

Abstract
Within the body, cellular recognition is mediated in large part by receptor–ligand interactions that result from the surface marker expression of the participant cells. In the case of immune cells, these interactions can be highly specific, enabling them to carry out their protective functions in fighting off infection and malignancy. In this work, we demonstrate the biomimetic targeting of antigen-specific immune cell populations by using nanoparticles functionalized with natural membrane derived from cells expressing the cognate antigen. Using red blood cell (RBC)-specific B cells as a model target, it is shown that RBC membrane-coated nanoparticles exhibit enhanced affinity compared with control nanoparticles. The concept is further demonstrated using murine models of alloimmunity and autoimmunity, where B cells elicited against RBCs can be positively labeled using the biomimetic nanoparticles. This strategy for antigen-specific immune cell targeting may have utility for the detection and treatment of various autoimmune conditions, and it may additionally have implications for the prevention of immune cell malignancies.
Keywords: biomimetic nanotechnology, membrane-coated nanoparticle, biointerfacing, B cell, autoimmune disease, lymphoma
Table of Contents (TOC) Graphic
1. INTRODUCTION
The continued advancement of nanomedicine over the past several decades has had a significant impact on how diseases are managed in the clinic.1–4 Nanomaterials have found wide use for a variety of applications, including drug delivery, detoxification, in vivo imaging, in vitro diagnostics, tissue engineering, and immunomodulation.5–11 Nanoparticulate systems are highly versatile, as they can be used to deliver a wide range of cargoes and are amendable to different surface engineering techniques for enhanced biointerfacing.12–14 For example, hydrophobic drugs, which normally have poor bioavailability, can be easily loaded into polymeric nanoparticles and released in a sustained or controllable manner once delivered.15 Surface functionalization of nanoparticles with stealth coatings, specific ligands, or immunomodulatory molecules can improve biocompatibility, prolong circulation, and facilitate targeted delivery.13, 14 A significant number of nanoparticle-based products, particularly liposomal, protein-based, and polymeric platforms, have been translated for clinical use, and countless others are in trials.16
The first generation of nanoformulations have relied largely on passive mechanisms such as the enhanced permeation and retention effect to achieve preferential accumulation in a site of interest.17, 18 Current research efforts are focused on therapeutics that utilize active targeting to further enhance potency while minimizing potential side effects.13, 19 In general, this is accomplished by surface functionalization strategies that incorporate ligands specific for receptors found on certain tissues or cell types. Conventional targeting ligands such as small molecules, peptides, aptamers, and antibodies can be identified through established screening processes.20 There has recently also been an emphasis on the utilization of biomimetic targeting strategies in which naturally occurring targeting ligands are employed.13 While these strategies have been highly effective at bestowing specificity based on differential cell surface marker expression, there are still some scenarios in which targeting is difficult to achieve.
One such case is the targeting of immune cell subsets within B cell or T cell populations, either of which can be further differentiated based on their antigen specificity. In response to infections and malignancies, clonal populations of effector cells will expand, and they are often of great therapeutic or diagnostic interest.21, 22 In the case of autoimmunity, where the standard of care is often broad immunosuppression that can have significant side effects or iatrogenic risks,23, 24 the ability to specifically target the offending cell population, while leaving non-pathological immune cells intact, would be of tremendous value. A significant number of autoimmune diseases are of B cell origin, and those such as Sjögren's syndrome, autoimmune thyroiditis, and autoimmune hemolytic anemia are also associated with an increased risk of non-Hodgkin’s lymphoma.25 While the factors that precipitate lymphoma are not well understood, the ability to effectively detect and treat autoimmune diseases may ultimately serve as a preventative measure against related types of cancer.
In terms of their surface marker expression, the only differentiating factor within B cell or T cell populations may be their B cell or T cell receptors (BCRs or TCRs), respectively, which provide antigenic or epitopic specificity. This makes targeting clonal subsets difficult, as traditional targeting ligands are not adept at differentiating among the countless number of minor variants that arise as a result of VDJ recombination.26 This is made even more difficult by the fact that oftentimes the exact antigen specificity of autoimmune cells is unknown.27 In this work, we evaluate a strategy for addressing the above issues by employing a cell membrane coating approach for nanoparticle functionalization.28 The cell membrane coting platform was initially developed using red blood cells (RBCs),29 and it was shown that RBC membrane-coated nanoparticles (RBC-NPs) express the same surface antigens as the original cells.30 The platform has since been expanded to include membrane coating sourced from numerous cell types,31–34 all of which can be leveraged to bestow unique nanoparticle biointerfacing capabilities.14 To apply this approach for the targeting of antigen-specific immune cells, we take advantage of the fact that, for many autoimmune conditions, such as autoimmune hemolytic anemia or immune thrombocytopenia, the target cell is known.35, 36 Accordingly, the membrane of these cells naturally carries the cognate antigens or peptide complexes of the BCRs or TCRs expressed by the pathological immune cells. We thus hypothesized that coating this membrane onto nanoparticles can be an effective means of bestowing targeting specificity towards antigen-specific autoimmune cells (Figure 1). To prove this concept, we first evaluated RBC-NP binding in vitro using an RBC-specific hybridoma-based model system. Then, targeting efficacy was demonstrated using animal models of both anti-RBC alloimmunity and autoimmunity. Overall, this approach demonstrated significant potential for enhancing nanoparticle affinity towards hard-to-target immune cell subsets and may be easily generalized to other cell types and conditions in the future.
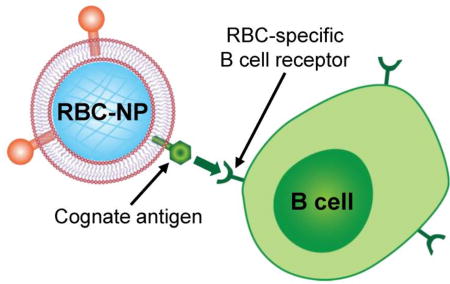
Figure 1.

Schematic of membrane-coated nanoparticles for the targeting of antigen-specific B cells. RBC-NPs naturally display the antigens targeted by RBC-specific B cells. By leveraging the affinity between the corresponding BCR and its cognate antigen, RBC-NPs can be used to target these B cells. In contrast, B cells that are not specific for RBC antigens will not be targeted by RBC-NPs.
2. MATERIALS AND METHODS
Synthesis and Characterization of RBC-NPs
To fabricate the nanoparticles, a previously reported membrane coating approach was employed.37 First, poly(lactic-co-glycolic acid) (PLGA; carboxy-terminated, 50:50, 0.67 dL/g; Lactel Absorbable Polymers) cores were prepared by precipitating the polymer dissolved at 10 mg/mL using acetone into water, followed by evaporation to remove the organic solvent. Human RBC (hRBC) membrane ghosts were obtained by the hypotonic lysis of human O-negative RBCs (BioreclamationIVT). Human RBC membrane-derived vesicles were prepared by brief sonication of hRBC ghosts using a Fisher Scientific FS30D bath sonicator. To make hRBC-NPs, the membrane and PLGA cores were mixed together, followed by sonication to induce membrane fusion. Size and zeta potential were measured by dynamic light scattering (DLS) using a Malvern ZEN 3600 Zetasizer. To visualize the nanoparticles, the hRBC-NPs were deposited onto a 400-mesh carbon-coated copper grid (Electron Microscopy Sciences), stained with 1 wt% uranyl acetate (Electron Microscopy Sciences), and imaged using a Zeiss Libra 120 PLUS EF-TEM transmission electron microscope.
Dot Blot Analysis
To assess the specificity of the hybridomas, 1 µg of hRBC membrane protein was spotted onto nitrocellulose membrane (Pierce) and allowed to dry. Then, cell culture supernatant from the anti-glycophorin A (GPA) hybridoma 10F7MN (HB-8162; American Type Culture Collection) or the anti-measles virus nucleoprotein hybridoma NP cl.120 (Sigma Aldrich) was used as the primary immunostain. A horseradish peroxidase (HRP)-conjugated anti-mouse IgG (Biolegend) was used for secondary staining. Membranes were developed with ECL western blotting substrate (Pierce) in an ImageWorks Mini-Medical/90 Developer.
In Vitro Targeting
To assess binding capacity, 250 µg of polyethylene glycol-coated nanoparticles (PEG-NPs) or hRBC-NPs loaded with 0.1 wt% of 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt (DiD; ex/em = 644/663 nm; Biotium) were incubated with either 5 × 105 anti-GPA hybridoma cells or control anti-measles hybridoma cells for 5 min at 4 °C. The cells were then washed 3× in phosphate buffered saline (PBS) and resuspended in 500 µL of PBS. Flow cytometry was performed on a Becton Dickinson FACSCanto-II flow cytometer to examine the percentage of DiD+ cells and the mean fluorescence intensity. Data was analyzed using Flowjo software.
Detection of Alloimmune Anti-hRBC B Cells
All animal studies were designed and proceeded in compliance to the University of California San Diego Institutional Animal Care and Use Committee (IACUC). To induce alloimmunity, 500 µg of hRBC membrane protein in PBS was injected intraperitoneally into male CD-1 mice (Envigo) every week for 3 weeks. An enzyme-linked immunosorbent assay (ELISA) was used to assess anti-hRBC titer levels periodically. To perform the ELISA, 96-well assay plates were coated with 1 µg of hRBC membrane protein per well. Blood was sampled from each mouse by submandibular puncture, and the serum was serially diluted in 5% milk (Apex BioResearch Products) in PBS containing 0.05% Tween 20 (National Scientific), referred to as PBST. The dilutions were then added to the hRBC membrane-coated assay plates and allowed to incubate for 2 h at room temperature. The plates were washed 4× with PBST. Subsequently, an HRP-conjugated anti-mouse IgG secondary antibody in 5% milk in PBST was added to each well and allowed to incubate for 2 h at room temperature. The plate was washed and TMB substrate (Life Technologies) was added to each well. After ~10 min, the reaction was stopped using 1 M HCl, and the absorbance was read at 450 nm using a Tecan Infinite M200 plate reader. Titers were calculated by fitting the data in Graphpad Prism and interpolating the dilution values at a constant absorbance threshold.
To assess the binding ability of hRBC-NPs to anti-hRBC B cells, whole blood was collected from the mice, and the RBCs were lysed using RBC lysis buffer (Biolegend) following the manufacturer’s protocol to obtain the leukocyte cell population. Following collection of the white blood cells, ~1 × 106 cells per sample were first incubated in 100 µM chlorpromazine (Alfa Aesar) for 1 h to prevent nonspecific internalization of nanoparticles. Then, 250 µg of DiD-loaded hRBC-NPs and FITC-conjugated anti-mouse CD19 antibody (Biolegend) were added and incubated for 20 min at 4 °C. The cells were then washed 3× in ice cold PBS and fixed with 10% formalin (Fisher). The cells were then examined using a Becton Dickinson FACSCanto-II flow cytometer. Data was analyzed using Flowjo software. Positive cells were determined by gating for FITC+ and DiD+ events.
Detection of Autoimmune Anti-mouse RBC (mRBC) B Cells
To induce autoimmune hemolytic anemia, female C57BL/6 mice (Envigo) were injected intraperitoneally with 500 µg of anti-CD25 antibody (Biolegend) per mouse. After 8 h, each mouse was injected intraperitoneally with 2 × 108 rat RBCs (rRBCs) (BioreclamationIVT). This process was repeated once a week for 3 weeks. Blood was collected every two weeks to assess the progress of autoimmunity development. IgG sensitization was examined by incubating ~1 × 106 washed mRBCs with a FITC-labeled anti-mouse IgG antibody (Biolegend). The cells were washed 3× and analyzed using a Becton Dickinson FACSCanto-II flow cytometer. Data was analyzed using Flowjo software. The percentage of mRBCs that were FITC+ was used to indicate the degree of IgG sensitization. To quantify the percentage of reticulocytes in the blood, 5 µL of whole blood was incubated with 1 mL of 0.5 mg/mL thiazole orange (ex/em = 510/530 nm; Sigma Aldrich) in PBS. The cells were washed 3× and analyzed using a Becton Dickinson FACSCanto-II flow cytometer. Data was analyzed using Flowjo software. The percentage of positive events in the FITC channel was quantified.
A protocol similar to that described for the alloimmunity experiments was used to evaluate the binding of mRBC-NPs to autoimmune anti-mRBC B cells. In this case, each sample was split into three groups to be incubated with different nanoparticle formulations, including DiD-loaded PEG-NPs and mRBC-coated NPs. For the blocked sample, the mRBC-NPs were first incubated with a polyclonal anti-mRBC antibody (Rockland).
3. RESULTS AND DISCUSSION
To fabricate hRBC-NPs, a previously reported membrane coating approach was employed. Polymeric cores were prepared first by dissolving PLGA in an organic phase, followed by precipitation into an aqueous phase to induce spontaneous nanoparticle formation. Afterwards, the organic phase was allowed to evaporate. Cell membrane was derived fresh from hRBCs by repeated hypotonic treatment to remove the intracellular hemoglobin. Subsequently, the PLGA cores and membrane were mixed together and then sonicated to promote fusion of the membrane onto the surface of the nanoparticulate cores. Using DLS measurements, it was determined that the final hRBC-NPs were approximately 90 nm in size, which was slightly larger than the bare PLGA cores (Figure 2a). In terms of zeta potential, the hRBC-NPs measured approximately −25 mV, similar in value to free hRBC membrane vesicles (Figure 2b). Taken together, this data was consistent with successful membrane coating.32 For further confirmation, the hRBC-NPs were negatively stained with uranyl acetate and imaged by transmission electron microscopy (Figure 2c). A core-shell structure was observed, confirming that there was a membrane layer coated around the polymeric cores.
Figure 2.

Characterization of human RBC-NPs. (a) Size of bare PLGA cores, RBC membrane-derived vesicles, and RBC-NPs as measured by DLS (n = 3; mean + SD). (b) Zeta potential of bare PLGA cores, RBC membrane-derived vesicles, and RBC-NPs as measured by DLS (n = 3; mean + SD). (c) Transmission electron micrograph of RBC-NPs negatively stained with uranyl acetate. Scale bar = 100 nm.
RBC-NPs have previously been shown to retain the same surface marker expression profile as their source RBCs.30 This includes membrane proteins such as CD47, which has been demonstrated to be an important self-marker that inhibits phagocytosis by macrophages.38 We thus sought to evaluate if RBC-NPs could be used to specifically target B cells that produce anti-RBC antibodies. As a model system, hybridomas, which are generated by the fusion of a B cell with a myeloma cell, were employed.39 As an RBC-specific hybridoma, we selected one that produces antibodies against GPA, an RBC glycoprotein that is a minor blood group determinant.40 For the negative control, an anti-measles antigen hybridoma was chosen. First, to confirm the specificity of the hybridomas against RBC-NPs, immunoblotting analysis was conducted using the culture supernatant of each type of cell as the primary immunostain (Figure 3). After staining with a secondary antibody and developing the blots, the sample probed using the anti-GPA hybridoma supernatant exhibited clear signal, whereas the one for the anti-measles hybridoma had no signal.
Figure 3.

Confirmation of B cell hybridoma specificity for human RBC-NPs. The culture supernatant from an anti-GPA hybridoma and a control anti-measles antigen hybridoma were used as the primary immunostains for dot blot analysis of RBC membrane.
After confirming the specificity of the hybridomas, the targeting ability of the hRBC-NPs was evaluated. B cells express antigen-specific receptors that are membrane-bound versions of the antibodies that they produce.41 As such, they should be amenable to targeting by nanoparticles that display their cognate antigen. To test this hypothesis, dye-labeled hRBC-NPs were incubated with both the anti-GPA hybridoma and the control anti-measles hybridoma, followed by flow cytometry analysis (Figure 4). In terms of the percentage of cells that were positive for nanoparticle signal, as well as the mean fluorescence intensity, the anti-GPA hybridoma was shown to bind much more efficiently to the hRBC-NPs compared with the anti-measles hybridoma. In contrast, when PEG-NPs that don’t display any RBC antigens were used, there was significantly less binding, especially when looking at the anti-GPA hybridomas. The results for this study suggested that hRBC-NPs, by displaying the appropriate cognate antigens, could be used to target B cells that generate antibodies against hRBCs.
Figure 4.

In vitro targeting by human RBC-NPs. Anti-GPA hybridoma cells or control anti-measles antigen hybridoma cells were incubated with DiD-labeled versions of either RBC-NPs or PEG-NPs. Afterwards, the cells were analyzed by flow cytometry and the percentage of nanoparticle-positive cells (a) and the mean fluorescence intensity (b) were determined (n = 3; mean + SD). **p < 0.01, ****p < 0.0001; Student’s t-test.
With the encouraging in vitro binding results, we next sought to evaluate if it was possible to distinguish antigen-specific B cells generated in vivo. As a first proof-of-concept, a murine anti-hRBC alloimmunity model was employed. To generate this model, mice were sensitized to hRBCs by intraperitoneal injections of hRBC membrane. As hRBCs are allogeneic, immunity should readily be generated against its antigens when used to sensitize a murine host. To confirm the presence of anti-hRBC immunity in the mice, titers were periodically assessed by ELISA (Figure 5). It was confirmed that anti-hRBC titers could be effectively generated in the mice, with a quick acceleration of antibody production in the first 2 weeks followed by slowing down of the kinetics by day 60.
Figure 5.

Induction of anti-hRBC alloimmunity. Mice were challenged with hRBC membrane every week for three weeks. The antibody titers against hRBC membrane were assessed periodically by ELISA (n = 30; min to max).
The successful generation of anti-hRBC titers in the mice indicated that there was an opportunity to target the corresponding B cells specific for the hRBC antigens. To test this, leukocytes were first isolated from the blood of both hRBC-sensitized and naïve mice. Subsequently, dye-labeled hRBC-NPs were used to probe the leukocytes, along with a fluorescent antibody against the B cell marker CD19 (Figure 6). Using flow cytometry, the percentage of cells positive for both the CD19 and the hRBC-NPs was assessed. The data indicated that, when the nanoparticles were incubated with leukocytes from mice sensitized to hRBCs, significantly more cells tested positive compared with leukocytes from naïve mice. In all, the results demonstrated that target-specific B cells could be identified using this approach in a manner that only requires knowledge of the target cell, rather than the specific antigens involved.
Figure 6.

Targeting of alloimmune B cells using hRBC-NPs. Leukocytes were isolated from mice sensitized to hRBC membrane or from naïve mice. Subsequently, the cells were incubated with DiD-labeled hRBC-NPs and analyzed by flow cytometry; a FITC-conjugated CD19 antibody was used to identify the B cell population during analysis (n = 5 for naïve, 15 for sensitized; mean ± SD). ****p < 0.0001; Student’s t-test.
To see if this could be translated to a more clinically relevant scenario, we evaluated the targeting potential of mRBC-NPs in a murine model of autoimmune hemolytic anemia.42 In order to generate autoimmunity in mice, rRBCs, along with anti-CD25 antibodies, were administered intraperitoneally. The similarity of the rRBCs to the endogenous mRBCs in mice allows for the production of antibodies with self-reactivity. In order to assess the development of autoimmunity, two relevant parameters were tracked over time. First, we tested for IgG sensitization, which measures the presence of autoantibodies bound to mRBCs (Figure 7a). Blood was sampled from the mice and purified, followed by staining with a fluorescent anti-mouse IgG antibody. From flow cytometric analysis, it was demonstrated that, by week 4 after induction, mice receiving rRBCs and anti-CD25 had significantly higher IgG sensitization compared with naïve mice. In the next assay, we assessed for the percentage of reticulocytes in the blood (Figure 7b). Using thiazole orange staining of whole blood followed by flow cytometric analysis, it was determined that sensitized mice had significantly higher reticulocyte counts, indicating that their bodies were attempting to compensate for the induced anemia.42
Figure 7.

Induction of anti-mRBC autoimmunity in mice. Mice were sensitized with rRBCs and anti-CD25 every week for three weeks. The degree of RBC IgG sensitization (a) and the reticulocyte counts (b) were then quantified periodically by flow cytometry (n = 4 for naïve, 15 for sensitized; mean + SEM). *p < 0.05, ****p < 0.0001; Student’s t-test.
After confirmation of anti-mRBC autoimmunity, the ability of mRBC-NPs to detect the corresponding autoimmune B cells was tested. Dye-labeled nanoparticles were incubated with leukocytes isolated from sensitized or naïve mice along fluorescent anti-CD19, followed by analysis using flow cytometry (Figure 8). As with the alloimmunity scenario, significantly more cells were positive for CD19 and mRBC-NPs in leukocytes from sensitized mice compared with those from naïve mice. In line with the in vitro results, the use of control PEG-NPs resulted in low detection values compared with the mRBC-NPs. Notably, when anti-mRBC antibodies were used to pre-block the mRBC-NPs, the detection capability of the nanoparticles was completely abrogated, demonstrating that the targeting effect most likely results from presentation of the mRBC membrane antigens found on the nanoparticle surface. Regarding future in vivo application, these results also suggest that excess nanoparticles may need to be given in order to deplete circulating autoantibodies before B cells can be targeted. In all, the data indicated that, by using the membrane of the same cells targeted by autoimmunity to functionalize nanoparticles, the resulting biomimetic nanoparticles could demonstrate specific affinity to rare populations of effector B cells.
Figure 8.

Targeting of autoimmune B cells using mRBC-NPs. Leukocytes were isolated from sensitized mice exhibiting autoimmune anemia or from naïve mice. Subsequently, the cells were incubated with DiD-labeled PEG-NPs, mRBC-NPs, or mRBC-NPs blocked with anti-mRBC antibodies and analyzed by flow cytometry; a FITC-conjugated CD19 antibody was used to identify the B cell population during analysis (n = 5 for naïve, 10 for sensitized; mean ± SEM). ***p < 0.001; one-way ANOVA.
4. CONCLUSIONS
In conclusion, we have demonstrated an approach for the specific targeting of immune cell populations by using nanoparticles expressing their cognate antigens. This was accomplished by taking advantage of a membrane coating technology that enables facile functionalization of nanoparticles with the same antigens that are present on the cells targeted by the immune cells. This was first demonstrated in vitro using B cell hybridomas, where RBC-NPs more efficiently bound to an RBC antigen-specific hybridoma. Then, in a murine model of alloimmunity, B cells specific for hRBCs could be identified. Finally, using a murine model of autoimmune hemolytic anemia, it was shown that nanoparticles functionalized with endogenous mRBC membrane could be used to identify a small population of autoimmune B cells. This approach has implications for the detection and treatment of various type II immune hypersensitivities, where oftentimes healthy cells are targeted and destroyed by autoantibodies.27 The ability to specifically eliminate autoimmune cell subsets may also help to prevent certain types of cancer, which have been shown to be strongly correlated with autoimmunity. Beyond the B cells studied in the present work, this approach in principle can be extended to autoimmune T cells by leveraging membrane-bound antigen presenting complexes. For rare cellular targets of autoimmunity, cell lines expressing similar antigenic profiles may be explored as a source of membrane coating. A major strength of employing the membrane coating approach is that it is highly generalizable and works in a manner that doesn’t require explicit identification of the individual antigens involved. Ultimately, this may enable the technology to be leveraged for the detection and therapy of a wide range of autoimmune conditions that are currently hard to manage in the clinic.
Acknowledgments
This work is supported by the National Institutes of Health under Award Number R01CA200574.
Footnotes
The authors declare no competing financial interest.
References
- 1.Min Y, Caster JM, Eblan MJ, Wang AZ. Clinical Translation of Nanomedicine. Chem. Rev. 2015;115:11147–11190. doi: 10.1021/acs.chemrev.5b00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anselmo AC, Mitragotri S. Nanoparticles in the Clinic. Bioeng. Transl. Med. 2016;1:10–29. doi: 10.1002/btm2.10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC. Nanoparticles in Medicine: Therapeutic Applications and Developments. Clin. Pharmacol. Ther. 2008;83:761–769. doi: 10.1038/sj.clpt.6100400. [DOI] [PubMed] [Google Scholar]
- 4.Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer Nanomedicine: Progress, Challenges and Opportunities. Nat. Rev. Cancer. 2017;17:20–37. doi: 10.1038/nrc.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang AZ, Langer R, Farokhzad OC. Nanoparticle Delivery of Cancer Drugs. Annu. Rev. Med. 2012;63:185–198. doi: 10.1146/annurev-med-040210-162544. [DOI] [PubMed] [Google Scholar]
- 6.Fang RH, Luk BT, Hu CM, Zhang L. Engineered Nanoparticles Mimicking Cell Membranes for Toxin Neutralization. Adv. Drug Deliv. Rev. 2015;90:69–80. doi: 10.1016/j.addr.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doane TL, Burda C. The Unique Role of Nanoparticles in Nanomedicine: Imaging, Drug Delivery and Therapy. Chem. Soc. Rev. 2012;41:2885–2911. doi: 10.1039/c2cs15260f. [DOI] [PubMed] [Google Scholar]
- 8.Cuenca AG, Jiang H, Hochwald SN, Delano M, Cance WG, Grobmyer SR. Emerging Implications of Nanotechnology on Cancer Diagnostics and Therapeutics. Cancer. 2006;107:459–466. doi: 10.1002/cncr.22035. [DOI] [PubMed] [Google Scholar]
- 9.Shi J, Votruba AR, Farokhzad OC, Langer R. Nanotechnology in Drug Delivery and Tissue Engineering: From Discovery to Applications. Nano Lett. 2010;10:3223–3230. doi: 10.1021/nl102184c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang RH, Kroll AV, Zhang L. Nanoparticle-Based Manipulation of Antigen-Presenting Cells for Cancer Immunotherapy. Small. 2015;11:5483–5496. doi: 10.1002/smll.201501284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fang RH, Zhang L. Nanoparticle-Based Modulation of the Immune System. Annu. Rev. Chem. Biomol. Eng. 2016;7:305–326. doi: 10.1146/annurev-chembioeng-080615-034446. [DOI] [PubMed] [Google Scholar]
- 12.Hu CM, Fang RH, Luk BT, Zhang L. Polymeric Nanotherapeutics: Clinical Development and Advances in Stealth Functionalization Strategies. Nanoscale. 2014;6:65–75. doi: 10.1039/c3nr05444f. [DOI] [PubMed] [Google Scholar]
- 13.Dehaini D, Fang RH, Zhang L. Biomimetic Strategies for Targeted Nanoparticle Delivery. Bioeng. Transl. Med. 2016;1:30–46. doi: 10.1002/btm2.10004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kroll A, Fang RH, Zhang L. Biointerfacing and Applications of Cell Membrane-Coated Nanoparticles. Bioconjug. Chem. 2016;28:23–32. doi: 10.1021/acs.bioconjchem.6b00569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamaly N, Yameen B, Wu J, Farokhzad OC. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev. 2016;116:2602–2663. doi: 10.1021/acs.chemrev.5b00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hassan S, Prakash G, Ozturk A, Saghazadeh S, Sohail MF, Seo J, Dockmeci M, Zhang YS, Khademhosseini A. Evolution and Clinical Translation of Drug Delivery Nanomaterials. Nano Today. 2017;15:91–106. doi: 10.1016/j.nantod.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iyer AK, Khaled G, Fang J, Maeda H. Exploiting the Enhanced Permeability and Retention Effect for Tumor Targeting. Drug Discov. Today. 2006;11:812–818. doi: 10.1016/j.drudis.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 18.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor Vascular Permeability and the EPR Effect in Macromolecular Therapeutics: A Review. J. Control. Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 19.Mishra B, Patel BB, Tiwari S. Colloidal Nanocarriers: A Review on Formulation Technology, Types and Applications Toward Targeted Drug Delivery. Nanomedicine. 2010;6:9–24. doi: 10.1016/j.nano.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 20.Steichen SD, Caldorera-Moore M, Peppas NA. A Review of Current Nanoparticle and Targeting Moieties for the Delivery of Cancer Therapeutics. Eur. J. Pharm. Sci. 2013;48:416–427. doi: 10.1016/j.ejps.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanz I, Lee FEB. B Cells as Therapeutic Targets in SLE. Nat. Rev. Rheumatol. 2010;6:326–337. doi: 10.1038/nrrheum.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crispin JC, Kyttaris VC, Terhorst C, Tsokos GC. T Cells as Therapeutic Targets in SLE. Nat. Rev. Rheumatol. 2010;6:317–325. doi: 10.1038/nrrheum.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schacke H, Docke WD, Asadullah K. Mechanisms Involved in the Side Effects of Glucocorticoids. Pharmacol. Ther. 2002;96:23–43. doi: 10.1016/s0163-7258(02)00297-8. [DOI] [PubMed] [Google Scholar]
- 24.Lallana EC, Fadul CE. Toxicities of Immunosuppressive Treatment of Autoimmune Neurologic Diseases. Curr. Neuropharmacol. 2011;9:468–477. doi: 10.2174/157015911796557939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mackay IR, Rose NR. Autoimmunity and Lymphoma: Tribulations of B Cells. Nat. Immunol. 2001;2:793–795. doi: 10.1038/ni0901-793. [DOI] [PubMed] [Google Scholar]
- 26.Schatz DG, Oettinger MA, Schlissel MS. V(D)J Recombination: Molecular Biology and Regulation. Annu. Rev. Immunol. 1992;10:359–383. doi: 10.1146/annurev.iy.10.040192.002043. [DOI] [PubMed] [Google Scholar]
- 27.Davidson A, Diamond B. Autoimmune Diseases. N. Engl. J. Med. 2001;345:340–350. doi: 10.1056/NEJM200108023450506. [DOI] [PubMed] [Google Scholar]
- 28.Fang RH, Jiang Y, Fang JC, Zhang L. Cell Membrane-Derived Nanomaterials for Biomedical Applications. Biomaterials. 2017;128:69–83. doi: 10.1016/j.biomaterials.2017.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu C-MJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang L. Erythrocyte Membrane-Camouflaged Polymeric Nanoparticles as a Biomimetic Delivery Platform. Proc. Natl. Acad. Sci. U. S. A. 2011;108:10980–10985. doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu CM, Fang RH, Luk BT, Chen KN, Carpenter C, Gao W, Zhang K, Zhang L. 'Marker-of-Self' Functionalization of Nanoscale Particles Through a Top-Down Cellular Membrane Coating Approach. Nanoscale. 2013;5:2664–2668. doi: 10.1039/c3nr00015j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thamphiwatana S, Angsantikul P, Escajadillo T, Zhang Q, Olson J, Luk BT, Zhang S, Fang RH, Gao W, Nizet V, Zhang L. Macrophage-Like Nanoparticles Concurrently Absorbing Endotoxins and Proinflammatory Cytokines for Sepsis Management. Proc. Natl. Acad. Sci. U. S. A. 2017;114:11488–11493. doi: 10.1073/pnas.1714267114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fang RH, Hu C-MJ, Luk BT, Gao W, Copp JA, Tai Y, O'Connor DE, Zhang L. Cancer Cell Membrane-Coated Nanoparticles for Anticancer Vaccination and Drug Delivery. Nano Lett. 2014;14:2181–2188. doi: 10.1021/nl500618u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao W, Fang RH, Thamphiwatana S, Luk BT, Li JM, Angsantikul P, Zhang QZ, Hu CMJ, Zhang L. Modulating Antibacterial Immunity via Bacterial Membrane-Coated Nanoparticles. Nano Lett. 2015;15:1403–1409. doi: 10.1021/nl504798g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu C-MJ, Fang RH, Wang K-C, Luk BT, Thamphiwatana S, Dehaini D, Nguyen P, Angsantikul P, Wen CH, Kroll AV, Carpenter C, Ramesh M, Qu V, Patel SH, Zhu J, Shi W, Hofman FM, Chen TC, Gao W, Zhang K, Chien S, Zhang L. Nanoparticle Biointerfacing by Platelet Membrane Cloaking. Nature. 2015;526:118–121. doi: 10.1038/nature15373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gehrs BC, Friedberg RC. Autoimmune Hemolytic Anemia. Am. J. Hematol. 2002;69:258–271. doi: 10.1002/ajh.10062. [DOI] [PubMed] [Google Scholar]
- 36.Cines DB, Blanchette VS. Immune Thrombocytopenic Purpura. N. Engl. J. Med. 2002;346:995–1008. doi: 10.1056/NEJMra010501. [DOI] [PubMed] [Google Scholar]
- 37.Copp JA, Fang RH, Luk BT, Hu C-MJ, Gao W, Zhang K, Zhang L. Clearance of Pathological Antibodies Using Biomimetic Nanoparticles. Proc. Natl. Acad. Sci. U. S. A. 2014;111:13481–13486. doi: 10.1073/pnas.1412420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a Marker of Self on Red Blood Cells. Science. 2000;288:2051–2054. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- 39.Zhang C. Hybridoma Technology for the Generation of Monoclonal Antibodies. Methods Mol. Biol. 2012;901:117–135. doi: 10.1007/978-1-61779-931-0_7. [DOI] [PubMed] [Google Scholar]
- 40.Reid ME. MNS Blood Group System: A Review. Immunohematology. 2009;25:95–101. [PubMed] [Google Scholar]
- 41.Dal Porto JM, Gauld SB, Merrell KT, Mills D, Pugh-Bernard AE, Cambier J. B Cell Antigen Receptor Signaling 101. Mol. Immunol. 2004;41:599–613. doi: 10.1016/j.molimm.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 42.Mqadmi A, Zheng X, Yazdanbakhsh K. CD4+CD25+ Regulatory T Cells Control Induction of Autoimmune Hemolytic Anemia. Blood. 2005;105:3746–3748. doi: 10.1182/blood-2004-12-4692. [DOI] [PMC free article] [PubMed] [Google Scholar]