

Abstract
Genetic variations in complement factor H (CFH) confer greater risk for age-related macular degeneration (AMD). In this issue of Immunity, Calippe et al. (2017) uncover a non-canonical role for CFH in the inhibition of mononuclear phagocyte elimination from sub-retinal lesions, providing insight into the pathophysiology of AMD associated with CFH variants.
Age-related macular degeneration (AMD) is a major source of vision loss in the aging human population. It is estimated that by the year 2020, close to 3 million people in the US alone will be legally blind because of AMD-related lesions (Friedman et al., 2004). AMD shows a strong genetic association with an allelic variant of complement (inhibitory) factor H, CFH(Y402H). Complement-inhibiting drugs are being developed and tested on the premise that complement activation underlies AMD pathology. Calippe et al. (2017) now provide new insight into the role of CFH in AMD that is as surprising as it is compelling, revealing that the role of CFH in AMD is non-canonical and separate from the complement pathway.
The macula is the central focal region of the posterior retina located directly opposite the pupil and responsible for high-resolution, detailed central vision. It is thought that the higher intensity of illumination that this area receives causes free radical formation and oxidative damage, which necessitates a high rate of repair. With age, dysregulation of tissue repair processes leads to formation of structures known as “drusen,” which appear as multiple white dots of varying sizes located between the retinal pigment epithelial (RPE) layer and Bruch’s membrane. Drusen contain cellular debris and secreted serum proteins such as complement, amyloid, immunoglobulin, apolipoprotein, and others. Phagocytic cells, believed to represent the “cleanup crew,” composed of resident microglia (MC) as well as recruited mononuclear phagocytes (MP), accumulate in the area, but appear to be unable to effectively clear the offending drusen material. Gradually, a state of low-grade chronic inflammation, known as para-inflammation, sets in, starting from the macular region and spreading radially to other parts of the posterior retina. Although descriptive studies abound, there is little mechanistic understanding of the processes involved in AMD.
The now classic genome-wide association study (GWAS) in AMD patients by Klein et al. (2005), and several subsequent studies (reviewed by Zhang and Baird, 2016), reported an increased risk of the disease in bearers of a SNP in the CFH gene that substitutes tyrosine with histidine at amino-acid position 402 (Y402H). Subsequent attention focused on the known canonical function of CFH in the alternative complement pathway. It was reported that the risk variant CFH(Y402H) decreased the inhibitory activity of CFH on the complement pathway, leading to hyperactivity and increased levels of complement components C3b, C3a, C5a, and C5b-C9 (Ambati et al., 2013). Not surprisingly, this led to the conclusion, which since then has become widely accepted, that poor control of complement activation by this CFH variant is a causative element in AMD pathogenesis.
Using two different mouse models of AMD (Cx3cr1−/− mice and TRE2 humanized transgenic mice) Calippe et al. (2017) dispel this notion, demonstrating a different role for CFH in AMD pathogenesis. First, they show that genetic deficiency of Cfh diminishes rather than aggravates AMD parameters, in contrast to what would be expected if its action were via complement inhibition. Deficiency of Cfh prevented age-associated progressive accumulation of MP and MC in the sub-retinal space and protected the macular region of the retina from loss of the light-sensing photoreceptor cells. The data supported the notion that (1) CFH acted locally in the eye and (2) its action was not mediated by complement activation either systemically or locally. Namely, restoration of C3 levels in Cfh−/− mice by liver-specific expression of CFH did not restore the AMD phenotype and Cfh sufficiency of the recipient did not alter retention of (adoptively transferred) MP/MC in the subretinal space. In addition, subretinal administration of neutralizing anti- C3b/iC3b/C3c antibody did not affect clearance of Cfh-sufficient MCs. The source of CFH protein had to be intrinsic, coming from the MPs and MCs themselves, as shown by the finding that transferred donor microglial cells that lack Cfh were cleared from the sub-retinal space faster than control Cfh-sufficient microglia. Thus, the contribution of CFH risk allele to AMD pathology could not be due to lack of conversion of C3b to iC3b and consequent inhibition of complement activity, but to a function of CFH outside the complement pathway.
Based on these findings, Calippe et al. (2017) explore non-canonical roles for CFH in AMD pathogenesis. CD11b (also known as CD18) is the natural receptor for CFH, and it is heavily expressed on the cell membranes of monocytes, macrophages, and microglial cells. Because their data indicated an intrinsic role of CFH, the authors investigated the possibility that CFH acts in an autocrine fashion by binding to its receptor, rather than through the complement pathway. Indeed, blocking of CD11b molecule on microglial cell surface by anti-CD11b antibody before injecting these cells into the sub-retinal space accelerated the process of clearance of MP and MC from the site of injection. Because the role of C3b (which also shares CD11b as its receptor) had been ruled out previously, it remained that CFH might delay elimination of accumulated MP and MC from AMD lesions by binding to CD11b.
Previous studies (Pfeiffer et al., 2001) had shown that CD11b on the cell membrane co-localizes with CD47, an integrin-associated protein whose ligation can promote cell death and clearance of monocytes and macrophages (Oldenborg, 2013; Poon et al., 2014). Based on these earlier findings, Calippe et al. (2017) hypothesized that CFH-CD11b complex on the microglial cell surface might physically interact with the membrane bound CD47 molecule, preventing ligation of the CD47 molecule by its natural ligands. A CD47 ligand known to be present in the ocular environment is TSP1, which is secreted by RPE cells into the subretinal space where the lesions are found (He et al., 2006). Using proximity ligation assay, the authors convincingly demonstrate a physical association between CD11b and CD47 on the retinal MC in the AMD models. Although CD47 was expressed in similar amounts, Cfh-sufficient phagocytes manifested slower rate of clearance from the subretinal space compared to the Cfh deficient cells. Signaling through CD47-TSP1 axis was found to enhance MC clearance rate as supported by (1) slower clearance of Cd47 or Tsp1 deficient MCs from subretinal space of wild-type recipients, (2) accelerated clearance of cells co-injected with TSP1 or CD47 activating peptide, and (3) inhibition of clearance by blocking antibody to TSP1. This role of CFH was also applicable to other models of inflammation: In a model of acute retinal stress induced by bright light, clearance of MC in the retina was slower in Cd47- or Tsp1-deficient mice, whereas in a model of acute peritonitis induced by injection of thioglycollate the clearance of recruited macrophages was accelerated by Cfh deficiency.
Finally, Calippe et al. (2017) took their findings one step further and demonstrated the clinical relevance of this mechanism. Recombinant CFH proteins encoding either the risk allele or the wild-type sequence were purified and co-injected together with Cfh-deficient phagocytic cells into the subretinal space. CFH(Y402H) inhibited MC clearance more than the non-risk variant of CFH.
Taken together, the elegant study of Calippe et al. (2017) demonstrates that CFH has a non-canonical role in regulating the clearance of mononuclear phagocytes from sites of inflammation, which is distinct from its well-known role in tissue protection through deactivation of C3b by converting it to iC3b. The risk variant of AMD CFH(Y402H) binds more effectively than the non-risk isoform to CD47-CD11b, inhibiting its interaction with TSP1 and dampening the rate of elimination of cellular debris (Figure 1). It remains to be investigated whether functional polymorphisms in the binding sites of CD11b or CD47 genes could also contribute to similar risks for AMD. Also, it is unknown whether more efficient association of CFH(Y402H) with CD47-CD11b has a parallel in its complement-inhibiting activity. Irrespective of these remaining questions, the findings of Calippe et al. (2017) open new avenues of research to develop therapeutic agents directed toward cell clearance that can prevent degenerative loss of central vision in the macular region.
Figure 1. CFH-CD11b Complex Binds to CD47 on Microglial Cells in the Retina, Competing with Binding of TSP1.
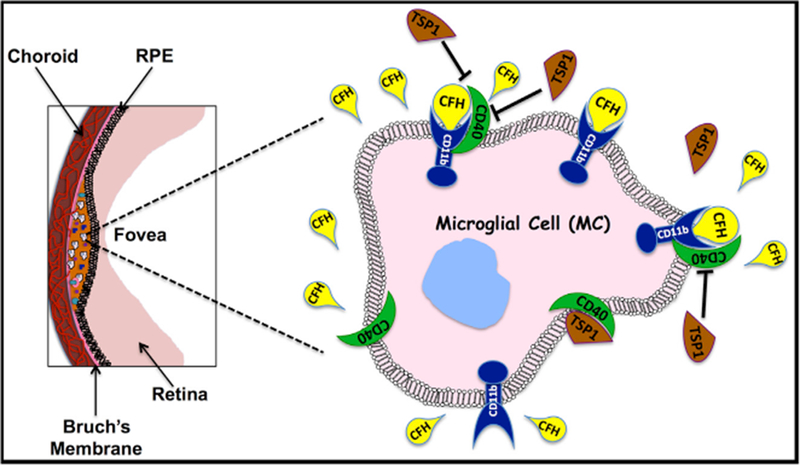
The consequence of this interaction is inhibition of CD47 driven clearance of mononuclear phagocytes, whose role is removal of physiological waste products, promoting inflammation characteristic of AMD.
ACKNOWLEDGMENTS
NIH/NEI Intramural grant ZIA EY000184-34 (R.R.C.) provided funding for this research.
REFERENCES
- Ambati J, Atkinson JP, and Gelfand BD (2013). Nat. Rev. Immunol 13, 438–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calippe B, Augustin S, Beguier F, Messance HC, Poupel L, Conart J-B, Hu S, Lavalette S, Fauvet A, Rayes J, et al. (2017). Immunity 46, this issue, 261–272. [DOI] [PubMed] [Google Scholar]
- Friedman DS, O’Colmain BJ, Muñoz B, Tomany SC, McCarty C, de Jong PT, Nemesure B, Mitchell P, and Kempen J; Eye Diseases Prevalence Research Group (2004). Arch. Ophthalmol 122, 564–572. [DOI] [PubMed] [Google Scholar]
- He S, Incardona F, Jin M, Ryan SJ, and Hinton DR (2006). Yan Ke Xue Bao 22, 265–274. [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, et al. (2005). Science 308, 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenborg PA (2013). ISRN Hematol 2013, 614619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer A, Böttcher A, Orsó E, Kapinsky M, Nagy P, Bodnár A, Spreitzer I, Liebisch G, Drobnik W, Gempel K, et al. (2001). Eur. J. Immunol 31, 3153–3164. [DOI] [PubMed] [Google Scholar]
- Poon IK, Lucas CD, Rossi AG, and Ravichandran KS (2014). Nat. Rev. Immunol 14, 166–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, and Baird PN (2016). Ophthalmic Genet Published online November 30, 2016 10.1080/13816810.2016.1227451. [DOI] [PubMed]