

Summary
CRSBP-1/LYVE-1 ligands (PDGF-BB, VEGF-A165 and hyaluronic acid) have been shown to induce opening of lymphatic intercellular junctions in vitro and in vivo by stimulating contraction of lymphatic endothelial cells (LECs). The mechanism by which CRSBP-1 ligands stimulate contraction of LECs is not understood. Here we demonstrate that CRSBP-1 is localized to the plasma membrane as well as intracellular fibrillar structures in LECs, including primary human dermal LECs and SVEC4–10 cells. CRSBP-1-associated fibrillar structures are identical to the ER network as evidenced by the co-localization of CRSBP-1 and BiP in these cells. CRSBP-1 ligands stimulate contraction of the ER network in a CRSBP-1-dependent and paclitaxel (a microtubule-stabilizing agent)-sensitive manner. These results suggest that ligand-stimulated ER contraction is associated with ligand-stimulated contraction in LECs.
Keywords: LYVE-1, PDGF-BB, VEGF-A165, hyaluonic acid, ER localization, ER contraction
Introduction
Cell-surface retention sequence (CRS) binding protein-1 (CRSBP-1), a membrane glycoprotein, was identified by its ability to interact with oligopeptides containing the CRS motifs of VEGF-A165 and PDGF-BB in human SK-Hep cells and bovine liver plasma membranes [1]. The CRS motifs of VEGF-A165 and PDGF-BB contain clustered basic amino acid residues (Arg, Lys and His) [2–5]. CRSBP-1 interacts with PDGF-BB in simian sarcoma virus (SSV)-transformed cells which express both CRSBP-1 and the autocrine sis oncogene product (PDGF-BB) [6]. CRSBP-1 is identical to lymphatic vessel endothel ial hyaluronic acid (HA) receptor-1 (LYVE-1) [7–9]. The primary localization of LYVE-1 in lymphatic endothelium implies the likely importance of the multiple ligand (CRS-containing growth factors/cytokines and HA) binding activity of CRSBP-1/LYVE-1 in regulating the function of lymphatic vessels. To determine the in vivo role of CRSBP-1, we generated CRSBP-1-null (CRSBP-1−/−) mice [10]. These mice are macroscopically normal but exhibit distended lumens of lymphatic vessels and constitutively increased transit of fluid from the interstitial space into lymphatic vessel lumens (termed interstitial-lymphatic transit). CRSBP-1 ligands are capable of increasing interstitial-lymphatic transit of fluid in wild-type mice but not in CRSBP-1−/− mice. These results suggest that CRSBP-1 and its ligands regulate the function of lymphatic intercellular junctions. They also support our hypothesis that the CRSBP-1-null mutation and binding of CRSBP-1 by its cognate ligands cause opening of lymphatic intercellular junctions, resulting in increased interstitial-lymphatic transit [10].
To determine the molecular mechanism by which CRSBP-1 and its ligands regulate the opening of lymphatic intercellular junctions, we investigated the effects of CRSBP-1 ligands on tyrosine phosphorylation and internalization of VE-cadherin, which forms button-like structures at lymphatic intercellular junctions [11] in LECs. We hypothesized that the CRSBP-1-null mutation and binding of CRSBP-1 by its cognate ligands would result in loss and “unbuttoning” of VE-cadherin-mediated buttons, respectively, leading to opening of lymphatic intercellular junctions [10,12]. We found that CRSBP-1 ligands (PDGF-BB, VEGF-A165, PDGF peptide, VEGF peptide and HA) stimulate tyrosine phophorylation of PDGF β-type receptor/β-catenin/VE-cadherin, internalization of VE-cadherin and cellular contraction in LEC monolayers [12]. This leads to opening of lymphatic intercellular junctions. These CRSBP-1 3 inhibitor or CRSBP-ligand-stimulated effects are abolished in LECs treated with a PDGF β-type receptor (PDGFβR) kinase 1 siRNA [12]. To determine the role of CRSBP-1 in ligand-stimulated LEC contraction, we investigated the subcellular localization of CRSBP-1 in LECs, which include primary human dermal LECs (HDLECs) and SVEC4–10 cells (a LEC-like cell line), treated with CRSBP-1 ligands. CRSBP-1 was previously localized at the plasma membrane (or cell surface) of CRSBP-1-transfected cells, SVEC4–10 cells and HDLECs by cell-surface cross-linking with125I-labeled specific peptide ligands of CRSBP-1 [9,12] and by indirect immunofluorescence staining [12]. In HDLECs, LYVE-1 was shown by Johnson et al. [13] to exhibit a perinuclear staining pattern, co-localizing with markers of ER and Golgi where its de novo synthesis occurs. The exact subcellular localization of CRSBP-1/LYVE-1 in LECs is not clear.
In this communication, we demonstrate that CRSBP-1 is localized to the plasma membrane as well as intracellular fibrillar structures. The CRSBP-1-associated fibrillar structure is identical to the ER network, as evidenced by its co-localization with binding immunoglobulin protein (BiP), an ER resident protein [14]. We also show that CRSBP-1 ligands stimulate contraction of CRSBP-1-associated ER network in a CRSBP-1-dependent and paclitaxel-sensitive manner.
Materials and Methods
Materials-
PDGF peptide (a 19-mer peptide containing the amino acid sequence YVRVRRPPKGKHRKFKHTH) and VEGF peptide (a 25-mer peptide containing the amino acid sequence KKSVRGKGKGQKRKRKKSRYKSWSV) were synthesized by C S Bio Co. (Menlo Park, CA). Rabbit anti-CRSBP-1 antiserum was prepared as described [6,9,12]. PDGF-BB was obtained from Abcam Inc. (Cambridge, MA) and R&D Systems (Minneapolis, MN). Anti-BiP/GRP78 antibody (sc-1051) was obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). VEGF-A165 was a gift from Dr. Daniel T. Connolly, Mosanto Company (St. Louis, MO). Paclitaxel (T7402) and most of the biochemical reagents were obtained from Sigma (St. Louis, MO). H1299/CRSBP-1 and H1299/vector cells were prepared as described [9]. SVEC4–10 cells and human dermal LECs (HDLECs) were obtained from American Type Culture Collection (Manassas, VA) and PromoCell (Heidelberg, Germany), respectively.
Immunofluorescence staining of CRSBP-1 and BiP in SVEC 4–10 cells and HDLECs-
Cells grown on coverslips for 2 days (~50% confluence) were treated with CRSBP-1 ligands (10 μM PDGF peptide, 10 μM VEGF peptide, 0.2 μM VEGF-A and 120 μg/ml HA) or vehicle only for 30 min or 1 h in serum-free DMEM. After treatment, cells were first washed with PBS twice, and then fixed in 100% methanol at −20°C for 10 min. Following rinsing with PBS, cells were quenched in 0.1% sodium borohydride in PBS for 5 min. Cells were washed with PBS and blocked with 0.2% gelatin in PBS at room temperature for 1 h. Cells were incubated for 1 h with a 1:200 or 1:500 dilution of anti-CRSBP-1 antiserum and anti-BiP antibody in a solution of 0.2% gelatin in PBS, followed by incubation with an 1:200 dilution of fluorescence-labeled secondary antibodies in a solution of 0.2% gelatin in PBS for 1 h. Coverslips were then mounted on glass slides. The slides were viewed under an Olympus AHBT3 fluorescence microscope using interference green filters. CRSBP-1 positive-associated fibrillar structures were analyzed and quantified using NIH software Image J. Using the function “Auto-Threshold”, a threshold value was arbitrarily chosen to highlight positively-stained fibrillar structures in an image. Lengths of the fibrillar structures of individual cells were then determined using the function “Analyze Particle.” The same threshold value was then used to analyze all images. A minimum of 25 cells from each condition was used for statistical analysis.
Immunofluorescence confocal microscopy of the CRSBP-1/BiP fibrillar structures in LECs-
SVEC4–10 cells and HDLECs were pretreated with paclitaxel (5 μM) or vehicle only at 37C for 2 h and then stimulated with CRSBP-1 ligands for 1 h and fixed as previously described [12]. Following blocking, cells were incubated overnight at 4°C with rabbit anti-CRSBP-1 serum and goat anti-BiP antibody at 1:200 dilution. To estimate colocalization of CRSBP-1 and BiP in SVEC4–10 cells, confluent cells grown on coverslips were treated with CRSBP-1 ligands. Following fixation and blocking, cells were incubated overnight with rabbit anti-CRSBP-1 serum and goat antibody to BiP. After extensive washing, cells were incubated with FITC-conjugated mouse anti-rabbit IgG antibody and rhodamine red-conjugated donkey anti-goat IgG antibody and at 1:200 dilution for 1 h. Images were acquired using a Leica TCS SP confocal microscope (Leica Microsystems Ltd., Heidelberg, Germany).
siRNA transfection-
Transfection of SVEC4–10 cells with control siRNA and CRSBP-1 siRNA was performed according to the manufacturer’s protocol (Santa Cruz Biotechnology) as previously described [12]. Briefly, cells grown on coverslips were washed once with transfection medium. Transfection medium (0.8 ml) (sc-36868) was added to the cells. A 0.2 ml aliquot of transfection medium containing 80 pmoles CRSBP-1 siRNA (sc-42902) or control siRNA (sc-37007) and 8 μl of transfection reagent (sc-29528) was incubated at room temperature for 30 min before adding to the cells. Following 48 h incubation at 37°C, transfected cells were washed and treated as described before.
Statistical analysis-
The values are presented as means ± s.d. (n = 3–5). Two-tailed unpaired Student’s t-test was used to determine the significance of differences between groups. P<0.05 was considered significant.
Results
CRSBP-1 localizes to the plasma membrane and intracellular fibrillar structures in LECs.
CRSBP-1 is known to be a cell-surface membrane protein but its subcellular localization in LECs has not been characterized. We previously generated H1299/CRSBP-1 cells for studying the cell biology of CRSBP-1 [9]. However, H1299 cells are a human lung carcinoma cell line and are not an ideal cell system for investigation of such properties of CRSBP-1 because CRSBP-1 is primarily localized to lymphatic capillary endothelial cells. For this reason, we identified SVEC4–10 cells as a candidate cell system for studying the cell biological functions of CRSBP-1. SVEC4–10 cells are a SV40-immortalized endothelial cell line derived from mouse axillary lymph nodes. These cells are thought to be derived from vascular endothelial cells [15,16]. However, Western blot analysis of SVEC4–10 cell revealed that they express CRSBP-1, podoplanin, PDGFβR and VEGFR3 [12]. All of these proteins have been reported to be specific markers for lymphatic capillary endothelial cells [17]. SVEC4–10 cells also exhibit ultrastructural features characteristic of LECs including sparse microvillous surface projections, overlapping intercellular junctions and abundant intermediate filaments [16]. They are LEC-like cells.
To define the cell biological role of CRSBP-1, we examined subcellular localization of CRSBP-1 in SVEC4–10 cells by immunolocalization using rabbit antiserum to the cytoplasmic tail of CRSBP-1 (anti-CRSBP-1 serum) [6,9,12]. After fixation and permeabilization of SVEC4–10 cells, CRSBP-1 was visualized with fluorescence microscopy after incubation of the fixed cells with preimmune serum (Fig. 1, panel A), anti-CRSBP-1 serum (Fig. 1, panel B), or anti-CRSBP-1 serum with 2 mM CRSBP-1 peptide antigen [1] (Fig. 1, panels C) followed by incubation with FITC-conjugated goat anti-rabbit IgG. As shown in Fig. 1 (panel B), CRSBP-1 is localized to the plasma membrane and also to intracellular fibrillar structures in SVEC4–10 cells. The fibrillar structures are not actin fibers, microtubules or intermediate filaments based on co-immunolocalization experiments (data not shown). In these cells, CRSBP-1 is associated with a fibrillar network extending from the perinuclear membrane to the leading edge of the plasma membrane (Fig. 1, panel B). The fibrillar staining pattern truly represents the subcellular localization of CRSBP-1 as the addition of CRSBP-1 peptide antigen [1] completely abolished anti-CRSBP-1 serum binding to CRSBP-1 (Fig. 1 panel C).
Fig. 1. CRSBP-1 is localized to the plasma membrane and intracellular fibrillar structures in SVEC4–10 cells.
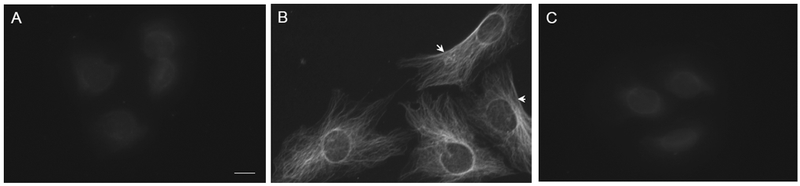
Cells were grown on cover slips and fixed with methanol at −200C for 10 min. Cells were stained with preimmune serum (panel A), anti-CRSBP-1 antiserum (panels B) or anti-CRSBP-1 antiserum with 2 mM CRSBP-1 peptide antigen (panel C) followed by FITC-conjugated goat anti-rabbit IgG and visualized with a fluorescence microscope. Arrowheads indicate the plasma membrane localization of CRSBP-1. Data are representative of four independent experiments.
CRSBP-1 ligands stimulate contraction of CRSBP-1-associated intracellular fibrillar structures in a time-dependent manner in LECs.
To determine the functional significance of the association of CRSBP-1 with the fibrillar structures, we determined the effects of CRSBP-1 ligands (VEGF peptide and HA) on the CRSBP-1-associated fibrillar structures in SVEC4–10 cells. VEGF peptide, a 25-mer peptide containing the CRS motif of VEGF-A165, is a specific CRSBP-1 ligand which does not interact with VEGFR2 [1]. The cells were treated with VEGF peptide (2 μM) or HA (100 μg/ml). After 2 h at 37°C, the cells were fixed with methanol at −20°C for 10 min and stained with anti-CRSBP-1 serum followed by FITC-conjugated goat anti-rabbit IgG. In cells treated with VEGF peptide (Fig. 2), the fibrillar structures became contracted as compared with the same structures in cells treated with vehicle only (Fig. 2, panel D vs. panel C). HA, also a ligand for CRSBP-1, exhibited moderate effects on the morphology of SVEC4–10 cells, causing them to assume a spindle (fibroblast-like) appearance which was due to changes in the CRSBP-1-asscoiated fibrillar structures (Fig. 2, panel E). However, the effects of both HA and VEGF peptide were abolished when cells were co-treated with HA and VEGF peptide (Fig. 2, panel F). The mechanism for this effect of co-treatment with HA and VEGF peptide is unknown, but VEGF peptide is known not to affect HA binding to CRSBP-1 [9]. This is likely due to an allosteric effect. The HA and CRS motif binding sites are located at distinct sites: the Link module (amino acid residues 54–130) and the acidic-amino-acid-rich region (amino acid residues 204–229), respectively, in human CRSBP-1 [18].
Fig. 2. CRSBP-1 ligands stimulate contraction of CRSBP-1-associated fibrillar structures in SVEC4–10 cells.
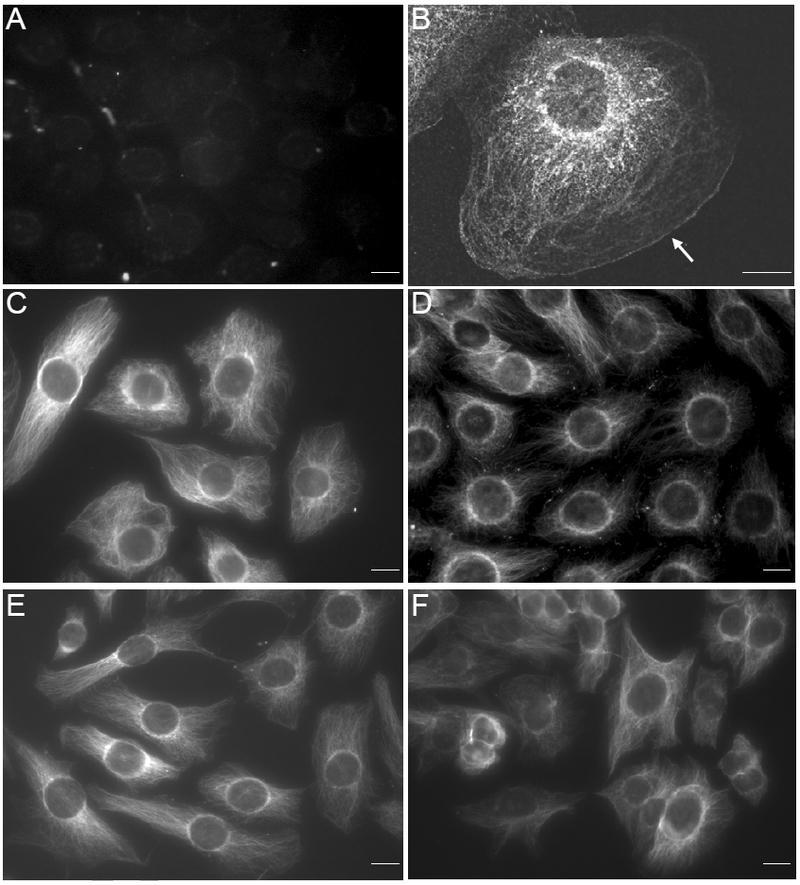
Cells were seeded on Petri dishes (panel B) and cover slips (panels A, C, D, E and F). Cells were treated with vehicle only (panels A, B and C), VEGF-A peptide (10 μM) (panel D), HA (100 μg/ml) (panel E) or HA (100 μg/ml) plus VEGF-A pep tid e (1 0 μM) (panel F). After 2 h at 37°C, cells were fixed with paraformaldehyde and then permeabilized by acetone (panel B) or fixed and permeabilized with methanol at −20°C for 10 min (panels A, C, D, E and F). CRSBP-1 was stained with anti-CRSBP-1 antiserum (panels B, C, D, E and F) or preimmune serum panel (panel A) followed by FITC-conjugated goat anti-rabbit IgG and visualized with a fluorescence microscope. The bar indicates a scale of 10 μm (panel A). The arrow in (panel B) indicates the edge of the plasma membrane. Control staining (with preimmune serum) of cells treated with CRSBP-1 ligands were negative like (panel A) (data not shown). Data are representative of five independent experiments.
To determine the functional significance of the contraction of CRSBP-1-associated fibrillar structures in SVEC4–10 cells, we examined the time dependence of the effects of CRSBP-1 ligands (PDGF peptide, VEGF peptide and HA) on CRSBP-1-associated fibrillar structures. PDGF peptide, a 19-mer peptide containing the CRS motif of PDGF-BB, is a specific CRSBP-1 ligand and does not interact with the PDGF β-type receptor (PDGFβR) [1,9]. SVEC4–10 cells were treated with PDGF peptide (10 μM), VEGF peptide (10 μM), or HA (100 μg/ml). After 0 h, 0.5 h, 1 h, and 2 h at 37°C, the cells were fixed with methanol at −20°C for 20 min and stained with rabbit antiserum to CRSBP-1 and rhodamine red-conjugated goat anti-rabbit IgG. The CRSBP-1-associated fibrillar structures appeared to be relatively stable and did not show any morphological changes after incubation with vehicle only at 37 °C for 2 h (Fig. 3, panel D vs. panel A). However, PDGF peptide, VEGF peptide and HA all stimulated maximal contraction of the fibrillar structures in SVEC4–10 cells after a 30-min incubation at 37 °C (Fig. 3, panels, F, J and N, respectively). CRSBP-1 ligand-stimulated contraction of the CRSBP-1-associated fibrillar structures was then quantified. As shown in Fig. 4 (panel A), PDGF peptide at 10 μM for 0.5 h stimulated contraction of the fibrillar structures by ~70%. Treatments with VEGF peptide (10 μM) and HA (100 μg/ml) for 0.5 h caused contraction of the fibrillar structures by ~ 40% and ~30%, respectively (Fig. 4, panels B and C).
Fig. 3. CRSBP-1 ligands stimulate contraction of CRSBP-1-associated fibrillar structures in a time-dependent manner in SVEC4–10 cells.
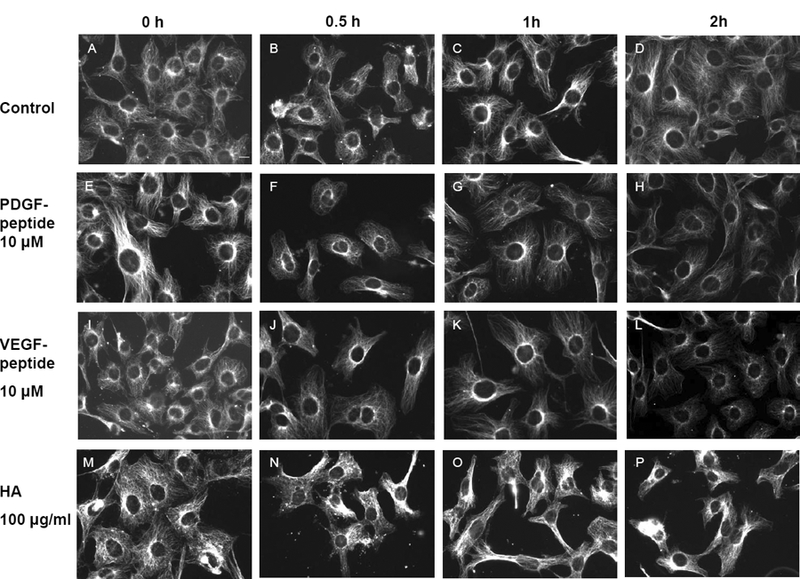
Cells were seeded on coverslips and treated with vehicle only (panels A to D), 10 μM PDGF peptide (panels E to H), 10 μM VEGF peptide (panels I to L), or 100 μg/ml HA (panels M to P) for 0 min, 0.5 h, 1 h, and 2 h. After incubation, cells were fixed with methanol at −20ºC for 10 min. Cells were stained with anti-CRSBP-1 serum followed by rhodoamine red-conjugated goat anti-rabbit IgG and visualized with a fluorescence microscope. The bar indicates a scale of 10 μm (panel A). Data are representative of four independent experiments.
Fig. 4. PDGF peptide, VEGF peptide and HA quantitatively stimulate contraction of CRSBP-1-associated fibrillar structures in SVEC4–10 cells.
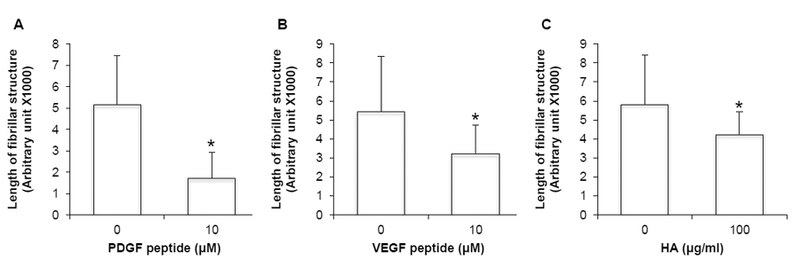
Cells were stimulated with vehicle only, 10 μM PDGF peptide (panel A), 10 μM VEGF peptide (panel B) and 100 μg/ml HA (panel C). Lengths of the CRSBP-1-associated fibrillar structures shown in Fig. 3 were quantified using the program NIH Image J. A minimum of 25 cells were examined for each experimental group. The same threshold was used for analysis in each group. The bar represents mean ± S.D. * Significantly lower then the vehicle control (ρ < 0.05). Data are representative of three independent experiments.
CRSBP-1 ligands stimulate contraction of the CRSBP-1-associated ER network in LECs.
Since immunostaining of CRSBP-1 does not colocalize with that of any of the known cytoskeleton proteins and since the CRSBP-1-associated fibrillar structures include the perinuclear membrane which is known to be associated with the endoplasmic reticulum (ER) network, we hypothesized that CRSBP-1-associated fibrillar structures are identical to the ER network. To test this hypothesis, we determined the co-localization of CRSBP-1 and BiP, which is a heat shock protein and a specific marker for the ER [14], using indirect immunofluorescence staining and confocal microscopy. As shown in Fig. 5A, the CRSBP-1-associated fibrillar structures co-localize with the staining pattern of BiP (panels A, B and C). While the immunostaining pattern of BiP lacked the plasma-membrane distribution, the intracellular staining pattern of BiP was completely identical to that of CRSBP-1 (panels C, F, I, L and O). To determine the effects of CRSBP-1 ligands on the CRSBP-1-associated ER network, SVEC4–10 cells were stimulated with 10 μM VEGF peptide, 10 μM PDGF peptide, 5 nM VEGF-A165, and 100 μg/ml HA. As shown in Fig. 5A, VEGF peptide (panels D, E and F), PDGF peptide (panels G, H and I), VEGF-A165 (panels J, K and L), and HA (panels M, N and O) stimulated contraction of the CRSBP-1-associated ER network when compared to vehicle only-treatment (panels A, B and C). VEGF-A165 is a putative physiological CRSBP-1 ligand for LECs which express very little endogenous VEGFR2, a specific receptor for VEGF-A165 in blood vascular endothelial cells [5,19,20]. These results suggest that CRSBP-1 ligands are capable of stimulating contraction of the CRSBP-1-associated ER network in SVEC4–10 cells.
Fig. 5. CRSBP-1 ligands stimulate contraction of the CRSBP-1-associated ER network in SVEC4–10 cells (A) and HDLECs (B).
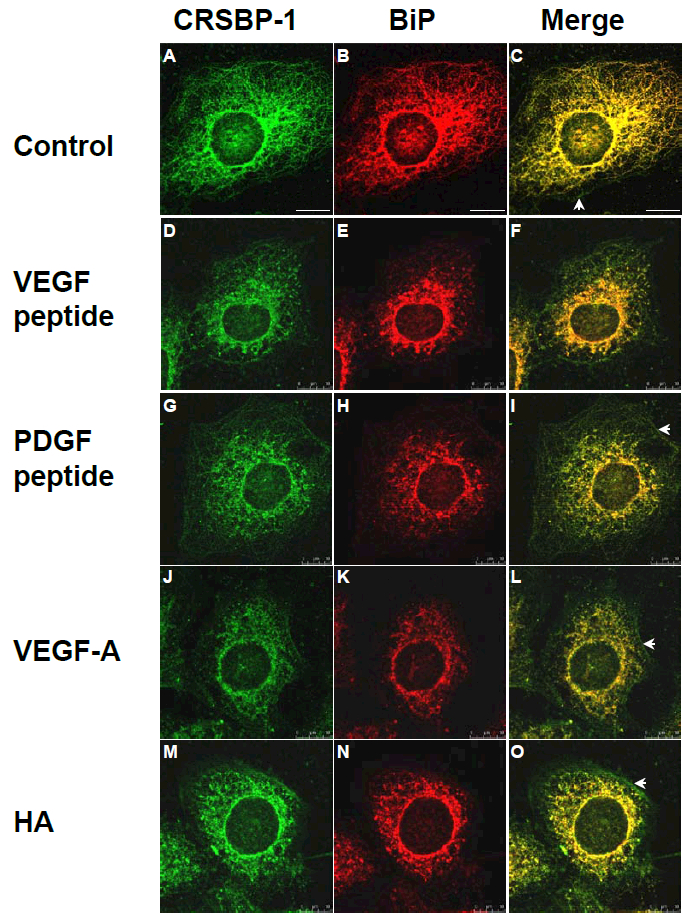
(A)SVEC4–10 cells were seeded on coverslips and treated with vehicle only (panels A, B and C), 10 μM VEGF peptide (panels D, E and F), 10 μM PDGF peptide (panels G, H and I), 100 ng/ml VEGF-A165 (panels J, K and L) or 100 μg/ml HA (panels M, N and O) for 1 h. After incubation, cells were fixed with methanol at −20°C for 10 min. Cells were stained with anti-CRSBP-1 serum (panels A, D, G, J and M) and anti-BiP (the ER marker) antibody (panels B, E, H, K and N), then visualized with a confocal microscope. Cells treated with vehicle only and CRSBP-1 ligands showed intracellular co-localization of CRSBP-1 and BiP (panels C, F, I, L and O). The bar indicates a scale of 10 μm (panel A). CRSBP-1 ligand treatment induced contraction of the CRSBP-1-associated ER network in SVEC4–10 cells. Arrowheads indicate the plasma membrane localization of CRSBP-1. Data are representative of four independent experiments.
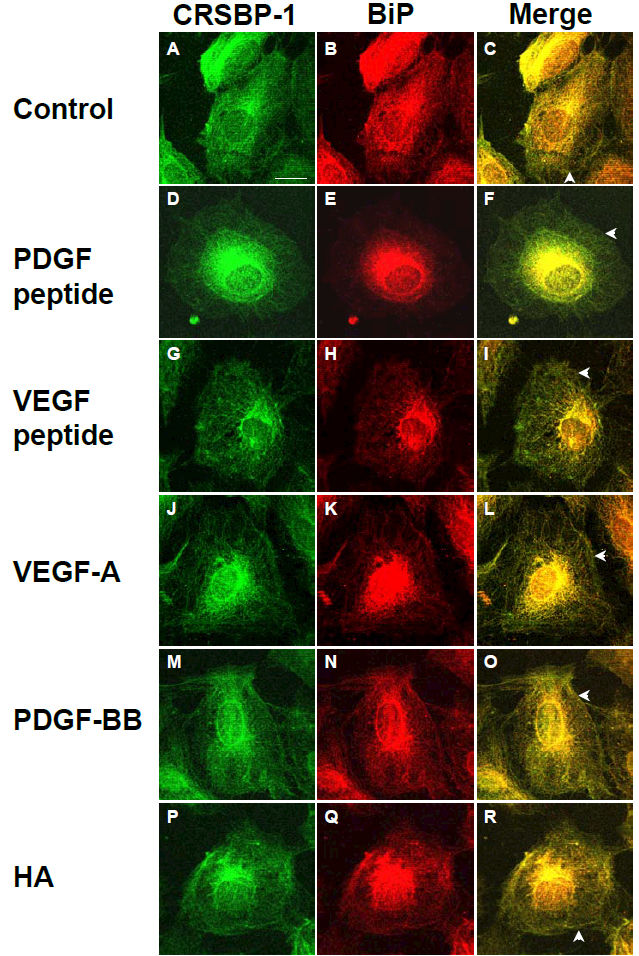
(B)HDLECs were seeded on coverslips and treated with vehicle only (control) (panels A, B and C), 10 μM PDGF peptide (panels D, E and F), 10 μM VEGF peptide (panels G, H and I), 100 ng/ml VEGF-A165 (panels J, K and L), 50 ng/ml PDGF-BB (panels M, N and O) or 100 μg/ml HA (panels P, Q and R) for 1 h. After incubation, cells were fixed with methanol at −20°C for 10 min. Cells were stained with anti-CRSBP-1 serum (panels A, D, G, J, M and P) and anti-BiP (ER marker) antibody (panels B, E, H, K, N and Q) and visualized with a confocal microscope. Cells treated with vehicle only and the same agents showed intracellular co-localization of CRSBP-1 and BiP (panels C, F, I, L, O and R). PDGF peptide, VEGF peptide, VEGF-A, PDGF-BB and HA treatment stimulated contraction of the CRSBP-1-associated ER network in SVEC4–10 cells (panels F, I, L, O and R). The bar indicates a scale of 10 μm (panel A). Arrowheads indicate the plasma membrane distribution of CRSBP-1. Data are representative of four independent experiments.
As described above, SVEC4–10 cells are SV40-transformed LECs [16]. To determine if CRSBP-1 ligand-stimulated effects observed in SVEC4–10 cells also occur in normal LECs, we performed the same set of experiments using primary-culture HDLECs isolated from human skin (PromoCell C-12216). HDLECs grown on coverslips were stimulated with 10 μM VEGF peptide, 10 μM PDGF peptide, 5 nM VEGF-A165, 2 nM PDGF-BB, or 100 μg/ml HA. Indirect immunofluorescence staining of stimulated cells using anti-CRSBP-1 serum and antibody to BiP followed by confocal microscopy analysis revealed intracellular co-localization of CRSBP-1 with BiP in the ER network of HDLECs as shown in Fig. 5B. As seen in SVEC4–10 cells, CRSBP-1 ligands stimulated contraction of the CRSBP-1-associated ER network in HDLECs (panels F, I, L, O and R). PDGF-BB (panels M, N and O) and HA (panels P, Q and R) caused less contraction of the CRSBP-1-associated ER network in HDLECs than PDGF peptide (panels D, E and F), VEGF peptide (panels G, H and I) or VEGF-A165 (panels J, K and L). VEGF-A165 contains the putative CRS motif [1,5,19,20]. These results suggest that CRSBP-1 is localized to the plasma membrane and the ER network in mouse and human LECs and that CRSBP-1 ligands stimulate contraction of the CRSBP-1-associated ER network in both cell types.
CRSBP-1 ligands stimulate contraction of the CRSBP-1-associated ER network in a CRSBP-1-dependent manner in SVEC4–10 cells.
To test the involvement of CRSBP-1 in ligand-stimulated contraction of the ER network, SVEC4–10 cells were transfected with control siRNA or CRSBP-1 siRNA as described previously [12]. These transfected cells were then treated with 10 μM of PDGF peptide or VEGF peptide for 1 h and then analyzed by indirect immunofluorescence staining using antiserum to CRSBP-1 and antibody to BiP. As shown in Fig. 6, cells transfected with control siRNA, like untransfected cells, exhibited responses to ligand-stimulated contraction of the CRSBP-1-associated ER network (panels D, E, F and panels G, H, I vs. panels A, B, C). However, down-regulation of CRSBP-1 expression by transfection of cells with CRSBP-1 siRNA abolished such ligand-stimulated contraction (Fig. 6, panels M, N, O and P, Q, R vs. panels J, K, L). Under the stated conditions [12], at 48 hours after CRSBP-1 siRNA transfection, CRSBP-1 protein levels were down-regulated by ~80% compared with levels in cells transfected with control siRNA. Interestingly, down-regulation of CRSBP-1 proteins by CRSBP-1 siRNA transfection also led to constitutive contraction of the ER network (panels J to R vs. panels A, B and C). These results suggest that CRSBP-1 is required for ligand-stimulated contraction of the ER network in SVEC4–10 cells. These results are also comparable to our recent findings that CRSBP-1 siRNA transfection abolishes ligand-stimulated permeability in SVEC4–10 cell monolayers and that down-regulation of CRSBP-1 protein by CRSBP-1 siRNA transfection leads to constitutively increased permeability in these cells [12].
Fig. 6. CRSBP-1 ligands stimulate contraction of the CRSBP-1-associated ER network in SVEC4–10 cells in a CRSBP-1-dependent manner.
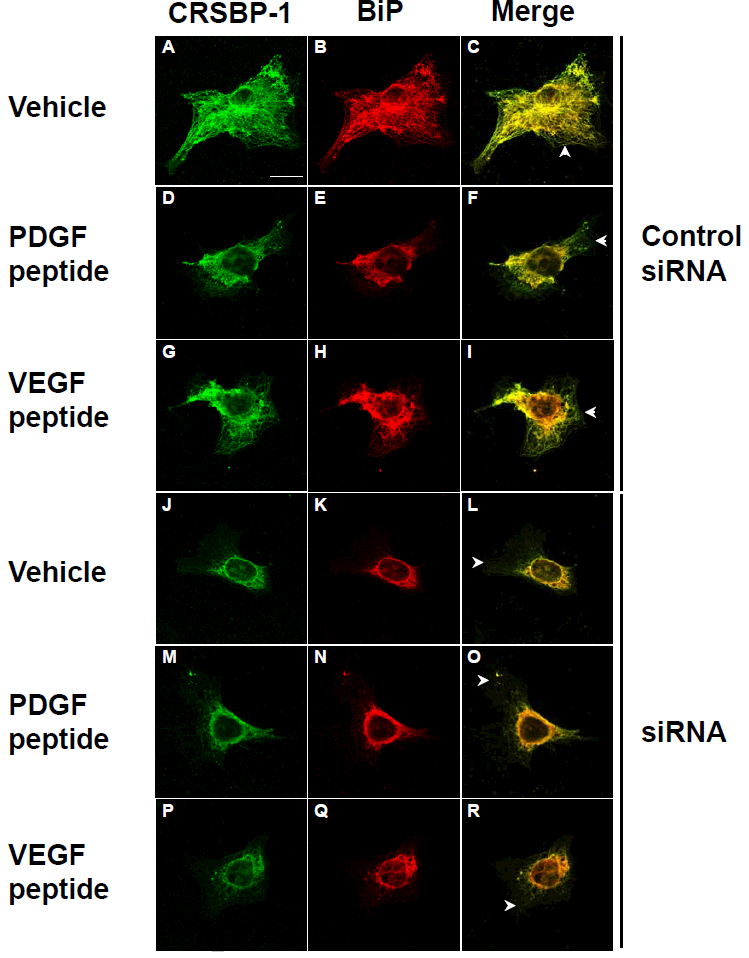
Cells transfected with control siRNA (panels A to I) and CRSBP-1 siRNA (panels J to R) were stimulated with vehicle only (panels A, B, C and panels J, K, L), 10 μM PDGF peptide (panels D, E, F and panels M, N, O) and 10 μM VEGF A peptide (panels G, H, I and panels P, Q, R) for 1 h. After stimulation, cells were fixed with methanol at −20°C for 10 min and stained with anti-CRSBP-1 serum (panels A, D, G, J, M and P) and anti-BiP (the ER marker) antibody (panels B, E, H, K, N and Q), then visualized with a confocal microscope. Cells treated with vehicle only and CRSBP-1 ligands showed intracellular co-localization of CRSBP-1 and BiP (panels C, F, I, L, O and R). As described previously (12), about 80% of CRSBP-1 protein was down-regulated by transfection of SVEC4–10 cells with CRSBP-1 siRNA compared with those of cells transfected with control siRNA. CRSBP-1 ligands stimulated contraction of the ER network in cells transfected with control siRNA (panels D to I vs panels A, B and C). Down regulation of CRSBP-1 protein by CRSBP-1 siRNA transfection abolished CRSBP-1 ligand-stimulated contraction of the ER network in these cells (panels M to R vs. panels J, K and L). Cells transfected with CRSBP-1 siRNA exhibited constitutive contraction of the ER network (panels J to R vs. panels A, B and C). Arrowheads indicate the plasma membrane localization of CRSBP-1.
Paclitaxel blocks ligand-stimulated contraction of the CRSBP-1-associated ER network in SVEC4–10 cells.
Rearrangement (polymerization and de-polymerization) of cytoskeletal proteins (e.g. microtubules) is usually involved in cellular contraction. It has been shown that contraction of the ER fibrillar network is associated with contraction of microtubules [21–24]. We hypothesize that CRSBP-1 ligand-induced contraction of the ER network is associated with CRSBP-1 ligand-stimulated cellular contraction in LECs. To test this, we determined the effect of paclitaxel [25], a microtubule polymer stabilizer, on CRSBP-1 ligand-induced ER network contraction in SVEC4–10 cells. Cells were pretreated with paclitaxel or vehicle only at 37C for 1 h and then stimulated with 10 μM PDGF peptide or VEGF peptide for 1 h. These stimulated cells were then analyzed by indirect immunofluorescence staining using antibodies to CRSBP-1 and BiP. As shown in Fig. 7, paclitaxel blocked the CRSBP-1 ligand-induced contraction of the ER network in these cells (panels M, N, O and panels P, Q, R vs. panels D, E, F and panels G, H, I). Paclitaxel alone did not affect the ER network (panels J, K and L vs. panels A, B and C). These results suggest that PDGF peptide and VEGF peptide stimulate contraction of the ER network in SVEC4–10 cells in a paclitaxel-sensitive manner.
Fig. 7. Paclitaxel blocks CRSBP-1 ligand-induced contraction of CRSBP-1-associated fibrillar structures in SVEC4–10 cells.
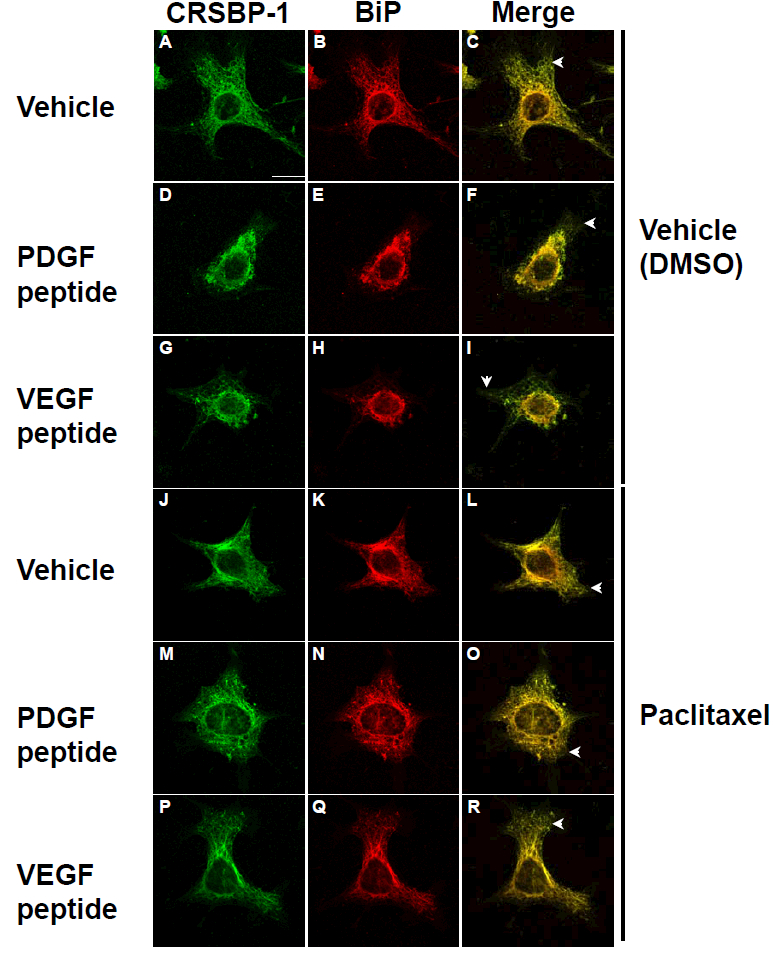
Cells were pretreated with vehicle only (DMSO) (panels A to I), 5 μM paclitaxel (panels J to R) for 1 h and then stimulated with vehicle only (panels A, B, C and panels J, K, L), 10 μM PDGF peptide (panels D, E, F and M, N, O) and 10 μM VEGF peptide (panels G, H, I and panels P, Q, R). After stimulation for 1 h, cells were fixed with methanol at −20°C for 10 min and stained with anti-CRSBP-1 serum and antibody to BiP. The bar indicates a scale of 10 μm (panel A). Arrowheads indicate the plasma membrane localization of CRSBP-1.
Discussion
The subcellular localization of CRSBP-1 in the plasma membrane and ER network suggests a unique property of CRSBP-1. Many type I membrane proteins are mainly localized in the plasma membrane and are not detected in the ER even though they traffic through it to the plasma membrane after synthesis. The ER retention mechanism of CRSBP-1 is unknown. CRSBP-1 does not contain the C-terminal di-lysine or di-arginine motif which has been shown to mediate ER retention of type I membrane proteins [26]. However, it contains a basic amino acid-rich motif (KSPSKTTVR) near its C-terminus. This motif is well conserved in all known mammalian CRSBP-1 congeners and may play a role in the ER localization of mammalian CRSBP-1 congeners (human, bovine and mouse). The functional significance of ER localization of CRSBP-1 in ligand-stimulated contraction of LECs is not known. Several lines of evidence indicate that ER localization of CRSBP-1 plays an important role in CRSBP-1 ligand-stimulated LEC contraction. These include: 1) CRSBP-1 ligands are unable to stimulate detectable contraction in SSV-transformed cells and H1299/CRSBP-1 cells, both of which express CRSBP-1 but do not exhibit the ER localization of CRSBP-1 [6,9]. 2) A specific kinase activity inhibitor of PDGFβR, which is a signaling partner of CRSBP-1, inhibits CRSBP-1 ligand-stimulated contraction in LECs [12]. This suggests that CRSBP-1 ligand-stimulated contraction of the CRSBP-1-associated ER network is a downstream effect of the CRSBP-1-PDGFβR-mediated signaling [12]. and 3) Contraction of the ER network is known to be associated with morphological changes in cells [23,24]. The localization and abundance of CRSBP-1 at both the plasma membrane and ER may explain why lymphatic endothelium in tissues can be strongly stained histochemically by anti-CRSBP-1 antibody.
The CRSBP-1 ligand-induced contraction of the ER network appears to be mediated by microtubules as evidenced by the observation that paclitaxel, a microtubule polymer stabilizer, effectively blocks CRSBP-1 ligand-induced contraction of the ER network. Since microtubule contraction is commonly associated with morphological changes in cells [21–24], we predicted that CRSBP-1 ligand-induced contraction of the ER network in SVEC4–10 cells and HDLECs would result in morphological changes in these cells. As predicted, we found that CRSBP-1 ligand stimulation does result in morphological (opening of intercellular junctions) and functional changes (increased permeability) at intercellular junctions in these LEC monolayers. The finding of paclitaxel as a potent inhibitor for CRSBP-1 ligand-stimulated contraction of CRSBP-1 ligand-associated ER network (and likely for CRSBP-1 ligand-stimulated opening of lymphatic intercellular junctionsas well) [12] has potentially important clinical implications. Physiological CRSBP-1 ligands (e.g., PDGF-BB) are hypothesized to be utilized by carcinoma cells in the interstitial space of tissues to enter lymphatic vessel lumens by inducing opening of lymphatic intercellular junctions during lymphatic metastasis. Paclitaxel and related compounds may prevent lymphatic metastasis in certain human cancers (e.g., lung cancer) [27] by blocking CRSBP-1 ligand-induced opening of lymphatic intercellular junctions.
Johnson et al. [13] demonstrated that most of the LYVE-1 (or CRSBP-1) intracellular staining is lost upon stimulation of HDLECs with TNF-α. This raises the possibility that CRSBP-1 ligands might induce contraction (or shortening) of CRSBP-1-asosciated ER network by down-regulating the cell-surface and intracellular levels of CRSBP-1, by analogy to the effect of TNF-α. However, we found that treatment with CRSBP-1 ligands does not diminish the cell-surface and total protein levels of CRSBP-1 in SVEC4–10 cells [28]. In contrast, TNF-α completely down-regulates cell-surface expression of CRSBP-1 in SVEC4–10 cells [28], as reported by Johnson et al. in HDLECs [13]. These results suggest that the effects of CRSBP-ligands on CRSBP-1/LYVE-1 expression are distinct from those of TNF-α in LECs. Here we demonstrate that down-regulation of CRBP-1 protein by transfection with CRSBP-1 siRNA abolishes lignd-stimulated contraction of the ER network in SVEC4–10 cells and that these cells transfected with CRSBP-1 siRNA exhibit constitutive contraction of the ER network. Together with previous observations [10,12], these results suggest that CRSBP-1 ligands stimulate contraction of the ER network and LECs in a CRSBP-1-dependent manner, resulting in opening of lymphatic intercellular junctions. This leads to increased permeability in LEC monolayers in vitro and increased interstitial–lymphatic transit in whole animals. Down-reulation of CRSBP-1 protein by transfection of cells with CRSBP-1 siRNA and CRSBP-1-null mutation in mice cause constitutive contraction of LECs and constitutively increased permeability in LEC monolayers, and formation of distended lymphatic vessel lumens (opening) and constitutive increase in interstitial-lymphatic transit in whole animals, respectively [10,12]. LYVE-1(CRSBP-1) is involved in the formation of VE-cadherin-mediated “button”-like structures at lymphatic intercellular junctions [11]. These results also support the hypothesis that CRSBP-1 siRNA transfection/CRSBP-1-null mutation and binding of CRSBP-1 by its cognate ligands result in loss and “unbuttoning” of VE-cadherin-mediated buttons, respectively, leading to opening of lymphatic intercellular junctions [10,12].
FEBS_35272.
CRSBP-1 ligands induce opening of lymphatic intercellular junctions.
We examine the mechanism by which CRSBP-1 ligands exert such activity.
CRSBP-1 is localized to the plasma membrane and ER network in endothelial cells.
CRSBP-1 ligands stimulate contraction of the ER network in a Taxol-sentive manner.
Ligand-induced ER contraction is associated with ligand-induced cell contraction.
Highlights.
CRSBP- 1 localizes to the plasma membrane as well as intracellular fibrillar structures in lymphatic endothelial cells (LECs).
The CRSBP-1-associated intracellular fibrillar structures are identical to the ER network as evidenced by co-localization with BiP, an ER protein.
CRSBP-1 ligands stimulate contraction of the CRSBP-1-associated ER network in LECs.
Acknowledgments
We thank Frank E. Johnson, M.D. for critically reviewing the manuscript and Thomas Heyduk, Ph.D. for providing assistance in confocal fluorescence microscopy. This work was supported by NIH grants HL 087463–01 and DK 078438 to Auxagen and AA 019233 to St. Louis University (JSH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimer sthat apply to the journal pertain.
Conflict of Interest
Jung San Huang and Shuan Shian Huang had equity positions in Auxagen, Inc. when the research was performed. Part of the research was supported by NIH grants awarded to Auxagen, Inc. (HL 087463–01 and DK 078438
References
- 1.Boensch C, Kuo MD, Connolly DT, Huang SS, and Huang JS (1995). Identification, purification and characterization of cell-surface retention sequence-binding proteins from human SK-Hep cells and bovine liver plasma membranes. J. Biol. Chem 270, 1807–1816. [DOI] [PubMed] [Google Scholar]
- 2.LaRochelle et al. , 1991; LaRochelle WJ, May-Siroff M, Robbins KC, and Aaronson SA (1991) A novel mechanism regulating growth factor association with the cell surface: identification of a PDGF retention domain. Genes Dev 5, 1191–1199. [DOI] [PubMed] [Google Scholar]
- 3.Ostman A, Andersson M, Betsholtz C, Westermark B, and Heldin C-H (1991) Identification of a cell retention signal in the B-chain of platelet-derived growth factor and in the long splice version of the A-chain. Cell Regul 2, 503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joukov V, Sorsa T, Kumar V, Jeltsch M, Claesson-Welsh L, Cao Y, Saksel O, Kalkkinen N, and Alitalo K (1997) Proteolytic processing regulates receptor specificity and activity of VEGF-C. EMBO J 16, 3898–3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carmeliet P, Ng YS, Nuyens D, Theilmeier G, Brusselmans K, Cornelissen I, Ehler E, Kakkar VV, Stalmans I, Mattot V, Nuyens D, Lutgens E, Clotman F, de Ruiter MC, Gittenberger-de Groot A, Poelmann R, Lupu F, Herbert JM, Collen D, and Dejana E (1999) Impaired myocardial angiogenesis and ischemic cardiomyopathy in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Nat. Med 5, 495–502. [DOI] [PubMed] [Google Scholar]
- 6.Boensch C, Huang SS, Connolly DT, and Huang JS (1999) Cell surface retention sequence binding protein-1 interacts with the v-sis gene product and platelet-derived growth factor beta-type receptor in simian sarcoma virus-transformed cells. J. Biol. Chem 274, 10582–10589. [DOI] [PubMed] [Google Scholar]
- 7.Banerji S, Ni J, Wang SX, Clasper S, Su J, Tammi R, Jones M, and Jackson DG (1999) LYVE-1, a new homologue of the CD44 glycoprotein, is a lymph-specific receptor for hyaluronan. J. Cell Biol 144, 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prevo R, Banerji S, Ferguson DJ, Clasper S, and Jackson DG (2001) Mouse LYVE-1 is an endocytic receptor for hyaluronan in lymphatic endothelium. J. Biol. Chem 276, 19420–19430. [DOI] [PubMed] [Google Scholar]
- 9.Huang SS, Tang FM, Huang YH, Liu I-H, Hsu SC, Chen ST, and Huang JS (2003) Cloning, expression, characterization, and role in autocrine cell growth of cell surface retention sequence binding protein-1. J. Biol. Chem 278, 43855–43869. [DOI] [PubMed] [Google Scholar]
- 10.Huang SS, Liu I-H, Smith T, Shah MR, Johnson FE, and Huang JS (2006) CRSBP-1/LYVE-1-null mice exhibited identifiable morphological and functional alterations of lymphatic capillary vessels. FEBS Letters, 580, 6259–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baluk P, Fuxe J, Hashizume H, Romano T, Lashnits E, Butz S, Vestweber D, Corada M, Molendini C, Dejana E, and McDonald DM (2007). Functionally specialized junctions between endothelial cells of lymphatic vessels. J. Exp. Med 204, 2349–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hou W-H, Liu I-H, Tsai CC, Johnson FE, Huang SS, and Huang JS (2011). CRSBP-1/LYVE-1 Ligands induce disruption of lymphatic intercellular adhesion by inducing tyrosine phosphorylation and internalization of VE-cadherin. J. Cell Sci 124, 1231–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson LA, Prevo R, Clasper S, and Jackson DG (2007) Inflammation-induced uptake and degradation of the lymphatic endothelial hyaluronan receptor LYVE-1. J. Biol. Chem, 282, 33671–33680. [DOI] [PubMed] [Google Scholar]
- 14.Shen J, Chen X, Hendershot L, and Prywes R (2002) ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 3, 99–111. [DOI] [PubMed] [Google Scholar]
- 15., Gavard J, and Gutkind JS (2006) VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat. Cell Biol 8, 1223–1234. [DOI] [PubMed] [Google Scholar]
- 16.O’Connell KA, and Edidin M (1990) A mouse lymphoid endothelial cell line immortalized by simian virus 40 binds lymphocytes and retains functional characteristics of normal endothelial cells. J. Immunol 144, 521–525. [PubMed] [Google Scholar]
- 17.Kato S, Shimoda H, Ji RC, and Miura M (2006) Lymphangiogenesis and expression of specific molecules as lymphatic endothelial cell markers. Anat. Sci. Int 81, 71–83. [DOI] [PubMed] [Google Scholar]
- 18.Chen W-H, Tseng W-F, Lin G-H, Huang FW, Schreiner, Chen H-R, Voigt M-M, Yuh C-H, Wu J-L, Huang SS, and Huang JS (2012). The ortholog of LYVE-1 is required for thoracic duct formation in zebrafish Submitted for publication. [Google Scholar]
- 19.Houck KA, Ferrara N, Winer J, Cachianes G, Li B, and Leung DW (1991) The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol. Endocrinol 5, 1806–1814. [DOI] [PubMed] [Google Scholar]
- 20.Ferrara N (2004) Vascular endothelial growth factor: basic science and clinical progress. Endocr. Rev 25, 581–611. [DOI] [PubMed] [Google Scholar]
- 21.Lee C, and Chen LB (1988) Dynamic behavior of endoplasmic reticulum in living cells. Cell 54, 37–46. [DOI] [PubMed] [Google Scholar]
- 22.Allan VJ, and Vale RD (1991) Cell cycle control of microtubule-based membrane transport and tubule formation in vitro. J. Cell Biol 113, 347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Terasaki M, Chen LB, and Fujiwara K (1986) Microtubules and the endoplasmic reticulum are highly interdependent structures. J. Cell Biol 103, 1557–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Waterman-Storer CM, and Salmon ED (1998) Endoplasmic reticulum membrane tubules are distributed by microtubules in living cells using three distinct mechanisms. Curr. Biol 8, 798–806 [DOI] [PubMed] [Google Scholar]
- 25.Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, and Woods CM (1995) Epothilones, a new class of microtubule-stabilizing agents with a taxol-like mechanism of action. Cancer Res 55, 2325–2333. [PubMed] [Google Scholar]
- 26.Pagny S, Lerouge P, Faye L, and Gomord V (1999) Signals and mechanisms for protein retention in the endoplasmic reticulum. J. Exp. Botany 50, 157–164. [Google Scholar]
- 27.Liu J, Meisner D, Kwong E, Wu XY, and Johnston MR 2009. Translymphatic chemotherapy by intrapleural placement of gelatin sponge containing biodegradable paclitaxel colloids controls lymphatic metastasis in lung cancer. Cancer Res. 69, 1174–1181. [DOI] [PubMed] [Google Scholar]
- 28.Chen Hsiao-Rong (2009) Role of CRSBP-1 in regulation of cell migration in lymphatic endothelial cells and its orthologue in zebrafish Master thesis, Institute of Systems Biology and Bioinformatics, National Central University, Choungli, Taiwan. [Google Scholar]