin (HA) protein (F-7; Santa Cruz Biotechnology) and rabbit antibody against caveolin-1 (N-20; Santa Cruz Biotechnology) at 1:100 dilution at 4°C in a humidified chamber. After extensive washing, cells were incubated with rhodamine-conjugated donkey anti-mouse antibody and FITC-conjugated goat anti-rabbit antibody at a 1:50 dilution for 1 h. Images were viewed using a Leica TCS SP confocal microscope (Leica Microsystems Ltd., Heidelberg, Germany). The measurements of co-localization rate were analyzed using a Leica Application Suite (LAS). The TβR-II-HA density in the cytoplasm of cells treated with and without DMSO was quantified using the NIH ImgeJ program (with views of 20–30 cells). DMSO (1.5%, v/v) treatment of cells depleted 70 ± 10% (n = 3) of TβR-II-HA-containing vesicles in the cytoplasm as compared to that (100%) in cells treated without DMSO.
STATISTICAL ANALYSIS
Statistical analysis was performed by comparing DMSO-treated cells versus untreated (control) cells using unpaired and two-tailed Student’s t-tests. The P values less than 0.05 were considered to be statistically significant.
RESULTS
DMSO ENHANCES TGF-β ACTIVITY MEDIATED BY CANONICAL SMAD-DEPENDENT SIGNALING IN Mv1Lu AND NMuMG CELLS
TGF-β receptors, including TβR-I and TβR-II, are expressed in all normal cell types studied except certain cancer cells [Heldin et al., 1997; Massagué, 1998; Roberts, 1998; Huang and Huang, 2005]. Canonical TGF-β signaling mediated by TβR-I and TβR-II hetero-oligomeric complexes is responsible for many important cell biological processes (e.g., cell growth, differentiation, apoptosis, epithelial-mesenchymal transition, and extracellular matrix production) induced by TGF-β in many cell types [Heldin et al., 1997; Massagué, 1998; Roberts, 1998]. This involves activation of TβR-I and TβR-II hetero-oligomeric complexes and downstream signal transducers Smad2/3 [Heldin et al., 1997; Massagué, 1998; Roberts, 1998; Huang and Huang, 2005]. Non-canonical TGF-β signaling includes a variety of intracellular signaling pathways activated by TGF-β independently of Smad2/3 activation [Heldin et al., 1997; Akhurst, 2012]. Mv1Lu cells have been used as a model cell system for studying canonical and non-canonical TGF-β signaling and cellular responses [Di Guglielmo et al., 2003; Huang and Huang, 2005; Chen et al., 2006, 2007, 2008, 2009]. Mv1Lu cells stably expressing a canonical TGF-β signaling-dependent PAI-1-luciferase reporter (PAI-l-luc) (termed MLE cells) [Abe et al., 1994; Chen et al., 2007, 2009] were utilized to determine the TGF-β enhancer activity of DMSO. MLE cells were treated with 50 pM TGF-β ± 1% (v/v) DMSO at 37°C for 0–5 h. The luciferase activity of the lysates from cells treated with and without DMSO was determined. As shown in Figure 1A, after treatment of MLE cells with DMSO at 37°C for 2 h, luciferase activity increased due to protein synthesis of the enzyme luciferase. It reached a maximum after a 4-h incubation. DMSO enhanced TGF-β-stimulated luciferase activity in a time-dependent manner similar to that of TGF-β treatment alone. DMSO also enhanced TGF-β-stimulated luciferase activity in a concentration-dependent manner (0, 0.5, 1, 1.5, 2, and 2.5%, v/v, at 37°C for 5 h) with a maximum of ~3–4-fold enhancement at 1.5% (v/v) DMSO (Fig. 1B). DMSO treatment also enhanced TGF-β-stimulated luciferase activity at all TGF-β concentrations tested (25, 50, 100, and 200 pM) (Fig. 1C). Furthermore, the DMSO-enhanced TGF-β-stimulated luciferase activity was completely inhibited by SB-505124, a specific inhibitor of TβR-I kinase activity [DaCosta Byfield et al., 2004] and a TGF-β receptor peptide antagonist [Huang et al., 1997; Huang and Huang, 2005; Singer et al., 2009] in these cells (Fig. 1D). The TGF-β receptor antagonist blocks binding of TGF-β to TβR-I-TβR-II hetero-oligomeric complexes in target cells [Huang et al., 1997; Huang and Huang, 2005; Singer et al., 2009].
Fig. 1.
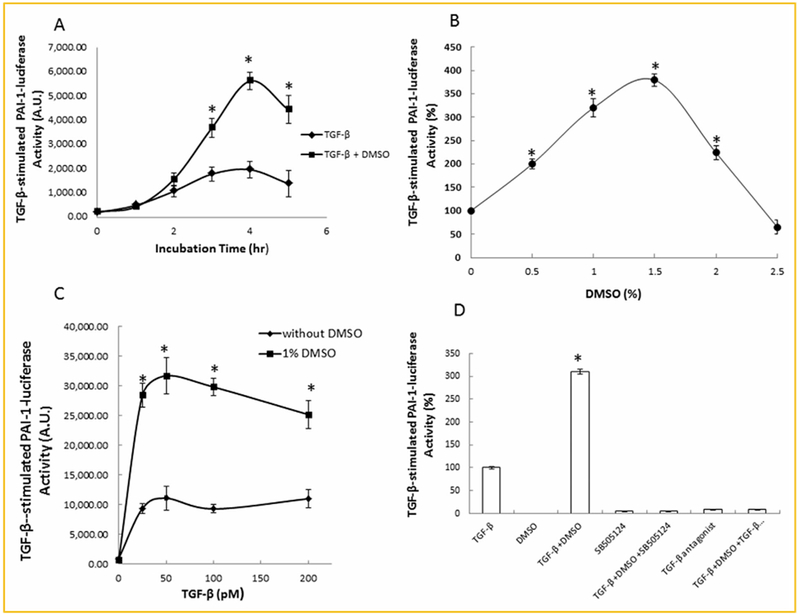
DMSO enhances TGF-β-stimulated PAI-1-luciferase activity in a time-dependent (A), concentration-dependent (B,C), and TGF-β receptor kinase inhibitor/antagonist-sensitive manner (D), in MLE cells. (A): Cells were treated with 50 pM TGF-β±1.0% (v/v) DMSO for several time periods (0–5 h). At each time period, the PAI-1-luciferase activity of lysates of treat cells was measured and expressed as arbitrary units (A.U.). (B,C): Cells were treated with 50 pM TGF-β in the presence of several concentrations of DMSO (0, 0.5, 1, 1.5, 2 and 2.5%, v/v) at 37°C for 5 h (B) or with 1% (v/v) DMSO in the presence of several concentrations of TGF-β (0, 25, 50, 100, and 200 pM) at 37°C for 5 h (C). The PAI-1-luciferase activity of lysates of treated cells was measured. It was taken as 100% in cells treated with TGF-β alone (B). The PAI-1-luciferase activity of lysates of treated cells was measured and expressed as arbitrary units (A.U.) (C). (D): MLE cells were treated with 50pM TGF-β and 1% (v/v) DMSO in the presence of 30 μM SB-431542 [Chen et al., 2009], a specific inhibitor of TβR-I kinase activity or a TGF-β receptor antagonist [Huang et al., 1997; Singer et al., 2009]. After 5 h at 37°C, the PAI-1-luciferase activity of lysates of treated ells was then measured. The PAI-1-luciferase activity in cells treated with TGF-β alone was taken as 100%. The experiments were performed in triplicate. The data are mean±s.d. *Significantly higher than that in cells treated with TGF-β alone (P < 0.05).
To further define the target gene specificity of DMSO on canonical TGF-β signaling, we determined the effect of DMSO on collagen-luciferase reporter expression in Mv1Lu cells transiently transfected with a collagen-luciferase reporter plasmid (COL1A2-luc). The collagen type I, α2 gene (COL1A2) is also one of the canonical TGF-β signaling targets [Poncelet et al., 1999]. The COL1A2-luc was constructed by linking the human COL1A2 promoter with the firefly luciferase gene [Poncelet et al., 1999]. As shown in Figure 2A, DMSO also enhanced TGF-β-stimulated COL1A2-luciferase activity in these cells in a concentration-dependent manner. Since P-Smad2 is an important signaling sensor of canonical TGF-β signaling [Heldin et al., 1997; Massagué, 1998; Roberts, 1998; Tsukazaki et al., 1998], we then determined the level of P-Smad2 protein by Western blot analysis in Mv1Lu cells treated with 50 pM TGF-β for 0.5 hr in the presence of several concentrations (0, 0.2, 0.5, 1, 1.5, and 2%, v/v) of DMSO. As shown in Figure 2B (top), DMSO enhanced TGF-β-stimulated phosphorylation of Smad2 with a maximum of ~3–4-fold at 1.5% (v/v). These results support the notion that DMSO specifically enhances canonical TGF-β signaling and cellular responses. This is further supported by the inability of DMSO to increase the level of phosphorylated-Erk1/2 (P-Erk1/2), which is an indicator of non-canonical TGF-β signaling, in Mv1Lu cells (Fig. 2B, bottom). PAI-1 is a transcriptional target of the canonical TGF-β signaling pathway [Abe et al., 1994; Chen et al., 2007, 2009]. To show the effect of DMSO on canonical TGF-β signaling-mediated PAI-1 transcription, we determined the level of PAI-1 mRNA by real-time RT-PCR in Mv1Lu cells treated with 50 pM of TGF-β and several concentrations (0. 0.5, 1.0, 1.5, 2, and 2.5%, v/v) of DMSO for 2 h. As shown in Figure 2C, DMSO enhanced TGF-β-stimulated PAI-1 mRNA expression in a concentration-dependent manner with a maximum of ~3–4-fold at 1.5% (v/v).
Fig. 2.
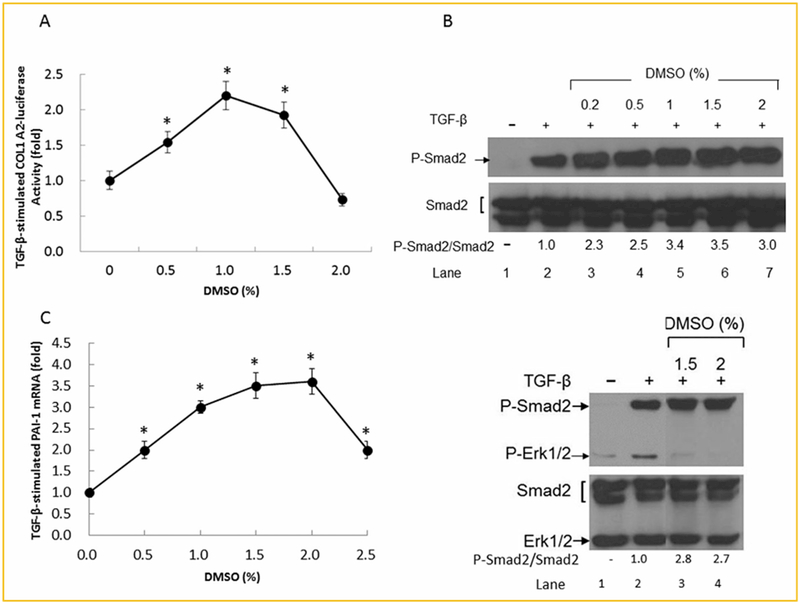
DMSO enhances TGF-β-stimulated expression of collagen-luciferase (A), P-Smad2 (B), and PAI-1 mRNA (C), as determined by COL1A2-luciferase activity assay, Western blot analysis and real-time RT-PCR, respectively, in Mv1Lu cells. Cells were treated with 50 pM TGF-β with and without 1% (v/v) DMSO at 37°C for 4, 0.5, and 2 h for assaying COL1A2-luciferase activity (A), and P-Smad2/Smad2 and P-Erk/Erk1/2 (B, top and bottom) levels and PAI-1 mRNA (C), respectively. The expression of COL1A2-luciferase was determined by assaying COL1A2-luciferase activity. The COL1A2-luciferase activity in cells treated with TGF-β alone was taken as onefold or 100% (A). The relative levels of P-Smad2 and Smad2, P-Erk1/2 and Erk1/2 were determined by quantitative Western blot analysis (B). Representative pictures of three Western blot analyses are shown. The ratio of P-Smad2/Smad2 in cells treated with TGF-β alone was taken as onefold (B). PAI-1 mRNA was determined in triplicate by real-time RT-PCR analysis. The relative level of PAI-1 mRNA normalized to β-actin mRNA was taken as onefold in cells treated with TGF-β alone (C). The data (A,C) are mean±s.d. *Significantly higher than that in cells treated with TGF-β alone (P < 0.05).
As described above, Mv1Lu cells are a model system for studying TGF-β-stimulated signaling and responses. We also performed experiments using other cell types (e.g., NMuMG cells) and different luciferase reporters (e.g., SBE4-luciferase) to examine the effects of DMSO on TGF-β-stimulated canonical signaling and responses. NMuMG cells are mouse mammary gland epithelial cells which have been used to study TGF-β-stimulated signaling (Piek et al., 2001). The expression of the SBE4-luciferase reporter is driven by four repeats of the CAGACA sequence identified as Smad binding element in the JunB promoter [Jonk et al., 1998]. TGF-β-stimulated activation of the SBE4-luciferase reporter is dependent on expression of Smad3, but not on expression of Smad2 (Piek et al., 2001). As shown in Figure 3A, DMSO stimulated luciferase activity in NMuMG cells transiently transfected with SBE4-luciferase reporter plasmid in a concentration-dependent manner. At 2.0% (v/v), DMSO stimulated SBE4-luciferase activity by threefold. In un-transfected NMuMG cells, DMSO, at 1.0% (v/v), stimulated phosphorylation of Smad2 by 1.5 ± 0.2-fold (n = 3) but inhibited phosphorylation of non-canonical signals such as Erk1/2 and p38 (Fig. 3B). This result was similar to that observed in Mv1Lu cells (Fig. 2B, bottom). It is important to note that TGF-β did not stimulate phosphorylation of JNK in these cells. The mechanisms by which DMSO inhibits TGF-β-stimulated phosphorylation of Erk1/2 and p38 are unknown. It is likely that DMSO exerts direct inhibitory effects on non-canonical signaling which utilizes lipid rafts/caveolae as a functional platform. DMSO (1%) was shown to inhibit cytokine-induced phosphorylation of p38 and Erk1/2 in chondrocytes [Kloesch et al., 2011].
Fig. 3.
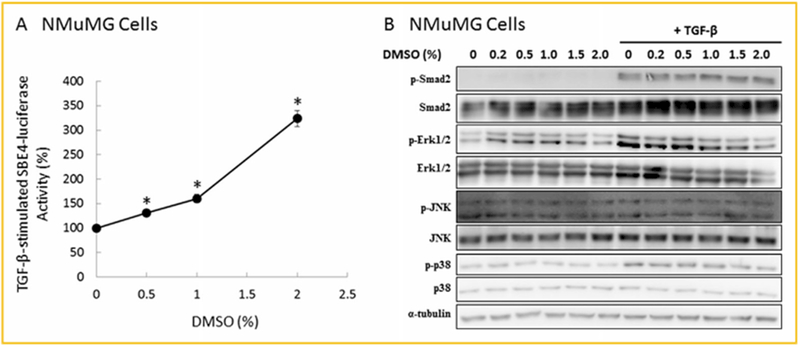
DMSO enhances TGF-β-stimulated expression of SBE-luciferase (A), and P-Smad2 (B), in NMuMG cells. NMuMG cells transiently transfected with SBE4-luciferase plasmid (A) or un-transfected NMuMG cells (B) were treated with 100pM TGF-β and several concentrations (0, 0.2, 0.5, 1.0, 1.5, and 2.0%, v/v) of DMSO for 4 and 0.5 h for assaying SBE4-luciferase activity (A), and P-Smad2/Smad2, P-Erk1/2/Erk1/2, P-JNK/JNK, P-p38/p38, and α-tubulin levels (B), respectively. SBE4-luciferase activity was determined by SBE4-luciferase activity assay. The SBE4-luciferase activity in cells treated with TGF-β alone was taken as 100% (A). The data (A) are mean±s.d. The relative levels of P-Smad2 and Smad2, P-Erk1/2 and Erk1/2, P-JNK/JNK, and P-p38/p38 were determined by quantitative Western blot analysis (B). Representative picture of three Western blot analyses is shown (B). *Significantly higher than that in cells treated with TGF-β alone (P < 0.05).
DMSO has a potent canonical TGF-β signaling-enhancing activity but has adverse effects on cells at higher concentrations. As shown in Figures 1B and 2A, DMSO at 2.5 and 2% (v/v), respectively, did not enhance but did inhibit TGF-β activity, as compared to the TGF-β activity of cells treated with TGF-β alone. This suggests that DMSO causes toxicity during the 5-h TGF-β-stimulated luciferase activity assays (Figs. 1B and2A). The optimal concentration of DMSO to enhance TGF-β activity is ~1.5% (v/v). Two % (v/v) DMSO or above are known to be toxic to cells [Pogorelaia et al., 1984; Deng et al., 2014]. However, DMSO at 2.5 and 2% (v/v) increased the TGF-β-stimulated levels of P-Smad2 (Fig. 2B) and PAI-1 mRNA (Fig. 2C) by three and twofold, respectively, as compared to those in cells treated with TGF-β only. The significant increase by 2 and 2.5% (v/v) DMSO in TGF-β-stimulated levels of P-Smad2 and PAI-1 mRNA is likely to be due to the brief time of incubation (0.5 and 2 h, respectively) in both Western blot analysis and real-time RT-PCR assay. The incubation time for the luciferase activity assay is 5 h. The longer time incubation may have caused toxic effects on cells [Pogorelaia et al., 1984; Deng et al., 2014]. This possibility is supported by the time course of luciferase activity assay in cells treated 1% (v/v) DMSO in the presence and absence of TGF-β (Fig. 1A). The 5-h incubation appeared to decrease the luciferase activity as compared to that seen in 4-h incubation (Fig. 1A).
DMSO has been used as a cryoprotectant when added to cell media. It reduces intracellular ice formation and thus prevents cell death during the freezing process [Brown and Nagle, 1965]. To see if other cryoprotectants exhibit the same TGF-β enhancer activity, we determined the effects of 1% (v/v) of DMSO, glycerol, ethylene glycol, propylene glycol, and formamide [Fahmy et al., 2014] on TGF-β-stimulated luciferase activity in MLE cells. Among these agents, only DMSO enhanced TGF-β-stimulated luciferase activity (data not shown). DMSO is the most commonly used cryoprotectant for storing cells [Brown and Nagle, 1965; Fahmy et al., 2014]. The TGF-β enhancer activity of DMSO toward different cell types [Brown and Nagle, 1965; Fahmy et al., 2014] would typically increase survival of these cells during the freezing and thawing processes. The solution commonly used for freezing cells consists of 30% fetal calf serum which contains TGF-β mainly derived from plasma platelets. TGF-β has been shown to exhibit anti-apoptosis activity and increase survival in several cell types [Ehata et al., 2007]. The TGF-β enhancer activity of DMSO may contribute to the efficacy (high cell viability) of DMSO as a cryoprotectant during freezing and thawing of cells.
DMSO INCREASES CELL-SURFACE EXPRESSION OF TβR-I AND TβR-II WITHOUT ALTERING THEIR CELLULAR LEVELS
As described above, DMSO treatment enhances TGF-β-stimulated luciferase activity at all TGF-β concentrations tested. This suggests that DMSO may enhance TGF-β activity by increasing cell-surface expression of TGF-β receptors without altering TGF-β binding affinity in these cells. To test this possibility, we determined the effect of DMSO treatment on cell-surface expression of TβR-I and TβR-II in Mv1Lu cells by cell-surface I125-labeled TGF-β (I125-TGF-β)-cross-linking at 0°C. I125-TGF-β-cross-linking has not only been used to determine cell-surface expression of TGF-β receptors but also has been used to determine the predominant location (>70%) of TβR-I and TβR-II hetero-oligomeric complexes in plasma-membrane microdomains [Huang and Huang, 2005]. In cells, TβR-I and TβR-II exist (in a ligand-independent manner) as hetero-oligomeric complexes at the cell surface. After I125-TGF-β-cross-linking in cells, when the ratio of I125-TGF-β-cross-linked TβR-II to I125-TGF-β-cross-linked TβR-I (TβR-II*/TβR-I*) is larger than one, this indicates that the majority of TβR-I and TβR-II hetero-oligomeric complexes are localized to non-lipid raft microdomains [Huang and Huang, 2005]. This is where they undergo clathrin-mediated endocytosis and transduce signaling following TGF-β stimulation [Di Guglielmo et al., 2003; Huang and Huang, 2005]. When the ratio of TβR-II*/TβR-I* is smaller than one, this indicates that the majority of TβR-I and TβR-II hetero-oligomeric complexes are localized to lipid rafts/caveolae where they undergo endocytosis and rapid degradation without transducing signaling following TGF-β stimulation [Di Guglielmo et al., 2003; Huang and Huang, 2005).
As shown in Figure 4A, DMSO increased cell-surface I125-TGF-β-cross-linking of TβR-I and TβR-II in a concentration-dependent manner with a maximum (~3–4-fold) at 1.5% (v/v). However, DMSO treatment increased cell-surface expression of TβR-I and TβR-II without altering the ratio (0.1) of TβR-II*/TβR-I*. This suggests that DMSO treatment does not alter the status of most TβR-I and TβR-II receptors in lipid rafts/caveolae but does increase localization of TβR-I and TβR-II hetero-oligomeric complexes to non-lipid raft microdomains as compared to controls. Furthermore, DMSO treatment (0.5, 1.0, 2.0, and 4.0%, v/v) did not alter the cellular levels of TβR-I and TβR-II in these cells, as determined with Western blot analysis (Fig. 4B). This suggests that Mv1Lu cells have large intracellular pools of TβR-I and/or TβR-II. Large intracellular pools of TβR-I and/or TβR-II have been found in many cell types studied, including fibroblasts [Massagué and Kelly, 1986; Doré et al., 2001; Asano et al., 2011]. These results suggest that DMSO enhances TGF-β activity by increasing cell-surface expression of TβR-I and/or TβR-II from their intracellular pools, which are not accessible to cell-surface I125-TGF-β-cross-linking (at 0°C).
Fig. 4.
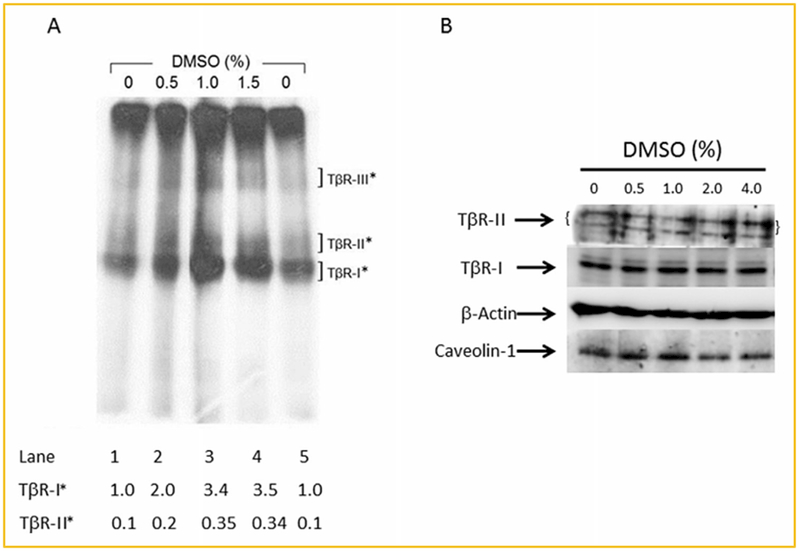
DMSO increases cell-surface expression of TβR-I and TβR-II without altering their cellular levels as determined by I125-TGF-β-cross-linking (A), and Western blot analysis (B), in Mv1Lu cells. (A): Cells were treated with several concentrations (0, 0.5, 1.0, and 1.5%) of DMSO at 37°C for 1.5 h. The cell-surface expression of TβR-I and TβR-II was determined by I125-TGF-β-cross-linking at 0°C followed by 7.5% SDS-PAGE and autoradiography as described [Chen et al., 2008]. I125-TGF-β-cross-linked TβR-I (TβR-I*) and I125-TGF-β-cross-linked TβR-II (TβR-II*) on the SDS-PAGE (dried gel) were quantified using PhosphoImager. I125-TGF-β-cross-linked type III TGF-β receptor (TβR-III*) was a 250-kDa protein on the autoradiogram. Free I125-TGF-β which migrated near the dye front, was cut off before autoradiography. Representative of three experiments is shown. The relative level of TβR-I* in cells treated without DMSO was taken as 1 (lanes 1 and 5). TβR-II* in control cells (lanes 1 and 5), and TβR-I* and TβR-II* in cells treated with 0.5, 1.0, and 1.5% DMSO (lanes 2–4, respectively) were expressed as the amounts relative to that (1.0) of TβR-I* in cells treated without DMSO (lanes 1 and 5). DMSO at 1.5% (v/v) increased cell-surface expression of TβR-I and TβR-II by ~3.5-fold. (B): Cells were treated with several concentrations (0. 0.5, 1.0, 2.0, and 4.0%, v/v) of DMSO at 37°C for 1.5 h. Western blot analysis of the cell lysates was performed using antibodies to TβR-I and TβR-II [Chen et al., 2007]. The relative amounts of TβR-I and TβR-II (with β-actin as an internal controls) were quantified by densitometry and were found to have no differences in cells treated with DMSO and without DMSO (0%).
DMSO INDUCES RECRUITMENT OF TβR-II, AND TβR-I AND TβR-II FROM INTRACELLULAR VESICLES AND LIPID RAFTS/CAVEOLAE, RESPECTIVELY, TO NON-LIPID RAFT MICRODOMAINS
The magnitude of TGF-β-induced canonical signaling and cellular responses in cells is known to be determined by the plasma-membrane microdomain (lipid rafts/caveolae or non-lipid raft microdomains) localization of TβR-I and TβR-II hetero-oligomeric complexes [Huang and Huang, 2005; Chen et al., 2006, 2007, 2008, 2009]. More localization of TβR-I and TβR-II hetero-oligomeric complexes in non-lipid raft microdomains occurs and more canonical signaling is induced [Huang and Huang, 2005; Chen et al., 2006, 2007, 2008]. We hypothesized that DMSO mainly enhances TGF-β activity by increasing localization of TβR-I and TβR-II to non-lipid microdomains. To test this hypothesis, we analyzed the microdomain localization of TβR-I and TβR-II in Mv1Lu cells treated with 1.5% (v/v) of DMSO and vehicle only (control) at 37°C for 1.5 h by sucrose density gradient ultracentrifugation fractionation followed by Western blot analysis using antibodies to TβR-II, TβR-I, caveolin-1 (as a lipid raft/caveolae marker), and EEA-1 (as a non-lipid raft microdomain marker) [Zhang et al., 2005]. As shown in Figure 5A (top: Western blot and bottom: quantitative analysis), treatment of Mv1Lu cells with 1.5% (v/v) DMSO increased more localization of TβR-II to non-lipid raft microdomains (fractions 8 and 9) than to lipid rafts/caveolae (fractions 4–6) by 350 ± 10 % (n = 4) and 130 ± 20% (n = 4), respectively, as compared to those (100%) in cells treated with vehicle only (control) (Fig. 5A, top: Western blot and bottom: quantitative analysis, fractions 8 and 9 and fractions 4–6, respectively). The increased amounts of TβR-II in these fractions are presumably derived from intracellular pools. It is important to note that DMSO treatment does not alter the status of most TβR-II receptors in lipid rafts/caveolae because it increases localization of TβR-II to both lipid rafts/caveolae and non-lipid raft microdomains (Fig. 5A, top: Western blot and bottom: quantitative analysis). This is consistent with the evidence that DMSO treatment does not alter the ratio of TβR-II*/TβR-I* (Fig. 4A). Interestingly, DMSO treatment did not alter the total protein level of TβR-I (Figs. 4B and 5B, top: Western blot and bottom: quantitative analysis) but did result in the recruitment of TβR-I from lipid rafts/caveolae to non-lipid raft microdomains (Fig. 5B, top: Western blot, fractions 5 and 6 vs. fraction 9). This suggests that DMSO induces recruitment of TβR-II (but not TβR-I) from its intracellular pool to lipid rafts/caveolae and non-lipid raft microdomains, presumably by inducing fusion of cytoplasmic vesicles, which contain intracellular TβR-II, a membrane protein, and the plasma membrane. The recruitment of TβR-I to non-lipid raft microdomains is likely mediated by TβR-II, which is newly recruited by DMSO from its intracellular pool to non-lipid raft microdomains, through its ability to form TβR-I hetero-oligomeric complexes.
Fig. 5.
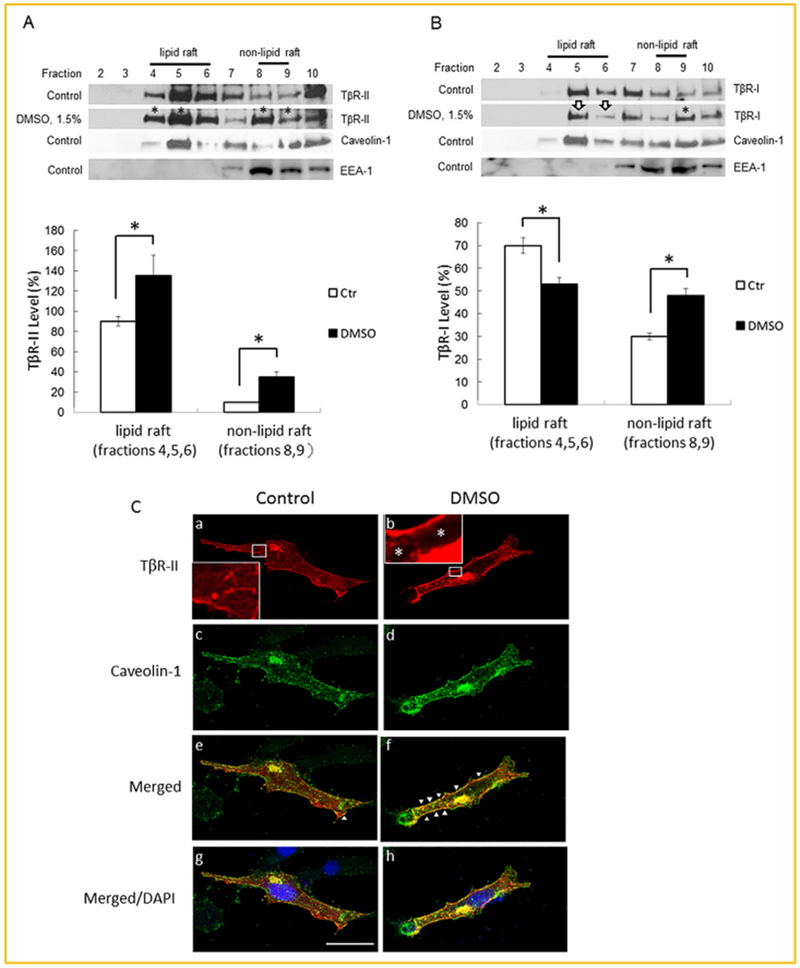
DMSO induces recruitment of TβR-II (A), and TβR-I (B), from intracellular pools and lipid rafts/caveolae, respectively, to non-lipid raft microdomains in Mv1Lu cells, and induces recruitment of TβR-II-HA (C), from cytoplasmic vesicles to the plasma membrane in Mv1Lu cells transiently expressing TβR-II-HA. (A,B): Mv1Lu cells were treated with 1.5% DMSO at 37°C for 1.5 h. The lipid raft/caveolae and non-lipid raft microdomain localization of TβR-II (A) and TβR-I (B) in DMSO-treated and -untreated (control) cells was then determined by sucrose gradient ultracentrifugation followed by Western blot analysis using antibodies to EEA-1, caveolin-1, TβR-I, and TβR-II. Fraction 1 (the top fraction), which did not contain protein, is not shown. Fraction 10 was the bottom fraction which mainly contained cell debris. Representative of four experiments is shown. Fractions 4–6, which contain caveolin-1, represent the location of lipid rafts/caveolae (A,B, top: Western blot) as described previously [Chen et al., 2007]. DMSO treatment did not affect the fraction distribution of caveolin-1. Fractions 8 and 9, which contain EEA-1, represent the location of non-lipid raft microdomains [Zhang et al., 2005]. Fractions 7–10 contain trace or small amounts of caveolin-1. This is due to the presence of mitochondria in these fractions. Caveolin-1 is known to be associated with mitochondria when cells (e.g., epithelial cells) are grown at sparse cell density (≤70% confluency) [Liu et al., 2002, Rashid-Doubell et al., 2007]. Caveolin-1 is mainly localized at the plasma membrane and the Golgi apparatus when cells are grown at confluence [Liu et al., 2002, Rashid-Doubell et al., 2007]. The * symbol indicates the increased amount of TβR-II or TβR-I in the fractions of cells treated with DMSO as compared with those of the untreated cells (A, top: Western blot: fractions 4, 5, 8, and 9; and B, top: Western blot, fraction 9). The open arrow indicates the decreased amount of TβR-I in the fractions of DMSO-treated cells as compared with those of the untreated cells (B, top: Western blot, fractions 5 and 6). The data are representative of a total of four independent analyses. The relative amounts of TβR-II and TβR-I in lipid rafts/caveolae and non-lipid raft microdomains in cells treated with and without 1.5% DMSO were quantified by densitometry using caveolin-1 as an internal control (A,B, bottom: quantitative analysis). The total level of TβR-II or TβR-I in lipid rafts/cavelolae and non-lipid raft microdomains in cells treated without DMSO was taken as 100%. The relative levels of TβR-II and TβR-I in lipid rafts/caveolae (lipid raft) and non-lipid raft microdomains (non-lipid raft) were estimated to be 90% and 10%, and 70% and 30%, respectively. (C): Mv1Lu cells transiently transfected with TβR-II-HA plasmid (from Addgene) were treated with and without 1.5% DMSO (panels b, d, and f and panels a, c, and e, respectively) at 37°C for 1.5 h. Cells were then subjected to indirect immunofluorescence staining using anti-HA (panels a, b, e, and f) and anti-caveolin-1 (panels c–h). DAPI (nuclear) staining (blue) was also performed (panels g and h). Merged staining is also shown (panels e and f). TβR-II-HA was present in the plasma membrane, cytoplasm and Golgi apparatus of cells treated without DMSO (panel a). The cytoplasm was full of the TβR-II-HA-containing vesicle-like network (inset, panel a). The TβR-II-HA density in the cytoplasm of cells treated with and without DMSO was quantified using the NIH ImageJ program. The presence of caveolin-1 staining in the cytoplasm may be due to its association with mitochondria (panels c and d). Arrowheads indicate the co-localization (yellow color) of TβR-II-HA and caveolin-1 at the plasma membrane (panels e and f). Size bar represents 10 microns.
To further define the intracellular pool of TβR-II and its DMSO-induced recruitment to plasma membrane microdomains, we performed immunofluorescence staining of hemagglutinin (HA) epitope-tagged TβR-II (TβR-II-HA) [Tsukazaki et al., 1998] and caveolin-1 using anti-HA tag and anti-caveolin-1 antibodies in MvlLu cells transiently transfected with TβR-II-HA after treatment of cells with and without 1.5% (v/v) DMSO. Anti-HA tag antibody, which is highly reliable and specific, is more suitable than anti-TβR-II (endogenous) antibody for detection of the antigen in the cytoplasm because of its very low non-specific immunofluorescence background in cells as compared with that using anti-TβR-II antibody. As shown in Figure 5C, before DMSO treatment of cells, TβR-II-HA was localized in the plasma membrane, cytoplasm (present as a vesicle-like network) (see inset in panel a) and the Golgi apparatus which is next to the nucleus (panels a and b). Caveolin-1 was localized in the plasma membrane, Golgi apparatus as well as in the cytoplasm (panels c and d). Cytoplasmic caveolin-1 is presumably associated with mitochondria [Liu et al., 2002; Helms and Zurzolo, 2004; Rashid-Doubell et al., 2007]. DMSO (1.5%,v/v) treatment appeared to deplete 70 ± 10% of TβR-II-HA vesicles from the cytoplasm (Fig. 5C, *inset in panel b) but also likely increased the TβR-II-HA density at the plasma membrane or cell surface as it did to endogenous TβR-II as shown by 125I-TGF-β-cross linking (Fig. 4A, lane 4 vs. lane 1). We presume that the depletion of TβR-II-HA from the cytoplasm would result in its recruitment to the cell surface or plasma membrane as shown in DMSO-induced co-localization of TβR-II-HA (which is derived from cytoplasmic vesicles) and caveolin-1 (which is present in the plasma membrane) at the plasma membrane (Fig. 5C, panel f). This suggests that DMSO treatment induces recruitment of TβR-II-HA from cytoplasmic vesicles to the plasma membrane likely by inducing fusion of cytoplasmic vesicles and the plasma membrane. The localization of TβR-II-HA to the Golgi apparatus supports the important role of the Golgi apparatus in the trafficking of TβR-II-HA-containing vesicles from the cytoplasm to the plasma membrane [Helms and Zurzolo, 2004]. Together with the result shown in Figure 5A, these results support our conclusion that DMSO enhances TGF-β activity by increasing localization of TβR-II and, TβR-I and TβR-II from cytoplasmic vesicles and lipid rafts/caveolae, respectively, to non-lipid raft microdomains where they form hetero-oligomeric complexes which mediate canonical signaling after TGF-β stimulation.
DISCUSSION
In this communication, we demonstrate that DMSO enhances TGF-β activity by increasing cell-surface localization of TβR-I and TβR-II to non-lipid raft microdomains where TβR-I-TβR-II hetero-oligomeric complexes transduce signaling following TGF-β stimulation. This is evidenced by: (1) DMSO increases cell-surface expression of TGF-β receptors by 3.5-fold in MvlLu cells, as determined by I125-TGF-β-cross-linking (at 0°), without altering their cellular levels. This suggests the presence of large intracellular pools of TβR-I and/or TβR-II in Mv1Lu cells. Large intracellular pools of TβR-I and/or TβR-II exist in a variety of cell types [Massagué and Kelly, 1986; Doré et al., 2001; Asano et al., 2011]. These intracellular receptors are believed to be present in recycling endosomes and/or exocytotic vesicles [Helms and Zurzolo, 2004]; (2) DMSO increases cell-surface expression of TGF-β receptors, canonical TGF-β signaling (e.g., increased levels of P-Smad2) and cellular responses (e.g., increased levels of luciferase reporter activity and PAI-1 mRNA) in a concentration-dependent manner, all with a maximum (~3–4-fold) at 1.5% (v/v); and (3) DMSO induces recruitment of TβR-II, and TβR-I and TβR-II from intracellular (cytoplasmic) vesicles and lipid rafts/caveolae, respectively, to non-lipid rail: microdomains where canonical TGF-β signaling occurs. This is mediated by DMSO to promote fusion of cytoplasmic vesicles (recycling endosomes and/or exocytotic vesicles) and plasma membranes in cells [Geron and Meiri, 1985; Faisst and Paweletz, 1987; Yu and Quinn, 1998].
The finding of DMSO-induced recruitment of TβR-II (but not TβR-I) from its intracellular vesicle pool to the cell surface raises an important question: does DMSO have similar effects on other membrane receptors or proteins? The answer is yes. It has been reported that treatment with 1% (v/v) DMSO induces recruitment of rat8, a membrane protein, presumably from an intracellular pool, to cell-surface lipid rafts/caveolae in rat mammary gland cells [Zucchi et al., 2004]. In fact, DMSO induces recruitment of rat8 from its intracellular pool to both lipid raft/caveolae and non-lipid raft microdomains in these cells as it does to TβR-II in Mv1Lu cells. Interestingly, DMSO induces more recruitment of rat8 and TβR-II to non-lipid raft microdomains than to lipid rafts/caveolae in mammary gland cells [Zucchi et al., 2004] and Mv1Lu cells, respectively. This suggests that DMSO-induced recruitment of membrane proteins from their intracellular vesicle pools to cell-surface or plasma-membrane lipid rafts/caveolae and non-lipid raft microdomains is not limited to TβR-II. Fusogenic activity of DMSO to induce fusion of intracellular vesicles and plasma membranes may play a pivotal role in its pharmacological actions involving membrane proteins which have large intracellular vesicle pools such as rat8 and TβR-II.
As described above, DMSO exhibits many biological activities depending on cell type. They may be mediated by DMSO-induced recruitment of various receptor proteins in addition to the TGF-β receptors from their intracellular vesicle pools to cell-surface microdomains (lipid rafts/caveolae and non-lipid raft microdomains). However, several lines of evidence support the notion that the TGF-β enhancer activity of DMSO is mainly mediated by TβR-I and TβR-II. These include: (1) TGF-β enhancer activity is determined using specific luciferase reporters whose expression is dependent on canonical TGF-β signaling (Smad-dependent); (2) DMSO induces increased expression of PAI-1 and P-Smad2. The gene of PAI-1 is one of the most TGF-β responsive genes [Sato et al., 1990]. P-Smad2 is an important indicator of canonical TGF-β signaling; and (3) The TGF-β enhancer activity of DMSO is completely inhibited by co-incubation with the TβR-I kinase inhibitor SB-505124 [DaCosta Byfield et al., 2004] or a specific TGF-β receptor antagonist [Huang et al., 1997; Singer et al., 2009].
DMSO is a well-known differentiation inducer for certain cultured cells such as murine Friend erythroleukemia cells [Friend and Freedman, 1978] and human HL 60 cells [Collins et al., 1978]. DMSO induces Friend erythroleukemia cells to differentiate along the erythroid developmental pathway. It can also induce HL60 to differentiate to granulocytes [Hino et al., 1989]. The TGF-β enhancer activity of DMSO may contribute to its ability to induce such differentiation. This possibility is supported by several lines of evidence, including: (1) DMSO induces differentiation (in the presence of fetal calf serum) of erythroid leukemia cells and HL 60 cells with optimal concentrations of 1–2% (v/v). These concentrations are similar to those of DMSO required to enhance canonical TGF-β signaling and cellular responses. Fetal calf serum contains a low but significant level of TGF-β; (2) Exogenous TGF-β has been shown to induce differentiation of these murine erythroleukemia cells and HL 60 cells [Collins et al., 1978; Hino et al., 1989]; and (3) Like DMSO, vitamin D3, a canonical TGF-β signaling enhancer [Brungs et al., 1994], is capable of inducing HL 60 cells to differentiate into mature granulocytes in vitro [Peiretti et al., 2001].
In addition to its ability to induce differentiation in cultured cells, DMSO has been shown to induce reversible G1 arrest in the medium (in the presence of serum which contains TGF-β) in many cell lines [Fiore et al., 2002; Mukherjee et al., 2010]. The TGF-β enhancer activity of DMSO appears to play an important role in its ability to induce G1 arrest. This possibility is supported by several lines of evidence: (1) TGF-β is a potent cell growth inhibitor which arrests cell growth at G1 [Roberts, 1998]; (2) The optimal concentrations (1-1.5%, v/v) of DMSO to induce growth arrest in target cells are similar to those which enhance TGF-β-stimulated canonical signaling; (3) Both DMSO and TGF-β effects are reversible.
We hypothesize that the TGF-β enhancer activity of DMSO is involved in its effects on the diseases and symptoms in which TGF-β plays a protective role. For example, intravesical therapy (bladder instillation therapy) with DMSO is effective for treating interstitial cystitis/painful bladder syndrome (IC/PBS) in human patients [Swanson, 1985]. The mechanisms by which DMSO attenuates the inflammation and pain in interstitial cystitis are not understood. However, the TGF-β enhancer activity of DMSO may be responsible, at least in part, for its anti-inflammatory effect on IC/PBS. TGF-β is a potent anti-inflammatory cytokine [Roberts, 1998]. This is supported by the observation that an inverse relationship exists between the expression of TGF-β and iNOS/NO2 -/NO3 in the urothelium of bladder in a rat model of acute interstitial cystitis [Tyagi et al., 2009]. NO is believed to play a pivotal role in the inflammation of interstitial cystitis in animals and human patients [Tyagi et al., 2009]. We also hypothesize that DMSO exerts its analgesic effects on IC/PBS, by inducing recruitment of analgesic-related G-protein coupled receptors (GPCR) from their intracellular pools to lipid rafts/caveolae where the GPCR mediates analgesic function. Increased expression of the μ-opioid receptor in lipid rafts/caveolae is known to enhance opioid analgesic effects in mice and humans [Zheng et al., 2012].
DMSO is known to be an anti-atherosclerotic agent in relevant animal models [Debons et al., 1987]. The TGF-β enhancer activity of DMSO is important in its anti-atherosclerotic activity. This is because TGF-β in the circulation is a protective cytokine against atherosclerosis [Grainger and Metcalfe, 1995]. This possibility is supported by the observations that other known TGF-β enhancers, such as dynasore and statins have been shown to be anti-atherosclerotic agents [Chen et al., 2009; Babelova et al., 2013]. Dynasore inhibits atherosclerosis without altering high plasma cholesterol levels in ApoE-null mice with hypercholesterolemia [Chen et al., 2009]. Statins, also canonical TGF-β signaling enhancers [Chen et al., 2009], exhibit anti-atherosclerotic activity [Babelova et al., 2013].
DMSO is often used as a vehicle for chemicals which are not soluble in water. The effect of DMSO on increasing cell-surface expression of membrane receptors (particularly TGF-β receptors) should be considered when one is interpreting the experimental results of tested agents dissolved in DMSO or when DMSO is present in the experimental system. TGF-β is a potent and dominant cytokine. The TGF-β enhancer activity of DMSO could complicate the interpretation of results in experiments using it as a solvent vehicle. For example, DMSO, when used as a solvent vehicle, has been found to contribute to a powerful protective effect against acetaminophen-induced liver injury in mice [Masson et al., 2008].
ACKNOWLEDGMENTS
We thank Frank E. Johnson, M.D. for critically reviewing the manuscript. We also thank Lai-Ping Yan and Ying Fei for their technical assistance. This work was supported by NIH grants HL 095261 and AR 052578 to Auxagen and AA019233 to Saint Louis University (J.S.H.). This work was also supported by the President’s Research Fund, Saint Louis University (J.S.H.) and the NSYSU-KMU Joint Research Project (NSYSUKMU104I004), National Sun Yatsen University (C.L.C.).
Grant sponsor: NIH; Grant numbers: HL 095261, AR 052578, AA019233; Grant sponsor: President’s Research Fund; Grant sponsor: NSYSU-KMU Joint Research Project; Grant number: NSYSUKMU104I004; Grant sponsor: National Sun Yatsen University (C.L.C).
Footnotes
Conflict of interest: The authors declare that they have conflict of interest. Jung San Huang and Shuan Shian Huang had an equity position in Auxagen Inc. during the time the research was carried out.
REFERENCES
- Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, Rifkin DB. 1994. An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal Biochem 216:276–284. [DOI] [PubMed] [Google Scholar]
- Akhurst RJ. 2012. The paradoxical TGF-β vasculopathies. Nat Genet 44:838–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano Y, Ihn H, Jinnin M, Tamaki K, Sato S. 2011. Altered dynamics of transforming growth factor β (TGF-β) receptors in scleroderma fibroblasts. Ann Rheum Dis 70:384–387. [DOI] [PubMed] [Google Scholar]
- Babelova A, Sedding DG, Brandes RP. 2013. Anti-atheroscerotic mechanisms of statin therapy. Curr Opin Pharmacol 13:260–264. [DOI] [PubMed] [Google Scholar]
- Brown BL, Nagle SC Jr. 1965. Preservation of mammalian cells in a chemically defined medium and dimethylsulfoxide. Science 149:1266–1267. [DOI] [PubMed] [Google Scholar]
- Brungs M, Rådmark O, Samuelsson B, Steinhilber D. 1994. On the induction of 5-lipoxygenase expression and activity in HL-60 cells: Effects of vitamin D3, retinoic acid, DMSO, and TGF-β. Biochem Biophys Res Commun 205:1572–1580. [DOI] [PubMed] [Google Scholar]
- Capriotti K, Capriotti JA. 2012. Dimethyl sulfoxide: History, chemistry, and clinical utility in dermatology. J Clin Aesthet Dermatol 5:24–26. [PMC free article] [PubMed] [Google Scholar]
- Chen CL, Huang SS, Huang JS. 2006. Cellular heparan sulfate negatively modulates transforming growth factor-β responsiveness. J Biol Chem 281:11506–11514. [DOI] [PubMed] [Google Scholar]
- Chen CL, Liu IH, Fliesler SJ, Han X, Huang SS, Huang JS. 2007. Cholesterol suppresses TGF-β responsiveness: Implications in atherogenesis. J Cell Sci 120:3509–35621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CL, Huang SS, Huang JS. 2008. Cholesterol modulates cellular TGF-β responsiveness by altering TGF-β binding to TGF-β receptors. J Cell Physiol 215:223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CL, Hou WH, Liu IH, Huang SS, Huang JS. 2009. Inhibitors of clarthrin-dependent endocytosis inhibitors enhance TGF-β signaling and responses. J Cell Sci 122:1863–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SJ, Ruscetti FW, Gallagher RE, Gallo RC. 1978. Terminal differentiation of human promyelocytic leukemia cells induced by dimethyl sulfoxide and other polar compounds. Proc Natl Acad Sci USA 75:2458–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DaCosta Byfield S, Major C, Laping NJ, Roberts AB. 2004. SB-505124 is a selective inhibitor of transforming growth factor-beta type I receptors ALK4, ALK5, and ALK7. Mol Pharmacol 65:744–752. [DOI] [PubMed] [Google Scholar]
- David NA. 1972. The pharmacology of dimethyl sulfoxide. Annu Rev Pharmacol 12:353–374. [DOI] [PubMed] [Google Scholar]
- Debons AF, Fani K, Jimenez FA, Maayan ML. 1987. Inhibition of cholesterol-induced atherosclerosis in rabbits by dimethyl sulfoxide. J Pharmacol Exp Ther 243:745–757. [PubMed] [Google Scholar]
- Deng R, Wang SM, Yin T, Ye TH, Shen GB, Li L, Zhao JY, Sang YX, Duan XG, Wei YQ. 2014. Dimethyl sulfoxide suppresses mouse 4T1 breast cancer growth by modulating tumor-associated macrophage differentiation. J Breast Cancer 17:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Akhurst RJ, Balmain A. 2001. TGF-β signaling in tumor suppression and cancer progression. Nat Genet 29:117–129. [DOI] [PubMed] [Google Scholar]
- Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. 2003. Distinct endocytic pathways regulate TGF-β receptor signaling and turnover. Nat Cell Biol 5:410–421. [DOI] [PubMed] [Google Scholar]
- Doré JJ Jr., Yao D, Edens M, Garamszegi N, Sholl EL, Leof EB 2001. Mechanisms of transforming growth factor-β receptor endocytosis and intracellular sorting differ between fibroblasts and epithelial cells. Mol Biol Cell 12:675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehata S, Hanyu A, Hayashi M, Aburatani H, Kato Y, Fujime M, Saitoh M, Miyazawa K, Imamura T, Miyazono K. 2007. Transforming growth factor-β promotes survival of mammary carcinoma cells through induction of antiapoptotic transcription factor D EC1. Cancer Res 67:9694–9703. [DOI] [PubMed] [Google Scholar]
- Fahmy MD, Almansoori KA, Laouar L, Prasad V, McGann LE, Elliott JA, Jomha NM. 2014. Dose-injury relationships for cryoprotective agent injury to human chondrocytes. Cryobiology 68:50–56. [DOI] [PubMed] [Google Scholar]
- Faisst S, Paweletz N. 1987. Erythrocyte ghost-mediated microinjection into cells in monolayer cultures: A highly efficient and low toxic technique. Cell Biol Int Rep 11:515–524. [DOI] [PubMed] [Google Scholar]
- Fiore M, Zanier R, Degrassi F. 2002. Reversible G1 arrest by dimethyl sulfoxide as a new method to synchronize Chinese hamster cells. Mutagenesis 17:419–424. [DOI] [PubMed] [Google Scholar]
- Fowler JE Jr. 1981. Prospective study of intravesical dimethyl sulfoxide in treatment of suspected early interstitial cystitis. Urology 18:21–26. [DOI] [PubMed] [Google Scholar]
- Friend C, Freedman HA. 1978. Effects and possible mechanism of action of dimethyl sulfoxide on friend cell differentiation. Biochem Pharmacol 27:1309–1313. [DOI] [PubMed] [Google Scholar]
- Geron N, Meiri H. 1985. The fusogenic substance dimethyl sulfoxide enhances exocytosis in motor nerve endings. Biochim Biophys Acta 819:258–262. [DOI] [PubMed] [Google Scholar]
- Gorog P, Kovacs IB. 1968. Effect on dimethyl sulfoxide (DMSO) on various experimental inflammations. Curr Ther Res 10:486–492. [PubMed] [Google Scholar]
- Grainger DJ, Metcalfe JC. 1995. A pivotal role for TGF-β in atherogenesis? Biol Rev Camb Philos Soc 70:571–596. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Miyazono K, ten Dijke P. 1997. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 390:465–471. [DOI] [PubMed] [Google Scholar]
- Helms JB, Zurzolo C. 2004. Lipids as targeting signals: Lipid rafts and intracellular trafficking. Traffic 5:247–254. [DOI] [PubMed] [Google Scholar]
- Hino M, Tojo A, Miyazono K, Miura Y, Chiba S, Eto Y, Shibai H, Takaku F. 1989. Characterization of cellular receptors for erythroid differentiation factor on murine. J Biol Chem 264:10309–10314. [PubMed] [Google Scholar]
- Huang SS, Liu Q, Johnson FE, Konish Y, Huang JS. 1997. Transforming growth factor-β peptide antagonists and their conversion to partial agonists. J Biol Chem 272:27155–27159. [DOI] [PubMed] [Google Scholar]
- Huang SS, Huang JS. 2005. TGF-β control of cell proliferation (review). J Cell Biochem 96:447–462. [DOI] [PubMed] [Google Scholar]
- Jonk LJ, Itoh S, Heldin CH, ten Dijke P, Kruijer W. 1998. Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor-beta, activin, and bone morphogenetic protein-inducible enhancer. J Biol Chem 273:21145–21152. [DOI] [PubMed] [Google Scholar]
- Kloesch B, Liszt M, Broell J, Steiner G. 2011. Dimethyl sulphoxide and dimethyl sulphone are potent inhibitors of IL-6 and IL-8 expression in the human chondrocyte cell line C-28/ I2. Life Sci 89:473–478. [DOI] [PubMed] [Google Scholar]
- Kulkarni AB, Ward JM, Yaswen L, Mackall CL, Bauer SR, Huh CG, Gress RE, Karlsson S. 1995. Transforming growth factor-β1 null mice. An animal model for inflammatory disorders. Am J Pathol 146:264–275. [PMC free article] [PubMed] [Google Scholar]
- Liu P, Rudick M, Anderson RG. 2002. Multiple functions of caveolin-1. J Biol Chem 277:41295–41298. [DOI] [PubMed] [Google Scholar]
- Massagué J, Kelly B. 1986. Internalization of transforming growth factor-β and its receptor in BALB/c 3T3 fibroblasts. J Cell Physiol 128:216–222. [DOI] [PubMed] [Google Scholar]
- Massagué J 1998. TGF-β signal transduction. Annu Rev Biochem 67:753–791. [DOI] [PubMed] [Google Scholar]
- Masson MJ, Carpenter LD, Graf ML, Pohl LR. 2008. Pathogenic role of natural killer T and natural killer cells in acetaminophen-induced liver injury in mice is dependent on the presence of dimethyl sulfoxide. Hepatology 48:889–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee P, Winter SL, Alexandrow MG. 2010. Cell cycle arrest by transforming growth factor-β1 near G1/S is mediated by acute abrogation of prereplication complex activation involving an Rb-MCM interaction. Mol Cell Biol 30:845–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piek E 1, Ju WJ, Heyer J, Escalante-Alcalde D, Stewart CL, Weinstein M, Deng C, Kucherlapati R, Bottinger EP, Roberts AB. 2001. Functional characterization of transforming growth factor beta signaling in Smad2- and Smad3-deficient fibroblasts. J Biol Chem 276:19945–19953. [DOI] [PubMed] [Google Scholar]
- Peiretti F, Lopez S, Deprez-Beauclair P, Bonardo B, Juhan-Vague I, Nalbone G. 2001. Inhibition of p70(S6) kinase during transforming growth factor-beta 1/vitamin D(3)-induced monocyte differentitiation ofHL-60 cells allows tumor necrosis factor-α to stimulate plasminogen activator inhibitor-1 synthesis. J Biol Chem 276:32214–32219. [DOI] [PubMed] [Google Scholar]
- Pogorelaia NKh, Fomina AF, Veselovskiĭ NS. 1984. Morphologic differentiation of neuroblastoma cells induced by dimethyl sulfoxide. Neirofiziologiia 16:519–527. [PubMed] [Google Scholar]
- Poncelet C, de Caestecker MP, Schnaper HW. 1999. The transforming growth factor-beta/SMAD signaling pathway is present and functional in human mesangial cells. Kidney Int 56:1354–1365. [DOI] [PubMed] [Google Scholar]
- Rashid-Doubell F, Tannetta D, Redman CWG, Sargent IL, Boyd CAR, Linton EA. 2007. Caveolin-1 and lipid rafts in confluent BeWo trophoblasts: Evidence for rock-1 association with caveolin-1. Placenta 28:139–151. [DOI] [PubMed] [Google Scholar]
- Roberts AB. 1998. Molecular and cell biology of TGF-β. Miner Electrolyte Metab 24:111–119. [DOI] [PubMed] [Google Scholar]
- Santos NC, Figueira-Coelho J, Martins-Silva J, Saldanha C. 2003. Multidisciplinary utilization of dimethyl sulfoxide: Pharmacological, cellular, and molecular aspects. Biochem Pharmacol 65:1035–1041. [DOI] [PubMed] [Google Scholar]
- Sato Y, Tsuboi R, Lyons R, Moses H, Rifkin DB. 1990. Characterization of the activation of latent TGF-β by co-cultures of endothelial cells and pericytes or smooth muscle cells: A self-regulating system. J Cell Biol 111:757–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See WA, Xia Q. 1992. Regional chemotherapy for bladder neoplasms using continuous intravesical infusion of doxorubicin: Impact of concomitant administration of dimethyl sulfoxide on drug absorption and antitumor activity. J Natl Cancer Inst 84:510–515. [DOI] [PubMed] [Google Scholar]
- Singer AJ, Huang SS, Huang JS, McClain SA, Romanov A, Rooney J, Zimmerman T. 2009. A novel TGF-β antagonist speeds reepithelialization and reduces scarring of partial thickness porcine burns. J Burn Care Res 30:329–334. [DOI] [PubMed] [Google Scholar]
- Swanson BN. 1985. Medical use of dimethyl sulfoxide (DMSO). Rev Clin Basic Pharm 5:1–33. [PubMed] [Google Scholar]
- Tsukazaki T, Chiang TA, Davison AF, Attisano L, Wrana JL. 1998. SARA, a FYVE domain protein that recruits Smad2 to the TGF-β receptor. Cell 95:779–791. [DOI] [PubMed] [Google Scholar]
- Tyagi P, Tyagi V, Yoshimura N, Witteemer E, Barclay D, Loughran PA, Zamora R, Vodovotz Y. 2009. Gender-based reciprocal expression of transforming growth factor-β1 and the inducible nitric oxide synthase in a rat model of cyclophosphamide-induced cystitis. J Inflamm (Lond) 6:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu ZW, Quinn PJ. 1998. The modulation of membrane structure and stability by dimethyl sulphoxide (review). Mol Membr Biol 15:59–68. [DOI] [PubMed] [Google Scholar]
- Zhang XL, Topley N, Ito T, Phillips A. 2005. Interleukin-6 regulation of transforming growth factor TGF-β receptor compartmentalization and turnover enhances TGF-β1 signaling. J Biol Chem 280:12239–12245. [DOI] [PubMed] [Google Scholar]
- Zheng H, Zou H, Liu X, Chu J, Zhou Y, Loh HH, Law PY. 2012. Cholesterol level influences opioid signaling in cell models and analgesia in mice and humans. J Lipid Res 53:1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucchi I, Prinetti A, Scotti M, Valsecchi V, Valaperta R, Mento E, Reinbold R, Vezzoni P, Sonnino S, Albertini A, Dulbecco R. 2004. Association of rat8 with Fyn protein kinase via lipid rafts is required for rat mammary cell differentiation in vitro. Proc Natl Acad Sci USA 101:1880–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]