

Abstract
Krüppel-like zinc finger proteins form one of the largest families of transcription factors. They function as key regulators of embryonic development and a wide range of other physiological processes, and are implicated in a variety of pathologies. GLI-similar 1–3 (GLIS1–3) constitute a subfamily of Krüppel-like zinc finger proteins that act either as activators or repressors of gene transcription. GLIS3 plays a critical role in the regulation of multiple biological processes and is a key regulator of pancreatic β cell generation and maturation, insulin gene expression, thyroid hormone biosynthesis, spermatogenesis, and the maintenance of normal kidney functions. Loss of GLIS3 function in humans and mice leads to the development of several pathologies, including neonatal diabetes and congenital hypothyroidism, polycystic kidney disease, and infertility. Single nucleotide polymorphisms in GLIS3 genes have been associated with increased risk of several diseases, including type 1 and type 2 diabetes, glaucoma, and neurological disorders. GLIS2 plays a critical role in the kidney and GLIS2 dysfunction leads to nephronophthisis, an end-stage, cystic renal disease. In addition, GLIS1–3 have regulatory functions in several stem/progenitor cell populations. GLIS1 and GLIS3 greatly enhance reprogramming efficiency of somatic cells into induced embryonic stem cells, while GLIS2 inhibits reprogramming. Recent studies have obtained substantial mechanistic insights into several physiological processes regulated by GLIS2 and GLIS3, while a little is still known about the physiological functions of GLIS1. The localization of some GLIS proteins to the primary cilium suggests that their activity may be regulated by a downstream primary cilium-associated signaling pathway. Insights into the upstream GLIS signaling pathway may provide opportunities for the development of new therapeutic strategies for diabetes, hypothyroidism, and other diseases.
Keywords: GLIS1, GLIS2, GLIS3, Krüppel-like zinc finger protein, Pancreatic beta cells, Diabetes, Insulin, Polycystic kidney disease, Nephronophthisis, Primary cilium, Reprogramming, Stem cells, Hypothyroidism, Spermatogenesis, Thyroid hormone biosynthesis, NIS, Development, HECT E3 ubiquitin ligase, TAZ, SUFU, ITCH, GWAS, Mutations
Introduction
GLI-Similar 1–3 (GLIS1–3) constitute a subfamily of Krüppel-like zinc finger transcription factors that contain a zinc finger domain (ZFD) that is highly conserved among GLIS1–3 as well members of the closely related GLI1–3 protein subfamily [1–8] (Fig. 1). The ZFDs of GLIS1 and GLIS3 exhibit the highest homology suggesting that they are evolutionary the most closely related. Outside their ZFD, GLIS1–3 exhibit little homology with each other or GLI proteins with the exception of an N-terminal, conserved region (NCR) of about 60 aa that GLIS3 has in common with GLI1–3 [9].
Fig. 1.

Schematic presentation of human GLIS1–3 proteins. The DNA binding domain (DBD) containing five zinc finger motifs, the activation domain (AD), and the N-terminal conserved region (NCR) are indicated. Numbers on the right indicate the size of the GLIS1–3 and GLI proteins. Lower panel: comparison of the amino acid sequence between the NCRs of GLIS3 and GLI1–3. Homologous amino acids are shown in blue. The ciliary localization signal consensus (CLS) and the SUFU binding domain are indicated. The CLS binds TNPO1, which guides transport into the primary cilium
Although several alternatively spliced GLIS transcripts and variants have been described, their physiological relevance has yet to be determined. GLIS homologs have been identified in all mammalian species, as well as Zebrafish, Medaka, Xenopus and Drosophila [10–12]. The GLIS homolog in Drosophila is referred to as gleeful (gfl) or lame duck (lmd).
GLIS1–3 play a critical role in the regulation of many physiological processes and are been implicated in a variety of pathologies, including neonatal diabetes, glaucoma, cystic kidney disease, neurological disorders, congenital hypothyroidism, and cancer [1, 4, 5, 13, 14]. Study of this subfamily of transcription regulators has provided greater mechanistic insights into the regulation of several physiological processes and the development of various pathologies, and might eventually lead to the discovery of novel therapeutic approaches for these diseases.
Transcriptional activity and protein interactions
Activator and repressor functions
GLIS1 and GLIS3 function primarily as transcriptional activators, whereas GLIS2 can activate transcription, but appears to function mainly as a transcriptional repressor [8, 15–18]. Deletion analysis revealed that GLIS1 and GLIS3 contain a strong activation domain at their C terminus that mediates their interaction with co-activators. GLIS proteins regulate gene transcription by interacting with GLIS-binding sites (GLISBS) in the regulatory region of target genes. Insulin (Ins2), cyclin D2 (Cdnd2), fibroblast growth factor 18 (Fgf18), neurogenin 3 (Ngn3), pendrin (Pds), and Na+/I− symporter (Nis) are among the genes directly regulated by GLIS3, while GLI1, the Wnt family member 4 (Wnt4) and Snail1 (Snai1) are target genes of GLIS2 [17–22]. The interaction of GLIS proteins with GLISBS is mediated by their ZFD, which consists of a tandem repeat of five C2H2 zinc finger motifs [1, 5, 8, 16, 19]. All five zinc finger motifs are required for optimal binding of GLIS proteins to GLISBS; loss of the first zinc finger has the least impact on this interaction. The in vivo consensus sequence of GLISBS derived from ChIP-Seq analysis exhibits high similarity with the GLISBS consensus sequence, (G/C)TGGGGG(A/C), identified by an in vitro screen [17, 23, 24]. Members of the closely related GLI and ZIC families also bind G-rich DNA response elements similar to GLISBS and therefore might compete for the same binding site and interfere with each other’s action in cells in which they are co-expressed. This is supported by a report showing that GLIS2 can inhibit GLISBS-dependent activation of a reporter gene by GLI1 and repress GLI1-induced transcriptional activation of Wnt4 by competing for the same binding site [17, 18]. In addition, different members of these protein families may interact by forming heterodimers as have been reported for other Krüppel-like zinc finger transcription factors, including members of the ZIC and GLI subfamilies [25].
GLIS proteins activate or repress transcription through the recruitment of co-activators or co-repressors, respectively. The co-activator, C-terminal binding protein (CBP), was shown to interact with GLIS3 and be part of a multiprotein co-activator complex [19]. Mass spectrometric analysis of GLIS3 protein complexes identified several phosphorylation and arginine methylation sites, and a number of GLIS3-interacting partners [9, 26]. Still very little is known about the role of GLIS3 phosphorylation and the upstream kinase signaling pathways that might regulate GLIS3 activity and function. The arginine methyltransferase PRMT5 and the lysine demethylase KDM4C were among the GLIS3 interacting proteins identified. PRMT5 catalyzes the formation of H3K4me3, which is associated with active chromatin, while KDM4C demethylates histone H3K9me3, which is associated with the repressed state of a gene. Thus, these (de)methylation activities correlate with transcriptionally active genes and are consistent with GLIS3 acting as an activator of gene transcription. Since GLIS3 was found to be methylated, PRTM5 might also be involved in mediating its methylation and as such modulate GLIS3 transcriptional activity or protein stability or alter its interactions with other proteins.
C-terminal binding protein 1 (CtBP1) functions as a co-repressor for a number of transcription factors, including GLIS2, by interacting with PXDLS consensus motifs, in which X is any amino acid [27]. CtBPs mediate transcriptional repression by recruiting histone deacetylases (HDACs) and histone methyltransferases. HDAC3 was found to be part of the GLIS2-CtBP1 transcription silencing complex. The latter is consistent with the concept that GLIS2 functions as a transcriptional repressor as indicated by data showing that GLIS2 represses the expression of several genes, including Gli1 and Wnt4 [18].
TAZ
GLIS3, but not GLIS1 or GLIS2, was found to interact with TAZ (WWTR1), a PDZ binding motif-containing transcriptional co-activator [28, 29]. TAZ is part of the Hippo signaling pathway that regulates its nuclear localization and activity [30]. The Hippo pathway plays a role in the regulation of many biological functions, including cell migration, differentiation, proliferation, and cell polarity. The WW-domain of TAZ recognizes a P/LPXY motif in the C terminus of GLIS3 [28]. Co-expression with GLIS3 promotes the translocation of TAZ from the cytoplasm to the nucleus where it co-localizes with GLIS3. TAZ enhances the transcriptional activity of GLIS3 and appears to function as a co-activator of GLIS3-mediated transcriptional activation. Whether GLIS3 activity in vivo is regulated by the Hippo signaling pathway is an attractive hypothesis that needs further study. Moreover, it is worthwhile noting that GLIS3-deficiency as well as loss of TAZ function leads to the development of polycystic kidney disease [28, 31]. These observations are consistent with a functional link between these two proteins and the development of polycystic kidney disease.
β-Catenin and p120 catenin
Yeast two-hybrid analysis identified β-catenin as a GLIS2-interacting partner [32]. The tetrahedral configuration of the first zinc finger of GLIS2 and the armadillo repeats of β-catenin are required for this interaction. GLIS2 inhibits β-catenin/T cell factor (TCF)-mediated transcription as well as the expression of the TCF target gene, cyclin D1 (Ccnd1) suggesting that it functions as a negative modulator of the Wnt/β-catenin-TCF signaling pathway consistent with GLIS2 repressor function.
GLIS2 also interacts with p120 catenin (CTNND1) and induces its translocation to the nucleus [33]. This interaction involved the 140 aa, N-terminal region of GLIS2 and the C terminus of p120 and was enhanced by the tyrosine kinase SRC. Interaction with p120 induces proteolytic cleavage of GLIS2 between ZF4 and ZD5 that results in the generation of a C-terminal-truncated GLIS2-lacking ZF5 (GLIS2∆C). Since ZF5 plays a critical role in the recognition of GLISBS by GLIS2, one would predict that GLIS2 is unable to bind this site and regulate GLISBS-dependent transcription. However, GLIS2∆C was still able to bind DNA possibly by recognizing a DNA sequence different from that of GLISBS or via interaction with another transcription factor. Moreover, GLIS2∆C was capable of inhibiting neural tube differentiation suggesting that it still has a function. Co-expression with E-cadherin, which sequesters p120, inhibited the p120–GLIS2 interaction.
Ubiquitin and SUMO ligases
Both GLIS2 and GLIS3 have been reported to be ubiquitinated and sumoylated [9, 34, 35]. Yeast two-hybrid and mass spectrometric analyses revealed that a number of WW-domain containing HECT E3 ubiquitin ligases, including ITCH, NEDD4, and SMURF1/2, interact with GLIS3 [9, 26]. These interactions with GLIS3 are mediated through their WW-domain, which recognizes a PPYP461 motif in GLIS3. ITCH was shown to enhance polyubiquitination of GLIS3 and promote its proteolytic degradation by proteasomes, and consequently reduce GLIS3 protein stability. ITCH inhibits the transcriptional activity of wild-type GLIS3, but not that of a PPYP mutant that is unable to interact with ITCH. This inhibition of GLIS3-mediated transactivation might be related to decreased GLIS3 protein levels. This study suggests that ubiquitination plays an important role in regulating GLIS3 protein stability and as a consequence its activity and function.
GLIS2 was found to interact with the E3 ubiquitin ligase, tripartite motif containing 32 (TRIM32, also named Bardet–Biedl syndrome 11 or BBS11), and BBS1 [35, 36], proteins that are part of the Bardet–Biedl syndrome (BSS) multiprotein complex, which plays a role in ciliogenesis and cilia maintenance. In zebrafish, deficiency in either TRIM32 or GLIS2 leads to the development of renal cysts. The 141–359 region of GLIS2 that includes the ZFD, interacts with the TRIM32 N -terminus containing the Ring and B-box domains. TRIM32 enhances K63-dependent polyubiquitination but inhibits K48-linked polyubiquitination of GLIS2, thereby inhibiting its proteasomal degradation. Unexpectedly, the catalytic activity of TRIM32 was not required for this increase in K63 ubiquitination suggesting the involvement of another ubiquitin ligase. TRIM32 also modifies GLIS2 transcriptional activity; it reversed the inhibition of β-catenin-mediated transcriptional activation by GLIS2 and inhibited the GLIS2-induced activation of the Ins2 promoter. In addition, TRIM32 changes the subnuclear localization of GLIS2 and instead of a diffuse pattern of expression it becomes localized to nuclear bodies [35], including PML bodies, which are involved in the regulation of multiple cellular functions, such as apoptosis, DNA repair, and cell cycle [37]. This co-localization and interaction with promyelocytic leukemia protein, PML (also named TRIM19) suggest that GLIS2 might have a role in these processes as well.
E3 SUMO ligase, PIAS4, was reported to also interact with GLIS2 and catalyze sumoylation of GLIS2 at multiple sites, including K195 [34]. This sumoylation was specific for SUMO3. The PIAS4-mediated sumoylation was shown to interfere with GLIS2 ubiquitination, thereby inhibiting its degradation by the proteasome and extending its half-life. Although mutation of K195R prevents ubiquitination and sumoylation at this site, the overall GLIS2 ubiquitination was increased and GLIS2 protein stability decreased. Increased sumoylation did not affect the inhibitory effect of GLIS2 on β-catenin-mediated gene activation, but inhibited its activation of the Ins2 promoter. These results suggest that sumoylation selectively influences GLIS2 transcriptional activity and function.
SUFU
As has been reported for GLI proteins [38], both GLIS2 and GLIS3 have been shown to interact with suppressor of fused (SUFU) [9, 18, 39]. GLIS3 interacts with SUFU through a VYGHF motif located within the NCR as well as with an undefined site at the C terminus of GLIS3. SUFU protects GLIS3 from proteasomal degradation, thereby stabilizing GLIS3 protein. The cullin-RING ubiquitin ligase cullin 3 (CUL3) also interacts with GLIS3 and promotes the ubiquitination and degradation of GLIS3. CUL3 interacts with the N terminus of GLIS3, a region that includes the SUFU binding motif. This raised the possibility that the stabilization of GLIS3 by SUFU may be due to its inhibition of GLIS3 interaction with CUL3. SUFU plays also an important role in the control of hedgehog/GLI signaling and regulates this pathway at multiple levels [40, 41]. In the case of GLIS3, co-expression of GLIS3 and SUFU causes translocation of SUFU to the nucleus, suggesting that GLIS3 and SUFU are part of a nuclear protein complex. Although SUFU was shown to moderately reduce GLIS3-mediated transcriptional activation, the precise function of SUFU in GLIS3-mediated transcriptional activation is not fully understood [9]. In contrast to GLIS3, the GLIS2–SUFU complex appears more susceptible to ubiquitination and degradation by the proteasome suggesting a different mode of regulation [18].
Genetic alterations in human GLIS1–3
GLIS3-associated mutations and variants
Genetic variations and mutations in the human GLIS3 gene, which maps to chromosome 9p24.2 (Fig. 2a), have been linked to a wide range of pathologies. Patients with loss-of-GLIS3-function mutations most consistently develop a syndrome referred to as neonatal diabetes and congenital hypothyroidism (NDH) and have a greatly reduced life span of a few days to several years [13, 42–48]. Abnormalities associated with GLIS3 mutations can extend to intrauterine growth retardation (IUGR), developmental delay, development of polycystic kidneys, congenital glaucoma, hepatic cholestasis, osteopenia, atrial septal defects, and minor facial dysmorphisms. Facial anomalies associated with GLIS3 mutations typically include depressed nasal bridge, bilateral low-set ears, long philtrum, large anterior fontanelle, and elongated, upslanted palpebral fissures. GLIS3-deficiency shows some phenotypic variability among patients; some patients do not develop cystic kidneys or skeletal abnormalities or show abnormal thyroid gland development. The phenotypic abnormalities observed in GLIS3-deficient mice largely mimic those observed in human patients [1, 4, 5, 22, 28, 49–51].
Fig. 2.

Schematic presentation of the genomic maps of the human GLIS1–3 genes. Missense and frame shift mutations, and deletions implicated in disease are indicated. SNPs associated with increased risk for several pathologies are also shown. a GLIS3. b GLIS2. c GLIS1
Mutations in human GLIS3 that cause loss of function, are extremely rare and were first described in several NDH patients from Saudi Arabia and France and subsequently in a few other patients [13, 42–47]. Genetic mutations in GLIS3 include deletions encompassing exons 5–9, 3–4, 9–11, 10–11, and several larger deletions covering regions > 100 kb that include exons 1–2 and part of intron 2 or exons 1–4 and part of intron 4 (Fig. 2a). Homozygous frameshift mutations, p.Arg780Profs*79, p.Gly311Alafs*15, and p.Pro772Leufs*35 resulting in an early termination codon and the loss of the GLIS3 transactivation domain, were identified in several NDH patients [43, 45, 46, 48]. In addition, several homozygous missense mutations, p.Arg589Trp, p.Cys536Trp and p.His561Tyr, within the DNA-binding domain of GLIS3, were found to be associated with NDH [43]. p.Cys536Trp and p.His561Tyr within the second ZF motif cause the collapse of the tetrahedral configuration of ZF2 and greatly diminishes the ability of GLIS3 to bind GLISBS. A patient with a p.Phe857Tyr missense mutation in GLIS3 was reported to develop neonatal diabetes and liver dysfunction and died of liver and kidney failure at 1.5 years of age [52]. A combination of heterozygous variants in GLIS3 (p.R720Q) and DUOX2 (p.R683L/p.L1343F) was found to be associated with congenital hypothyroidism [47].
Genome wide association studies (GWAS) have uncovered associations between single nucleotide polymorphisms (SNPs) in GLIS3 and several pathologies. The GLIS3-associated SNPs, rs7034200, rs7875253, rs7041847, and rs10814916, have been linked to elevated fasting glucose levels, altered β cell function, and increased risk for type 2 diabetes (T2D) [53–71] (Fig. 2a). The GLIS3 SNP rs2380949 was found to be associated with reduced insulin clearance, a predictor of T2D incidence [72], while the GLIS3 SNP, rs180867004, exhibited a sex-specific association with a higher risk for early onset of T2D in male American Pima Indians [68].
More than 40 genes have been identified as a susceptibility locus for type 1 diabetes (T1D); however, GLIS3 is one of a handful of genes that have also been linked to both T1D and T2D. The GLIS3 SNPs, rs7020673 and rs10758593, have been reported to be significantly associated with increased risk for T1D [54, 63, 73–76]. However, in a study of diabetic patients in Brazil no significant association was observed between these SNPs and T1D [77]. The low-frequency p.Ala908Val variant of GLIS3 was found to be strongly associated with resistance to T1D [78].
GLIS3 variations have been linked to a number of additional pathologies. Certain susceptibility loci are shared between different autoimmune disorders. For the T1D-associated GLIS3 SNP rs7020673, a significant association was found with rheumatoid arthritis risk [79]. Rs10116772 was found to be associated with osteoarthritis [80], while rs736893, was identified as a risk factor for primary angle closure glaucoma (PACG) [81, 82]. A rare GLIS3 duplication was reported to be associated with congenital heart defects [83]. Rs476155 was identified as a susceptibility locus for low HDL-cholesterol levels, a risk factor for coronary artery disease in ethnic Arabs [84]. A significant association was also found between the GLIS3 SNP, rs514716, and elevated levels of cerebrospinal fluid (CSF) tau and phosphorylated tau (ptau181), established biomarkers for AD [85, 86]. No significant correlation was found between T2D-associated GLIS3 SNP, rs10814916, and risk of Parkinson’s or Alzheimer’s disease (AD) [87].
GLIS2-associated mutations and variants
GLIS2 maps to chromosome 16p13.3 (Fig. 2b). Loss of GLIS2 function in humans as well as in mice lead to the development of type 7 nephronophthisis (NPHP7) [2, 88–91]. Nephronophthisis is a rare autosomal recessive cystic kidney disease characterized by renal atrophy, fibrosis, and interstitial infiltration of inflammatory cells. It is the most common genetic cause of end-stage renal disease in children and young adults. A homozygous transversion at the acceptor splice site of intron 6 (IVS5 + 1G > T) in GLIS2 was found to lead to loss of GLIS2 function and the development nephronophthisis [88]. A homozygous missense mutation, p.Cys175Arg (rs587777353), in exon 6 of GLIS2 was identified in a patient with nephronophthisis [89, 91]. This mutation, which is within the first ZF, destroys its tetrahedral configuration and thereby the ability of GLIS2 to optimally bind GLISBS and regulate GLISBS-dependent transcription. This mutation also changed the subcellular localization of GLIS2 from nucleus to cytoplasm, but had no effect on GLIS2 protein stability or its interaction with CTBP1, HDAC3 or β-catenin [89].
GLIS1-associated mutations and variants
The human GLIS1 gene maps to chromosome 1p32.3 (Fig. 2c). GLIS1 variants have been reported to be a risk factor for several neural pathologies. The intergenic SNP, rs185031519, near the GLIS1 locus was found to be associated with lower cerebrospinal fluid (CSF) amyloid-beta1-42 (Aβ42) levels and higher tau and phosphorylated tau (ptau181) levels [86]. Lower CSF Aβ42 levels and higher tau and ptau181 correlate with the number of neurofibrillary tangles and plaque load and are well-established AD endophenotypes. Rs185031519 was found to be associated with faster disease progression and increased AD risk.
A different GWAS study found an association between two GLIS1 SNPs, rs12082358 and rs12080993, and increased risk of autism spectrum disorder in a Taiwanese Han population [92]. An additional SNP in GLIS1 (rs797906) was identified as a susceptibility locus for late-onset Parkinson’s disease (PD) in a Han Chinese population [93]. And although individual studies did not find a significant correlation between this SNP and PD in Caucasian populations, pooled data appear to support an association between PD and rs797906 [93–97]. Another GLIS1-associated SNP (rs6663966) was found to be linked to increased severity of coronary artery calcified plaques and risk for cardiovascular disease in African Americans with T2D [98].
Molecular and physiological functions of GLIS1–3
GLIS as reprogramming factors
Several studies have established roles for GLIS proteins in reprogramming. Somatic cells can be reprogrammed into induced pluripotent stem cells (iPSCs) by ectopic expression of several reprogramming factors, OCT3/4 (POU5F1), SOX2, KLF4, and c-MYC (OSKM) [99]. GLIS1 significantly promotes the reprogramming of both human and mouse fibroblasts into iPSCs when co-expressed with OSK (referred to as OSKG), whereas down-regulation of GLIS1 by shRNAs reduced reprogramming efficiency [14, 100–102]. iPSCs generated by OSKG were shown to be fully able to produce germline competent chimaeras.
The mechanism by which GLIS1 enhances reprogramming efficiency is not fully understood. Gene profiling analysis demonstrated that GLIS1 enhanced the expression of several pluripotent genes, including NANOG, ESSRB, LIN28A, MYCN, MYCL1, and several WNT ligands, such as WNT3 and WNT8A. Most of these genes appeared to be indirectly regulated by GLIS1. Mesenchymal–epithelial transition (MET) is a critical element in the reprogramming of somatic cells. GLIS1 regulates several genes implicated in MET, including increased expression of the transcription factor FOXA2, a repressor of epithelial–mesenchymal transition (EMT), which as a consequence would promote MET and IPSC generation. GLIS1 was found to be in a protein complex with OCT4, KLF4, and SOX2. The N terminus and ZFD of GLIS1 are required for the interaction with KLF4. Whether this interaction has a role in mediating the increase in programming efficiency by GLIS1 needs further study [100].
The ability of GLIS1 to function as an efficient reprogramming factor was also demonstrated by transfecting human primary foreskin fibroblasts with a modified version of a non-infectious Venezuelan equine encephalitis (VEE) virus RNA replicon expressing OCT4, KLF4, SOX2, and GLIS1 [102]. Moreover, OCT4, KLF4, SOX2, GLIS1 and c-MYC, robustly generated iPSCs from human fibroblasts from older adults, age 54–77 [103]. A different study showed efficient reprogramming of human urine-derived cells using a high-efficiency episomal system containing OCT4, GLIS1, KLF4, SOX2, L-MYC, and the miR-302 cluster [104].
GLIS1 also enhances reprogramming induced by an alternative pathway. C-JUN functions as a powerful inhibitor of somatic cell reprogramming by inducing the expression of EMT genes, including TGFB and SNAIL, and inhibiting the expression of pluripotent genes, such as SOX2 and NANOG [105]. In contrast, JUN dimerization protein 2 (JDP2), which represses JUN-mediated transactivation, promotes reprogramming in conjunction with KLF4, SOX2 and MYC. JDP2 also induces reprogramming of MEFs into iPSCs via an alternative pathway in combination with the expression of GLIS1, lysine demethylase KDM2B, and the transcription factors ID1/3, SALL4, and LRH1 (NR5A2) [105].
A recent study demonstrated that GLIS3 can promote reprogramming in human somatic cells as efficiently as GLIS1 [106]. In contrast to GLIS1 and -3, GLIS2 had a negative effect on reprogramming [106, 107]. However, knockdown of GLIS2 expression in human ESCs by siRNA abolished the expression of OCT4, SOX2, and NANOG and loss of stem cell phenotype [107]. This was accompanied by differentiation and expression of multiple genes associated with endodermal and extraembryonic lineages. Knockdown of OCT4 also induced differentiation that was associated with decreased expression of GLIS2. OCT4 was found to regulate GLIS2 transcription directly by binding to its promoter region. Thus, these observations suggest that GLIS2 may have a role in maintaining the pluripotent state of hESCs. GLIS3, together with several other transcription factors, also promotes the reprogramming of human fibroblasts into retinal pigmented epithelial cells [108].
GLIS3 and thyroid hormone biosynthesis
Loss of GLIS3 function in humans and mice causes congenital or neonatal hypothyroidism [4, 43–46, 50, 109]. Congenital hypothyroidism is the most common inborn endocrine disorder and is caused by abnormalities in either thyroid development (thyroid dysgenesis) or thyroid hormone (T3/T4) biosynthesis (dyshormonogenesis) [110, 111]. The thyroid phenotype in GLIS3-deficient patients is quite variable ranging from aplasia/dysplasia to dyshormonogenesis [13] suggesting that GLIS3 might have a role in both thyroid gland development and thyroid hormone biosynthesis.
The development of neonatal hypothyroidism in GLIS3-deficient mice has been serving as a model to study the mechanisms underlying this pathology. GLIS3 expression in the thyroid gland is restricted to the thyroid follicular cells [24]. The basic morphology of the thyroid gland and the expression of several genes critical for thyroid development, such as Pax8, Ttf1 (Nkx2.1), and Ttf2 (FoxE1), are not greatly affected in PND7 Glis3-KO mice, suggesting that abnormal thyroid gland development does not play a major role in the development of hypothyroidism in these Glis3-KO mice, but has a major function in regulating thyroid hormone biosynthesis and homeostasis (Fig. 3).
Fig. 3.
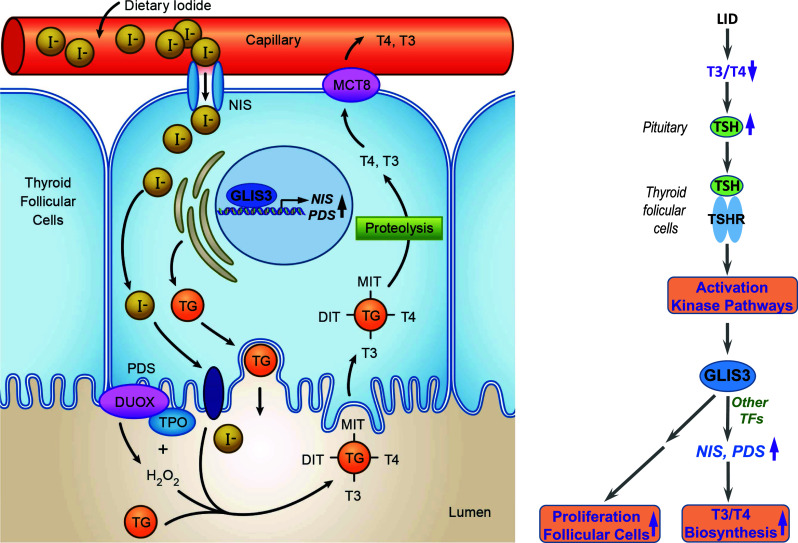
GLIS3 is essential for thyroid hormone biosynthesis. a GLIS3 expression is restricted to thyroid follicular cells where it directly regulates the transcription of several genes required for thyroid hormone biosynthesis, particularly the two iodide transporter genes, NIS and PDS. Loss of GLIS3 causes a dramatic decrease in NIS and PDS expression and iodide transport, and consequently reduced thyroid hormone biosynthesis and hypothyroidism. b Low iodide diet (LID) greatly elevates blood TSH levels. Interaction of TSH with the TSHR in thyroid follicular cells activates several kinase pathways and expression of the thyroid biosynthetic genes, including NIS and PDS, and cell cycle genes. However, these genes are not induced by TSH in GLIS3-deficient thyroid follicular cells suggesting that GLIS3 mediates the downstream effects of TSH/TSHR
Thyroid hormone biosynthesis is controlled by the hypothalamic–pituitary–thyroid axis, in which low T3/T4 leads to increased production of thyrotropin-releasing hormone (TRH) by the hypothalamus and thyroid-stimulating hormone (TSH) by the pituitary, which subsequently increases thyroid hormone production and blood levels [112] (Fig. 3). Increasing blood levels of thyroid hormone subsequently act as a negative feedback loop. Human and mice deficient in GLIS3 function exhibit low blood levels of T3 and T4, and highly elevated levels of thyroid-stimulating hormone (TSH). These observations indicate that the development of hypothyroidism is not due to the inability of the pituitary to respond to low thyroid hormone levels or to produce TSH.
Study of gene expression profiles of thyroid glands from wild-type and GLIS3-deficient mice revealed that GLIS3 regulates the expression of a selective set of genes critical for thyroid hormone biosynthesis. The expression of particularly the iodide transporters, Nis (Slc5a5) and pendrin (Pds, Slc26a4), was greatly repressed in the thyroid gland of GLIS3-deficient mice (Fig. 3). Previous studies reported that mutations in NIS that result in a nonfunctional NIS protein [113], as well as mutations in PDS [114], cause congenital hypothyroidism in humans. Thus, the suppression of these genes appears to be a major contributory factor to the impaired T3/T4 biosynthesis and the development of neonatal hypothyroidism in GLIS3-deficiency.
In addition to controlling T3/T4 biosynthesis, TSH regulates thyroid follicular proliferation. These actions are mediated through the interaction of TSH with the TSH-receptor (TSHR), a G-protein coupled receptor consisting of Gsα and Gq, and the subsequent activation of several kinase pathways, including cAMP- and phosphoinositol-dependent kinase, and the mTORC1/RPS6 pathway [115, 116]. In addition to the activation of several thyroid hormone biosynthetic genes, including Nis and Pds, persistent high TSH levels induce activation of many cell proliferation regulatory genes, including cyclins A2, B1 and B2 (Ccna2, Ccnb1, Ccnb2), and cell division cycle-associated protein 2 (Cdca2). Blood TSH levels are persistently elevated in low iodide diet resulting in increased proliferation of thyroid follicular cells and a significantly enlarged thyroid gland (goiter) (Fig. 3). In contrast to wild-type mice fed a low iodide diet, elevated TSH levels in GLIS3-deficient mice fed either a normal or low iodide diet do not induce expression of Nis and Pds or cell proliferation regulatory genes, such as Ccna2, Ccnb1, and Cdca2, and mice do not develop an enlarged thyroid gland. The lack of a TSH response is not due a loss of TSHR since Tshr expression was not altered in GLIS3-deficient mice. Together, these observations suggest that GLIS3 acts downstream of TSH/TSHR and mediates the transcriptional activation of Nis and Pds by TSH (Fig. 3). The TSH-dependent induction of cell cycle genes involves activation of the mTORC1/RPS6 pathway. The repression of most cell cycle genes in GLIS3-deficient mice does not involve direct transcriptional regulation by GLIS3, but appears to involve inhibition of RPS6 kinase activation.
GLIS3 and pancreas development
Studies demonstrating that loss of GLIS3 function in humans and mice causes neonatal diabetes indicated a regulatory role for GLIS3 in pancreatic β cells [1, 4, 5, 13, 22, 28, 42–50]. The development of hyperglycemia and hypoinsulinemia in GLIS3 deficiency was found to be related to aberrant pancreatic β cell generation and insulin production, suggesting that GLIS3 may have a role in the regulation of pancreas development. In the mouse, pancreas organogenesis starts at about E9.5 when multipotential pancreatic cells (MPCs) emerge from the foregut endoderm and form the dorsal and ventral pancreatic buds, which subsequently undergo extensive expansion and branching (Fig. 4) [117, 118]. By E12.5, the MPCs expressing PDX1, PTF1A, NKX6.1, HNF1β, and SOX9 become restricted to the branch tips, where they give rise to preacinar cells and subsequently mature acinar cells, while the trunk regions differentiate into PDX1+PTF1A−NKX6.1+HNF1β+SOX9+ bipotent progenitors (BPs). GLIS3 protein is first observed in BPs and is not detectable in MPCs or acinar cells [119]. The BPs subsequently give rise to neurogenin 3+ (NGN3+) endocrine progenitors and preductal cells, which then mature into ductal cells. The endocrine progenitors delaminate from the trunk via a still poorly understood process that involves EMT, and differentiate along the five endocrine cell lineages, α, β, γ, δ, and PP cells that produce glucagon, insulin, somatostatin, ghrelin, and pancreatic polypeptide (PP), respectively, thereby leading to the formation of pancreatic islets. GLIS3 protein remains expressed in (pre)ductal cells, NGN3+ endocrine progenitors, and in insulin+ and PP+ cells, but is repressed in α, γ, and δ cells (Fig. 4). This differential pattern of GLIS3 expression raises the question whether GLIS3 might have a role in endocrine lineage determination.
Fig. 4.

Schematic of the multiple functions of GLIS3 in the pancreas. GLIS3 protein is first detectable in bipotent cells (BPCs) and is not expressed in multipotent progenitor cells (MPCs) or acinar cells. GLIS3 protein remains expressed in preductal and ductal cells and in endocrine progenitors. When the endocrine progenitors differentiate into the five endocrine cell types, it remains expressed in β and PP cells, but is repressed in α, γ, and ∂ cells. GLIS3 deficiency inhibits differentiation of BPCs into NGN3+ endocrine progenitors and as a consequence the generation of endocrine cells. In addition, loss of GLIS3 function inhibits β cell maturation and greatly reduces insulin gene expression. GLIS3 deficiency also causes dilation of the pancreatic ducts and repression of Ppy expression in PP cells
In human patients with GLIS3 dysfunction, as well as in Glis3-deficient mice, the size of pancreatic islets and the number of β cells are greatly reduced leading to the development of neonatal diabetes [28, 45, 50]. This was found to be at least in part related to a reduction in the generation of NGN3+ endocrine progenitors during the secondary transition stage (E13.5–E15.5) of pancreas development [5, 14, 28]. The decrease in endocrine progenitors appears to be in part due to suppression of NGN3 expression, a key transcription factor that is required for the commitment of BPs to endocrine progenitors [117, 118, 120]. GLIS3 was shown to regulate Ngn3 transcription directly by binding to GLISBS in its promoter region [21, 22], likely in collaboration with other transcription factors, such as PDX1, SOX9, and HNF1β [121, 122]. The inhibition of NGN3+ endocrine progenitors is at least in part responsible for the observed decrease in pancreatic β cell generation, smaller islet size, and the development of neonatal diabetes in GLIS3-deficient mice. In addition to endocrine progenitor and pancreatic β cell generation, evidence is accumulating that in postnatal mouse pancreas GLIS3 regulates the maintenance and maturation of β cells [14, 19, 20, 28, 119]. GLIS3 transcriptionally regulates the expression of Ins1, Ins2, and Glut2 (Slc2a2), and the maturation markers, urocortin 3 (Ucn3) and MafA [123, 124], all of which are significantly down-regulated in postnatal GLIS3-deficient pancreas (Fig. 4). In addition to its role in β cells, GLIS3 regulates duct morphogenesis as well as the expression pancreatic polypeptide (Ppy) in PP cells [28]. GLIS3 deficiency causes significant dilation of pancreatic ducts and greatly reduces Ppy expression. Together these studies indicate that GLIS3 has multiple critical regulatory functions during pancreas development and in the adult pancreas: regulating the development of endocrine progenitors, the generation and maturation of pancreatic β cells, regulation of insulin and Ppy expression, as well as in maintaining normal duct morphology (Fig. 4).
Although global knockout of Glis3 results in neonatal diabetes, a pancreas-specific knockout of Glis3 in Glis3fl/fl/RipCre mice, expressing Cre under control of the rat insulin promoter, does not cause a diabetic phenotype [125, 126]. However, Pdx1Cre-mediated knockout of Glis3 yielded a mixed phenotype depending on the efficiency of the Pdx1Cre with 30% of the mice developing severe diabetes around PND21, 55% became mildly diabetic with aging, while 15% did not develop diabetes. The diabetic phenotype correlated with the degree by which GLIS3 was knocked out indicating that it is dose dependent. These observations are consistent with the conclusion that GLIS3 is required for the maintenance of postnatal pancreatic beta cells.
Several studies have provided evidence for a possible role of GLIS3 in the control of pancreatic β cell proliferation and apoptosis. This is indicated by studies showing that Glis3 heterozygous mice exhibit increased susceptibility to high fat diet-induced hyperglycemia and age-induced diabetes that appears to be associated with impaired proliferation and expansion of pancreatic β cells [126]. Tamoxifen-inducible, β cell-specific knockout of GLIS3 in Glis3fl/fl/Pdx1CreERT2 mice led to hypoinsulinemia and hyperglycemia and was associated with decreased insulin expression and an increase in apoptotic β cells [125–127]. Using a model of pancreatic β cell-selective endoplasmic reticulum (ER) stress in NOD mice, an association was found between increased apoptosis and the down-regulation of Glis3 and the anti-apoptotic gene, Manf [127]. This study further showed that GLIS3 heterozygous mice exhibit increased sensitivity to ER-stress induced apoptosis and diabetes. A different study reported that down-regulation of GLIS3 expression by siRNAs in rat pancreatic β cells INS-1E enhanced cell death via the intrinsic apoptotic pathway that involved modulation of the alternative splicing of the pro-apoptotic favoring the expression of BIMs, its pro-apoptotic form BIM [128]. Further studies are needed to understand the molecular mechanisms by which GLIS3 regulates various pancreatic β cell functions and how this relates to the development of diabetes in GLIS3-deficiency.
GLIS3 and regulation of insulin transcription
In addition to regulating pancreatic β cell generation, GLIS3 plays a critical role in the regulation of mouse insulin (mIns2) gene expression [1, 19, 20, 28, 45, 119]. The β cells in postnatal pancreas from GLIS3-deficient mice contain fewer and smaller insulin-containing granules than WT cells and exhibit a greatly reduced expression of mIns2 mRNA [119]. This is further supported by data showing that ectopic expression of GLIS3 can induce Ins2 expression in β cell lines. Ins2 expression is under complex control and a number of transcription factors, including Pdx1, MafA, and NeuroD1, which bind, respectively, A-, C-, and E-boxes in the proximal Ins2 promoter, have been implicated in the regulation of Ins2 transcription [123, 129]. Ins2 promoter analysis showed that GLIS3 can efficiently activate the Ins2 promoter by binding two GLISBS in its proximal promoter region. Mutation of both GLISBS sites abolishes the GLIS3-mediated activation [19]. The transcriptional activation by GLIS3 was mediated through its interaction with the co-activator CBP/p300. ChIP-Seq analysis supported the conclusion that GLIS3 regulates Ins2 transcription directly. Moreover, GLIS3 binding is required for optimal activation of the Ins2 promoter by PDX1, MAFA, and NEUROD1 [19, 20]. We proposed that GLIS3–CBP/p300 coactivator complex might help recruiting PDX1, MAFA, and NEUROD1 or stabilize their interaction with their respective DNA binding sites. GLIS3 was also shown to directly regulate MafA expression [20, 28]. Thus, the down-regulation of MAFA in GLIS3-KO islets likely contributes to the repression of Ins2 activation. Together, these data suggest that GLIS3 appears to regulate Ins2 transcription through several different mechanisms.
GLIS3 is essential for spermatogenesis
Glis3 is highly expressed in the testis where it plays a critical role in the regulation of early postnatal spermatogenesis [16, 130]. In the mouse testis at PND1, nondividing gonocytes migrate to the basal compartment of the seminiferous tubule where they differentiate into self-renewing spermatogonial stem cells (SSCs), which are distinguished by the high expression of inhibitor of DNA binding 4 (Id4) [131–134]. The SSCs sequentially differentiate into several subsets of spermatogonial progenitor cells (SPCs) that are classified as A-single (As), A-paired (Apr), and A-aligned spermatogonia (Aal4, Aal8, Aal16) consisting of 1, 2, 4, 8 and 16 cell(s), respectively (Fig. 5). The SSCs and SPCs together are referred to as undifferentiated spermatogonia and share the expression of a number of genes implicated in SSC maintenance and self-renewal, and PGC development, including Pax7, Etv5, Gfra1, Zbtb16 (Plzf), Lhx1, Sall4, Lin28, and Nanos2. The Apr and Aal SPCs are transient amplifying cells that become irreversibly committed to differentiation into type B spermatogonia [131–134]. This is accompanied by down-regulation of the expression of various genes, including Gfra1, Zbtb16, E-cadherin (Cdh1), Nanos2, and FoxO1 and induction of cKit. The type B spermatogonia then give rise to meiotic spermatocytes, which after meiosis is completed, differentiate into spermatids and ultimately mature spermatozoa. During postnatal spermatogenesis, GLIS3 protein is detectable in gonocytes, and most GFRA1+ and PLZF+ SSCs and SPCs, but not in differentiated c-Kit+ spermatogonia and germ cells at later stages [130] (Fig. 5). GLIS3 expression is lost during the Aal stages, after the down-regulation of GFRA1, but before the suppression of PLZF. GLIS3 is not detectable in Sertoli or Leydig cells. The number of undifferentiated PLZF+ spermatogonia is significantly decreased in testes from PND1 and PND7 GLIS3-deficient mice [130]. This decrease appeared not to be due to increased apoptosis, but to inhibition of differentiation and/or cell proliferation of SSCs and SPCs. As a consequence, the number of differentiated c-Kit+ spermatocytes, as well as the number of HSPA2+ cells, representing meiotic and post-meiotic cells, was greatly diminished in testes of Glis3-KO mice. Histological observations showed that spermatids and spermatozoa were largely absent in 3–5 weeks old Glis3-KO testes. This was accompanied by a dramatically reduced expression of genes associated with meiosis, spermatids and spermatozoa, including aurora kinase C (Aurkc), protamines 1 and 2 (Prm1, Prm2), sperm motility kinase 2a (Smok2a), and spermatogenesis associated 3 (Spata3) [130]. Together, these studies identify GLIS3 as a key regulator of early stages of postnatal spermatogenesis and demonstrate that loss of GLIS3 function greatly impairs the generation of spermatozoa, thereby rendering the Glis3-KO males infertile.
Fig. 5.
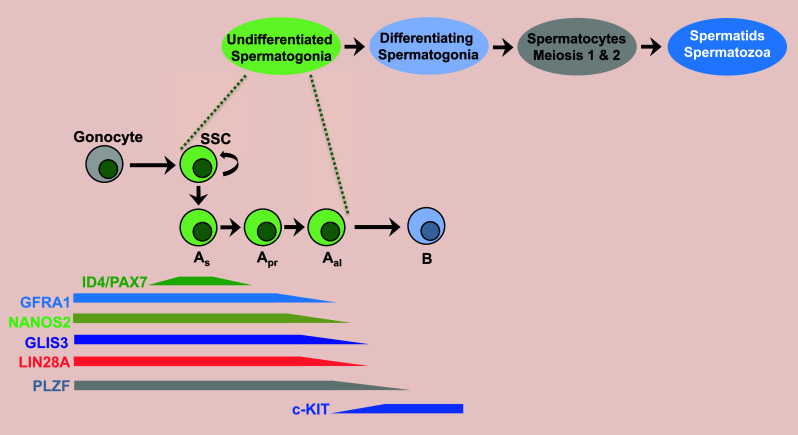
GLIS3 is required for early postnatal spermatogenesis. At PND1, non-mitotic gonocytes differentiate into spermatogonial stem cells (SSCs), which give rise to several spermatogonial progenitor (SPCs) subpopulations (As, Apr, Aal) with decreasing self-renewal capacity. These cells subsequently give rise to c-KIT+ differentiated spermatogonia and eventually mature spermatozoa. GLIS3 expression is restricted to gonocytes, SSCs, and SPCs. Loss of GLIS3 expression affects self-renewal of SSCs and early differentiation and impairs the generation of c-KIT+ differentiated spermatogonia leading to infertility
GDNF produced by Sertoli cells plays a critical role in regulating the proliferation and differentiation of SSCs and SPCs through action of its receptor, the GFRA1–RET complex [131, 133]. The expression of GFRA1 and RET was significantly decreased in Glis3-KO testis as was the expression of several GDNF-dependent genes that are important in SCC maintenance, including Etv5, Lhx1, and Brachyury homolog (T) [130]. The expression of the transcription factors, Cxcr4, Zbtb16, Ngn3 and Dmrt1, and the RNA binding proteins, Nanos2, Nanos3 and Lin28a, which are involved in the regulation of the maintenance proliferation, and differentiation of undifferentiated spermatogonia was also diminished in Glis3-KO testis. NANOS2 and LIN28A have been reported to be important in regulating self-renewal of SSCs and proliferation of SPCs, respectively [135, 136]. The expression of Piwi Like RNA-Mediated Gene Silencing 4 (Piwil4), which is expressed in gonocytes and SSCs [137], as well as several other genes involved in retrotransposon silencing suggest that GLIS3 might be involved in regulating the repression of transposable elements [130]. These studies suggest that GLIS3 may play a critical role in regulating the dynamics of the gonocytes–SSC–SPC transition and the proliferation and differentiation of these cells. The observation that the translocation of FoxO1 to the nucleus, which marks the gonocyte-to-SSC transition and is required for SSC proliferation [138], was significantly inhibited in Glis3-KO testes consistent with the hypothesis that GLIS3 might positively regulate the gonocyte-to-SSC transition and SCC self-renewal [130]. This is supported by data showing that the expression of FoxO1 target genes, Lhx1 and Sall4, and that of Id4, a specific marker of SSCs, were greatly reduced in Glis3-KO testes.
GLIS1 functions
Relatively little is known about the physiological functions of GLIS1. In postnatal mouse testis, Glis1 is expressed in spermatogonia, but not in pachytene spermatocytes and spermatids [15]. Glis1 null mice are fertile and display abnormalities in spermatogenesis upon aging. Glis1 is also highly expressed in mouse and bovine oocytes and 1-cell embryos. Glis1 mRNA expression is significantly reduced in 2-cell embryos and is no longer detectable in 4-cell mouse embryos and blastocysts or 8-cell bovine embryos [100, 139]. Down-regulation of GLIS1 in 4-cell bovine embryos by siRNAs resulted in defective development of embryos beyond the 16-cell stage, a time at which the developmental control by maternal transcription factors is shifted to embryonic control, a process referred to as zygotic gene activation (ZGA). The expression of PDHA1 and HSPA8, which are induced after ZGA, was significantly decreased in Glis1-downregulated bovine embryos suggesting that ZGA is not proceeding [139]. Together these observations suggest that GLIS1 is important for preimplantation development; however, whether GLIS1 is required for ZGA or whether it affects development at a preceding stage needs further study.
A recent study reported a positive role for GLIS1 in brown adipocyte differentiation [140]. This was indicated by data showing that exogenous expression of GLIS1 in mouse myoblast C2C12 cells decreases the expression of myogenic markers and enhances adipogenic marker expression, whereas down-regulation of GLIS1 by siRNAs has the inverse effects. This association was supported by the observed increase in Glis1 expression during brown adipocyte differentiation in C2C12 cells in which Kdm1a (Lsd1) expression was down-regulated.
Functions of GLIS2 in the kidney
During embryonic kidney development, Glis2 is well expressed in the ureteric bud, which subsequently forms the collecting ducts, but at low levels in S-shaped bodies that will form the nephrons [2, 88]. However, at later stages of development Glis2 is also detectable in renal tubules. Glis2 is highly expressed in the adult kidney [8]. As discussed above, loss of GLIS2 function causes nephronophthisis, a cystic kidney disease characterized by cyst formation, renal atrophy, inflammation, and fibrosis [2, 88–91]. GLIS2 deficiency in mice is also associated with an increase in apoptotic tubular cells and interstitial infiltration of inflammatory cells [2]. Progression of the disease leads to the development of proteinuria, elevated blood levels of urea nitrogen and creatine, and ultimately end-stage renal disease. Gene expression profiling showed that kidneys from Glis2-null mice exhibited increased expression of several inflammatory genes (e.g., Cxcl10, Ccl2, Cx3cl1) and genes related to fibrosis (e.g., Col1a1, Col3a1, Ltbp1), apoptosis (e.g., Casp4), and EMT (e.g., Snai1, Fsp1/S100A4, Ctgf, Tgfβ1) [2, 88]. The increased expression of these genes at least partially explains the development of inflammation, fibrosis, and renal atrophy in Glis2-deficient kidneys. The increased inflammatory response likely contributes significantly to the progression of nephronophthisis.
Observations showing that kidney tubule cells in Glis2-knockout mice express a number of mesenchymal markers is consistent with the concept that they undergo EMT [2, 88]. This is further supported by studies showing that down-regulation of GLIS2 expression in murine inner medullary collecting duct IMCD3 cells, in which GLIS2 is abundantly expressed, leads to increased cell migration and changes in gene expression that are characteristic of EMT [18]. This includes suppression of E-cadherin and induction of FSP1. In addition, down-regulation of GLIS2 significantly increased the expression of Gli1 as well as two GLI1 target genes, Snai1 and Wnt4. The transcription factor SNAI1 promotes EMT by inhibiting E-cadherin (Cdh1) transcription and inducing the expression of mesenchymal markers, such as vimentin and fibroblast-specific protein 1 (FSP1 or S100A4). ChIP analysis demonstrated that GLIS2 was associated with the upstream promoter regulatory region of Snai1 and Wnt4 and repressed the activation of the Snai1 and Wnt4 promoter. Together, these findings led to the hypothesis that GLIS2 negatively regulates Snai1 and Wnt4 transcription through both an indirect and direct mechanism by, respectively, repressing the expression of the positive regulator Gli1 and by directly binding to GLISBS in the Snai1 and Wnt4 promoters thereby competing with GLI1 for the same sites [18]. Thus, GLIS2 acts as a suppressor of the SHH/GLI signaling pathway and thereby helping to maintain the epithelioid phenotype of renal tubule cells.
A recent study showed that deficiency in GLIS2 function suppresses the growth of cysts in Kif3a knockout kidneys [141]. Kidney-specific knockout of Kif3a (Ksp-CreKif3af/−), one of the kinesins required for primary cilium formation, causes loss of primary cilia and development of polycystic kidneys [142]. This leads to increased blood urea nitrogen and serum creatinine levels, renal failure and death around the age of 6 weeks. Crossing these mice with Glis2-deficient mice suppressed proliferation of renal tubule cells, decreased DNA damage and apoptosis, and led to reduced growth of renal cysts and improved kidney function. Thus, GLIS2 deficiency was able to partially rescue the polycystic phenotype in Ksp-CreKif3af/− mice [141]. Immortalized tubular epithelial cells derived from these Kif3a null mice also exhibited accelerated cell cycle progression, decreased p53 stability, and increased DNA damage and apoptosis, while down-regulation of GLIS2 by shRNA reversed this phenotype. In addition, loss of GLIS2 function in Kif3a null as well as wild-type kidney tubule cells induced senescence as indicated by the positive staining for the senescence markers, H3K9me3 and β-galactosidase.
Unilateral ureteral obstruction, a mouse model of kidney injury, caused more severe injury and dilated tubules in kidneys of heterozygous Glis2 null mice than WT mice [143]. Glis2 mutant tubular epithelial cells appeared less differentiated and interstitial collagen depositions were increased, which was at least in part related to enhanced proliferation of pericytes and interstitial fibroblasts due to increased expression of SHH in heterozygous Glis2-deficient kidneys.
Functions of GLIS3 in the kidney
Glis3 is highly expressed in the kidney, where it localizes to the epithelial cells of renal tubules, collecting ducts, and Bowman’s capsule [2]. Although not in all human patients, GLIS3 deficiency in humans and mice leads to the development of polycystic kidney disease [5, 13, 45, 46, 49]. Glis3-KO mice develop prominent glomerulocysts as well as dilated distal tubules and collecting ducts. Deficiency in Zinc Finger E-Box Binding Homeobox 2 (ZEB2), a SMAD-interacting transcription factor, also causes glomerulocystic kidney disease [144]. This is associated with decreased GLIS3 expression suggesting that it might be part of the mechanism by which ZEB2 deficiency causes glomerulocyst formation. Similarly, knockout of hepatocyte nuclear factor 1b (HNF1b) in mice causes polycystic kidney disease as well as cyst formation in pancreatic ducts [121, 145]. The down-regulation of Glis3 expression in HNF1b-KO pancreas may be causally related to the development of dilated ducts.
MRI studies of polycystic kidneys from GLIS3-deficient mice showed that kidney volume, cyst volume and cyst-to-kidney volume ratio, increased significantly with age and progression of the disease [51]. Treatment of Glis3 null mice with rapamycin inhibited the progression of polycystic kidney disease, but did not improve renal functions significantly. The molecular mechanism(s) involved in polycystic kidney disease are still poorly understood. Changes in cell proliferation, planar cell polarity, and defects in primary cilium structure or signaling have been implicated in polycystic kidney disease [146–150]. Inflammation and fibrosis also play an important role in the pathogenesis of cystic renal disease [151].
GLIS proteins, the primary cilium, and cystic kidney disease
The primary cilium is a non-motile, microtubule-based organelle that protrudes from the surface of many eukaryotic cells [150, 152–155] (Fig. 6). It constitutes a subcellular compartment that allows enrichment of membrane receptors and their downstream signaling molecules, thereby serving as an important sensory organelle and signaling hub. An increasing number of membrane receptors for a wide variety of signals as diverse as peptide hormones, neurotransmitters, odorants, lipids, and light, have been associated with the primary cilium. These include many G protein-coupled receptors (GPCRs), including receptors for Sonic Hedgehog (SHH), WNT, and PDGFA, somatostatin, dopamine, and serotonin [152, 156, 157]. In addition to GPCRs, several Ca2+ channels have been reported to localize to the primary cilium, including the transient receptor potential (TRP) channels, TRPM4 and TRPV4, and the calcium-activated chloride channel ANO1 [158–160]. The primary cilium plays a key role in the regulation of embryonic development, proliferation, and autophagy. Disruption of the primary cilium or associated signaling pathways has been implicated in a broad range of genetic disorders, collectively referred to as ciliopathies. This includes autosomal recessive and autosomal dominant polycystic kidney disease (ARPKD and ADPKD, respectively), nephronophthisis (NPHP), Bardet–Biedl syndrome, Joubert syndrome and Meckel–Gruber syndrome, cancer, and several neuropsychiatric disorders [152, 153, 161–164]. Polycystin PC2, a member of the TRP family, forms with polycystin PC1 a receptor-channel complex that is localized to the primary cilium [147, 165]. Mutations in PC1 and PC2 are implicated in the majority of autosomal dominant polycystic kidney disease PKD1 and PKD2, respectively, while PKHD1, encoding fibrocystin, is associated with autosomal recessive polycystic kidney disease.
Fig. 6.

Regulation of GLIS3 activity and function by an upstream primary cilium-associated signaling pathway. Several studies have provided evidence for the localization of GLIS3 to the primary cilium. TNPO1-GLIS3 interaction allows GLIS3 to enter the primary cilium, where it is transported to the tip of the primary cilium by the anterograde intraflagellar transport (IFT) system. We hypothesize that activation of a GPCR localized to the ciliary membrane by an external signal modulates GLIS3 activity possibly through posttranslational modification or proteolytic cleavage. This may lead to the generation of either a GLIS3 activator (GLIS3A) or GLIS3 repressor (GLIS3R). The retrograde IFT mediates the transportation of GLIS3 out of the primary cilium. GLIS3 then regulates target gene transcription after its translocation to the nucleus
Study of the subcellular localization of GLIS3 protein has indicated that it largely localizes to the nucleus in cells in vivo and when it is ectopically expressed in cultured cells [16, 23, 24, 119]. Immunohistochemistry using proximal tubule TKPTS cells ectopically expressing GLIS3-EGFP and kidney sections from transgenic mice expressing an GLIS3-EGFP fusion protein, showed that in addition to its nuclear localization, GLIS3 is also detectable in the tip of the primary cilium [49]. This is supported by proteomics identifying GLIS3 as a primary cilium-associated protein [166]. A recent study showed that the localization of GLI1–3 proteins to the primary cilium is mediated by transportin 1 (TNPO1, also named importin 1 or karyopherin beta-2), which recognizes a primary cilium localization signal (CLS) in the NCR of GLI1–3 proteins [167]. Interestingly, GLIS3 also contains this CLS consensus motif within its NCR (Fig. 1) suggesting that its primary cilium localization might be mediated by a similar mechanism. It is further interesting to note that GLIS3 interacts with SUFU, a protein that has been demonstrated to interact and co-localize with GLI proteins to the tip of the primary cilium [9, 10, 28, 166]. The ciliary localization of GLIS3 raises the question whether GLIS3 activity is under the control of an external signal that acts through a specific primary cilium-associated GPCR, as has been demonstrated for the hedgehog-GLI pathway [4, 155, 168]. We hypothesize that GLIS3 activity is regulated by an as-yet unknown external signal that activates a distinct GPCR signaling pathway that subsequently modulates GLIS3 activity and promotes GLIS3 nuclear localization [4] (Fig. 6). This modulation of GLIS3 activity might involve posttranslational modification (e.g., phosphorylation) and/or proteolytic cleavage of GLIS3. Further studies are needed to identify such upstream signal(s) to understand the role of the primary cilium in the control of GLIS3 activity. The identification of such a signal might open possibilities to regulate GLIS3 activity in vivo and as such might provide opportunities for the development of new therapeutic strategies for diabetes, hypothyroidism, and other diseases.
Deficiency in GLIS2 function leads to the development of nephronophthisis, a different type of renal ciliopathy [49, 88]. Evidence has been provided indicating that GLIS2 interacts with SUFU and might localize to the primary cilium [18]; however, GLIS2 does not contain a CLS consensus sequence similar to that of GLIS3. Whether GLIS2 localizes to the primary cilium in vivo and whether this is part of a physiologically relevant signaling pathway needs further study. Nothing is known about the potential ciliary localization of GLIS1.
Summary and conclusions/concluding remarks
Recent studies revealed that the GLIS1–3 transcription factors function as critical regulators of embryonic development and many biological processes. Significant molecular insights have been obtained about the mechanisms by which GLIS3 regulates of thyroid hormone biosynthesis, pancreatic β cell generation and insulin expression, and spermatogenesis, as well as the role of GLIS2 in the maintenance of normal kidney functions. These studies have provided not only a better understanding of these biological processes, but also provided insights into the mechanisms by which GLIS3 dysfunction leads to hypothyroidism, diabetes, and infertility and GLIS2-deficiency results in nephronophthisis. Future studies are needed to elucidate the roles of GLIS1–3 in other biological processes and pathologies, such as the role of GLIS3 in the development cystic kidney disease, as well as their emerging regulatory functions of GLIS1–3 in different stem/progenitor populations. Progress would highly benefit from the availability of good GLIS antibodies, which have been a limiting factor in these studies. In addition, future studies have to determine the roles of different posttranslational modifications and GLIS-interacting proteins in the modulation of GLIS1–3 activity and function. Increasing evidence showing that GLIS2 and GLIS3 localize to the primary cilium and might be part of a primary cilium-associated GPCR signaling pathway is very exciting. Further insights into the nature of this upstream signaling pathway may allow regulation of GLIS activity by small molecules and provide opportunities for the development of new therapeutic strategies for diabetes, hypothyroidism, cystic kidney disease and other pathologies.
Acknowledgements
This research is supported by the Intramural Research Program of the National Institute of Environmental Health Sciences, the National Institutes of Health [Z01-ES-100485]. I would like to thank Drs. Hong Soon Kang and David Scoville for their comments on the manuscript.
References
- 1.Kang HS, ZeRuth G, Lichti-Kaiser K, Vasanth S, Yin Z, Kim YS, Jetten AM. Gli-similar (Glis) Krüppel-like zinc finger proteins: insights into their physiological functions and critical roles in neonatal diabetes and cystic renal disease. Histol Histopathol. 2010;25:1481–1496. doi: 10.14670/hh-25.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim YS, Kang HS, Herbert R, Beak JY, Collins JB, Grissom SF, Jetten AM. Krüppel-like zinc finger protein Glis2 is essential for the maintenance of normal renal functions. Mol Cell Biol. 2008;28:2358–2367. doi: 10.1128/MCB.01722-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lamar E, Kintner C, Goulding M. Identification of NKL, a novel Gli-Krüppel zinc-finger protein that promotes neuronal differentiation. Development. 2001;128:1335–1346. doi: 10.1242/dev.128.8.1335. [DOI] [PubMed] [Google Scholar]
- 4.Lichti-Kaiser K, ZeRuth G, Jetten AM. Transcription factor Gli-similar 3 (Glis3): implications for the development of congenital hypothyroidism. J Endocrinol Diabetes Obes. 2014;2:1050–1059. [PMC free article] [PubMed] [Google Scholar]
- 5.Lichti-Kaiser K, ZeRuth G, Kang HS, Vasanth S, Jetten AM. Gli-similar proteins: their mechanisms of action, physiological functions, and roles in disease. Vitam Horm. 2012;88:141–171. doi: 10.1016/B978-0-12-394622-5.00007-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakashima M, Tanese N, Ito M, Auerbach W, Bai C, Furukawa T, Toyono T, Akamine A, Joyner AL. A novel gene, GliH1, with homology to the Gli zinc finger domain not required for mouse development. Mech Dev. 2002;119:21–34. doi: 10.1016/S0925-4773(02)00291-5. [DOI] [PubMed] [Google Scholar]
- 7.Zhang F, Jetten AM. Genomic structure of the gene encoding the human Gli-related, Krüppel-like zinc finger protein Glis2. Gene. 2001;280:49–57. doi: 10.1016/S0378-1119(01)00764-8. [DOI] [PubMed] [Google Scholar]
- 8.Zhang F, Nakanishi G, Kurebayashi S, Yoshino K, Perantoni A, Kim YS, Jetten AM. Characterization of Glis2, a novel gene encoding a Gli-related, Krüppel like transcription factor with transactivation and repressor functions. Roles in kidney development and neurogenesis. J Biol Chem. 2002;277:10139–10149. doi: 10.1074/jbc.M108062200. [DOI] [PubMed] [Google Scholar]
- 9.ZeRuth GT, Yang XP, Jetten AM. Modulation of the transactivation function and stability of Krüppel-like zinc finger protein Gli-similar 3 (Glis3) by suppressor of fused. J Biol Chem. 2011;286:22077–22089. doi: 10.1074/jbc.M111.224964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hashimoto H, Miyamoto R, Watanabe N, Shiba D, Ozato K, Inoue C, Kubo Y, Koga A, Jindo T, Narita T, Naruse K, Ohishi K, Nogata K, Shin IT, Asakawa S, Shimizu N, Miyamoto T, Mochizuki T, Yokoyama T, Hori H, Takeda H, Kohara Y, Wakamatsu Y. Polycystic kidney disease in the medaka (Oryzias latipes) pc mutant caused by a mutation in the Gli-similar3 (Glis3) gene. PLoS ONE. 2009;4:e6299. doi: 10.1371/journal.pone.0006299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duan H, Skeath JB, Nguyen HT. Drosophila Lame duck, a novel member of the Gli superfamily, acts as a key regulator of myogenesis by controlling fusion-competent myoblast development. Development. 2001;128:4489–4500. doi: 10.1242/dev.128.22.4489. [DOI] [PubMed] [Google Scholar]
- 12.Furlong EE, Andersen EC, Null B, White KP, Scott MP. Patterns of gene expression during Drosophila mesoderm development. Science. 2001;293:1629–1633. doi: 10.1126/science.1062660. [DOI] [PubMed] [Google Scholar]
- 13.Dimitri P. The role of GLIS3 in thyroid disease as part of a multisystem disorder. Best Pract Res Clin Endocrinol Metab. 2017;31:175–182. doi: 10.1016/j.beem.2017.04.007. [DOI] [PubMed] [Google Scholar]
- 14.Scoville DW, Kang HS, Jetten AM. GLIS1–3: emerging roles in reprogramming, stem and progenitor cell differentiation and maintenance. Stem Cell Investig. 2017;4:80. doi: 10.21037/sci.2017.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim YS, Lewandoski M, Perantoni AO, Kurebayashi S, Nakanishi G, Jetten AM. Identification of Glis1, a novel Gli-related, Krüppel-like zinc finger protein containing transactivation and repressor functions. J Biol Chem. 2002;277:30901–30913. doi: 10.1074/jbc.M203563200. [DOI] [PubMed] [Google Scholar]
- 16.Kim YS, Nakanishi G, Lewandoski M, Jetten AM. Glis3, a novel member of the Glis subfamily of Krüppel-like zinc finger proteins with repressor and activation functions. Nucleic Acids Res. 2003;31:5513–5525. doi: 10.1093/nar/gkg776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vasanth S, ZeRuth G, Kang HS, Jetten AM. Identification of nuclear localization, DNA binding, and transactivating mechanisms of Krüppel-like zinc finger protein Gli-similar 2 (Glis2) J Biol Chem. 2011;286:4749–4759. doi: 10.1074/jbc.M110.165951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li B, Rauhauser AA, Dai J, Sakthivel R, Igarashi P, Jetten AM, Attanasio M. Increased Hedgehog signaling in postnatal kidney results in aberrant activation of nephron developmental programs. Hum Mol Genet. 2011;20:4155–4166. doi: 10.1093/hmg/ddr339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.ZeRuth GT, Takeda Y, Jetten AM. The Krüppel-like protein Gli-similar 3 (Glis3) functions as a key regulator of insulin transcription. Mol Endocrinol. 2013;27:1692–1705. doi: 10.1210/me.2013-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang Y, Chang BH, Samson SL, Li MV, Chan L. The Krüppel-like zinc finger protein Glis3 directly and indirectly activates insulin gene transcription. Nucleic Acids Res. 2009;37:2529–2538. doi: 10.1093/nar/gkp122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim YS, Kang HS, Takeda Y, Hom L, Song HY, Jensen J, Jetten AM. Glis3 regulates neurogenin 3 expression in pancreatic beta-cells and interacts with its activator, Hnf6. Mol Cells. 2012;34:193–200. doi: 10.1007/s10059-012-0109-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Y, Chang B, Yechoor V, Chen W, Li L, Tsai M, Chan L. The Krüppel-like zinc finger protein Glis3 transactivates neurogenin 3 for proper fetal pancreatic islet differentiation in mice. Diabetologia. 2011;54:2595–2605. doi: 10.1007/s00125-011-2255-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beak JY, Kang HS, Kim YS, Jetten AM. Functional analysis of the zinc finger and activation domains of Glis3 and mutant Glis3(NDH1) Nucleic Acids Res. 2008;36:1690–1702. doi: 10.1093/nar/gkn009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang HS, Kumar D, Liao G, Lichti-Kaiser K, Gerrish K, Liao XH, Refetoff S, Jothi R, Jetten AM. Glis3 is indispensable for TSH/TSHR-dependent thyroid hormone biosynthesis and follicular cell proliferation. J Clin Invest. 2017;127:4326–4337. doi: 10.1172/JCI94417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koyabu Y, Nakata K, Mizugishi K, Aruga J, Mikoshiba K. Physical and functional interactions between Zic and Gli proteins. J Biol Chem. 2001;276:6889–6892. doi: 10.1074/jbc.C000773200. [DOI] [PubMed] [Google Scholar]
- 26.ZeRuth GT, Williams JG, Cole YC, Jetten AM. HECT E3 ubiquitin ligase itch functions as a novel negative regulator of Gli-similar 3 (Glis3) transcriptional activity. PLoS ONE. 2015;10:e0131303. doi: 10.1371/journal.pone.0131303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim SC, Kim YS, Jetten AM. Krüppel-like zinc finger protein Gli-similar 2 (Glis2) represses transcription through interaction with C-terminal binding protein 1 (Ctbp1) Nucleic Acids Res. 2005;33:6805–6815. doi: 10.1093/nar/gki985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang HS, Kim YS, ZeRuth G, Beak JY, Kilic G, Jensen J, Sosa-Pineda B, Jetten AM. Transcription factor Glis3: a novel critical player in the regulation of pancreatic β-cell development. Mol Cell Biol. 2009;29:6366–6379. doi: 10.1128/MCB.01259-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meng Z, Moroishi T, Guan KL. Mechanisms of Hippo pathway regulation. Genes Dev. 2016;30:1–17. doi: 10.1101/gad.274027.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hansen CG, Moroishi T, Guan KL. YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol. 2015;25:499–513. doi: 10.1016/j.tcb.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hossain Z, Ali SM, Ko HL, Xu J, Ng CP, Guo K, Qi Z, Ponniah S, Hong W, Hunziker W. Glomerulocystic kidney disease in mice with a targeted inactivation of Wwtr1. Proc Natl Acad Sci USA. 2007;104:1631–1636. doi: 10.1073/pnas.0605266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim YS, Kang HS, Jetten AM. The Krüppel-like zinc finger protein Glis2 functions as a negative modulator of the Wnt/beta-catenin signaling pathway. FEBS Lett. 2007;581:858–864. doi: 10.1016/j.febslet.2007.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hosking CR, Ulloa F, Hogan C, Ferber E, Figueroa A, Gevaert K, Birchmeier W, Briscoe J, Fujita Y. The transcriptional repressor Glis2 is a novel binding partner for p120 catenin. Mol Biol Cell. 2007;18:1918–1927. doi: 10.1091/mbc.e06-10-0941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramachandran H, Herfurth K, Grosschedl R, Schafer T, Walz G. Sumoylation blocks the ubiquitin-mediated degradation of the nephronophthisis gene product Glis2/NPHP7. PLoS ONE. 2015;10:e0130275. doi: 10.1371/journal.pone.0130275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramachandran H, Schafer T, Kim Y, Herfurth K, Hoff S, Lienkamp SS, Kramer-Zucker A, Walz G. Interaction with the Bardet–Biedl gene product TRIM32/BBS11 modifies the half-life and localization of Glis2/NPHP7. J Biol Chem. 2014;289:8390–8401. doi: 10.1074/jbc.M113.534024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim YH, Epting D, Slanchev K, Engel C, Walz G, Kramer-Zucker A. A complex of BBS1 and NPHP7 is required for cilia motility in zebrafish. PLoS ONE. 2013;8:e72549. doi: 10.1371/journal.pone.0072549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sahin U, Lallemand-Breitenbach V, de Thé H. PML nuclear bodies: regulation, function and therapeutic perspectives. J Pathol. 2014;234:289–291. doi: 10.1002/path.4426. [DOI] [PubMed] [Google Scholar]
- 38.Merchant M, Vajdos F, Ultsch M, Maun H, Wendt U, Cannon J, Desmarais W, Lazarus R, de Vos A, de Sauvage F. Suppressor of fused regulates Gli activity through a dual binding mechanism. Mol Cell Biol. 2004;24:8627–8641. doi: 10.1128/MCB.24.19.8627-8641.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 40.Han Y, Shi Q, Jiang J. Multisite interaction with Sufu regulates Ci/Gli activity through distinct mechanisms in Hh signal transduction. Proc Natl Acad Sci USA. 2015;112:6383–6388. doi: 10.1073/pnas.1421628112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Z, Shen L, Law K, Zhang Z, Liu X, Hua H, Li S, Huang H, Yue S, Hui CC, Cheng SY. Suppressor of fused chaperones Gli proteins to generate transcriptional responses to Sonic Hedgehog signaling. Mol Cell Biol. 2017;37:e00421-16. doi: 10.1128/MCB.00421-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dimitri P, De Franco E, Habeb AM, Gurbuz F, Moussa K, Taha D, Wales JK, Hogue J, Slavotinek A, Shetty A, Balasubramanian M. An emerging, recognizable facial phenotype in association with mutations in GLI-similar 3 (GLIS3) Am J Med Genet A. 2016;70:1918–1923. doi: 10.1002/ajmg.a.37680. [DOI] [PubMed] [Google Scholar]
- 43.Dimitri P, Habeb AM, Gurbuz F, Millward A, Wallis S, Moussa K, Akcay T, Taha D, Hogue J, Slavotinek A, Wales JK, Shetty A, Hawkes D, Hattersley AT, Ellard S, De Franco E. Expanding the clinical spectrum associated with GLIS3 mutations. J Clin Endocrinol Metab. 2015;100:E1362–E1369. doi: 10.1210/jc.2015-1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dimitri P, Warner J, Minton J, Patch A, Ellard S, Hattersley A, Hawkes D, Wales J, Gregory J. Novel GLIS3 mutations demonstrate an extended multisystem phenotype. Eur J Endocrinol. 2011;164:437–443. doi: 10.1530/EJE-10-0893. [DOI] [PubMed] [Google Scholar]
- 45.Senee V, Chelala C, Duchatelet S, Feng D, Blanc H, Cossec JC, Charon C, Nicolino M, Boileau P, Cavener DR, Bougneres P, Taha D, Julier C. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet. 2006;38:682–687. doi: 10.1038/ng1802. [DOI] [PubMed] [Google Scholar]
- 46.Taha D, Barbar M, Kanaan H, Williamson Balfe J. Neonatal diabetes mellitus, congenital hypothyroidism, hepatic fibrosis, polycystic kidneys, and congenital glaucoma: a new autosomal recessive syndrome? Am J Med Genet A. 2003;122A:269–273. doi: 10.1002/ajmg.a.20267. [DOI] [PubMed] [Google Scholar]
- 47.Fu C, Luo S, Long X, Li Y, She S, Hu X, Mo M, Wang Z, Chen Y, He C, Su J, Zhang Y, Lin F, Xie B, Li Q, Chen S. Mutation screening of the GLIS3 gene in a cohort of 592 Chinese patients with congenital hypothyroidism. Clin Chim Acta. 2017;476:38–43. doi: 10.1016/j.cca.2017.11.011. [DOI] [PubMed] [Google Scholar]
- 48.Alghamdi KA, Alsaedi AB, Aljasser A, Altawil A, Kamal NM. Extended clinical features associated with novel GLIS3 mutation: a case report. BMC Endocr Disord. 2017;17:14. doi: 10.1186/s12902-017-0160-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kang HS, Beak JY, Kim YS, Herbert R, Jetten AM. Glis3 is associated with primary cilia and WWTR1/TAZ and implicated in polycystic kidney disease. Mol Cell Biol. 2009;29:2556–2569. doi: 10.1128/MCB.01620-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watanabe N, Hiramatsu K, Miyamoto R, Yasuda K, Suzuki N, Oshima N, Kiyonari H, Shiba D, Nishio S, Mochizuki T, Yokoyama T, Maruyama S, Matsuo S, Wakamatsu Y, Hashimoto H. A murine model of neonatal diabetes mellitus in Glis3-deficient mice. FEBS Lett. 2009;583:2108–2113. doi: 10.1016/j.febslet.2009.05.039. [DOI] [PubMed] [Google Scholar]
- 51.Xie L, Qi Y, Subashi E, Liao G, Miller-DeGraff L, Jetten AM, Johnson GA. 4D MRI of polycystic kidneys from rapamycin-treated Glis3-deficient mice. NMR Biomed. 2015;28:546–554. doi: 10.1002/nbm.3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cao B, Gong C, Wu D, Lu C, Liu F, Liu X, Zhang Y, Gu Y, Qi Z, Li X, Liu M, Li W, Su C, Liang X, Feng M. Genetic analysis and follow-up of 25 neonatal diabetes mellitus patients in China. J Diabetes Res. 2016;2016:6314368. doi: 10.1155/2016/6314368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barker A, Sharp SJ, Timpson NJ, Bouatia-Naji N, Warrington NM, Kanoni S, Beilin LJ, Brage S, Deloukas P, Evans DM, Grontved A, Hassanali N, Lawlor DA, Lecoeur C, Loos RJ, Lye SJ, McCarthy MI, Mori TA, Ndiaye NC, Newnham JP, Ntalla I, Pennell CE, St Pourcain B, Prokopenko I, Ring SM, Sattar N, Visvikis-Siest S, Dedoussis GV, Palmer LJ, Froguel P, Smith GD, Ekelund U, Wareham NJ, Langenberg C. Association of genetic loci with glucose levels in childhood and adolescence: a meta-analysis of over 6,000 children. Diabetes. 2011;60:1805–1812. doi: 10.2337/db10-1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C, Plagnol V, Pociot F, Schuilenburg H, Smyth DJ, Stevens H, Todd JA, Walker NM, Rich SS, Type 1 Diabetes Genetics C Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boesgaard TW, Grarup N, Jorgensen T, Borch-Johnsen K, Hansen T, Pedersen O. Variants at DGKB/TMEM195, ADRA2A, GLIS3 and C2CD4B loci are associated with reduced glucose-stimulated beta cell function in middle-aged Danish people. Diabetologia. 2010;53:1647–1655. doi: 10.1007/s00125-010-1753-5. [DOI] [PubMed] [Google Scholar]
- 56.Cho YS, Chen CH, Hu C, Long J, Ong RT, Sim X, Takeuchi F, Wu Y, Go MJ, Yamauchi T, Chang YC, Kwak SH, Ma RC, Yamamoto K, Adair LS, Aung T, Cai Q, Chang LC, Chen YT, Gao Y, Hu FB, Kim HL, Kim S, Kim YJ, Lee JJ, Lee NR, Li Y, Liu JJ, Lu W, Nakamura J, Nakashima E, Ng DP, Tay WT, Tsai FJ, Wong TY, Yokota M, Zheng W, Zhang R, Wang C, So WY, Ohnaka K, Ikegami H, Hara K, Cho YM, Cho NH, Chang TJ, Bao Y, Hedman AK, Morris AP, McCarthy MI, Takayanagi R, Park KS, Jia W, Chuang LM, Chan JC, Maeda S, Kadowaki T, Lee JY, Wu JY, Teo YY, Tai ES, Shu XO, Mohlke KL, Kato N, Han BG, Seielstad M. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in East Asians. Nat Genet. 2012;44:67–72. doi: 10.1038/ng.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dou HY, Wang YY, Yang N, Heng ML, Zhou X, Bu HE, Xu F, Zhao TN, Huang H, Wang HW. Association between genetic variants and characteristic symptoms of type 2 diabetes: a matched case–control study. Chin J Integr Med. 2016;23:415–424. doi: 10.1007/s11655-015-2290-3. [DOI] [PubMed] [Google Scholar]
- 58.Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, Wheeler E, Glazer NL, Bouatia-Naji N, Gloyn AL, Lindgren CM, Mägi R, Morris AP, Randall J, Johnson T, Elliott P, Rybin D, Thorleifsson G, Steinthorsdottir V, Henneman P, Grallert H, Dehghan A, Hottenga JJ, Franklin CS, Navarro P, Song K, Goel A, Perry JR, Egan JM, Lajunen T, Grarup N, Sparsø T, Doney A, Voight BF, Stringham HM, Li M, Kanoni S, Shrader P, Cavalcanti-Proença C, Kumari M, Qi L, Timpson NJ, Gieger C, Zabena C, Rocheleau G, Ingelsson E, An P, O’Connell J, Luan J, Elliott A, McCarroll SA, Payne F, Roccasecca RM, Pattou F, Sethupathy P, Ardlie K, Ariyurek Y, Balkau B, Barter P, Beilby JP, Ben-Shlomo Y, Benediktsson R, Bennett AJ, Bergmann S, Bochud M, Boerwinkle E, Bonnefond A, Bonnycastle LL, Borch-Johnsen K, Böttcher Y, Brunner E, Bumpstead SJ, Charpentier G, Chen YD, Chines P, Clarke R, Coin LJ, Cooper MN, Cornelis M, Crawford G, Crisponi L, Day IN, de Geus EJ, Delplanque J, Dina C, Erdos MR, Fedson AC, Fischer-Rosinsky A, Forouhi NG, Fox CS, Frants R, Franzosi MG, Galan P, Goodarzi MO, Graessler J, Groves CJ, Grundy S, Gwilliam R, Gyllensten U, Hadjadj S, Hallmans G, Hammond N, Han X, Hartikainen AL, Hassanali N, Hayward C, Heath SC, Hercberg S, Herder C, Hicks AA, Hillman DR, Hingorani AD, Hofman A, Hui J, Hung J, Isomaa B, Johnson PR, Jørgensen T, Jula A, Kaakinen M, Kaprio J, Kesaniemi YA, Kivimaki M, Knight B, Koskinen S, Kovacs P, Kyvik KO, Lathrop GM, Lawlor DA, Le Bacquer O, Lecoeur C, Li Y, Lyssenko V, Mahley R, Mangino M, Manning AK, Martínez-Larrad MT, McAteer JB, McCulloch LJ, McPherson R, Meisinger C, Melzer D, Meyre D, Mitchell BD, Morken MA, Mukherjee S, Naitza S, Narisu N, Neville MJ, Oostra BA, Orrù M, Pakyz R, Palmer CN, Paolisso G, Pattaro C, Pearson D, Peden JF, Pedersen NL, Perola M, Pfeiffer AF, Pichler I, Polasek O, Posthuma D, Potter SC, Pouta A, Province MA, Psaty BM, Rathmann W, Rayner NW, Rice K, Ripatti S, Rivadeneira F, Roden M, Rolandsson O, Sandbaek A, Sandhu M, Sanna S, Sayer AA, Scheet P, Scott LJ, Seedorf U, Sharp SJ, Shields B, Sigurethsson G, Sijbrands EJ, Silveira A, Simpson L, Singleton A, Smith NL, Sovio U, Swift A, Syddall H, Syvänen AC, Tanaka T, Thorand B, Tichet J, Tönjes A, Tuomi T, Uitterlinden AG, van Dijk KW, van Hoek M, Varma D, Visvikis-Siest S, Vitart V, Vogelzangs N, Waeber G, Wagner PJ, Walley A, Walters GB, Ward KL, Watkins H, Weedon MN, Wild SH, Willemsen G, Witteman JC, Yarnell JW, Zeggini E, Zelenika D, Zethelius B, Zhai G, Zhao JH, Zillikens MC, DIAGRAM Consortium. GIANT Consortium. Global BPgen Consortium. Borecki IB, Loos RJ, Meneton P, Magnusson PK, Nathan DM, Williams GH, Hattersley AT, Silander K, Salomaa V, Smith GD, Bornstein SR, Schwarz P, Spranger J, Karpe F, Shuldiner AR, Cooper C, Dedoussis GV, Serrano-Ríos M, Morris AD, Lind L, Palmer LJ, Hu FB, Franks PW, Ebrahim S, Marmot M, Kao WH, Pankow JS, Sampson MJ, Kuusisto J, Laakso M, Hansen T, Pedersen O, Pramstaller PP, Wichmann HE, Illig T, Rudan I, Wright AF, Stumvoll M, Campbell H, Wilson JF, Anders Hamsten on behalf of Procardis Consortium. MAGIC investigators. Bergman RN, Buchanan TA, Collins FS, Mohlke KL, Tuomilehto J, Valle TT, Altshuler D, Rotter JI, Siscovick DS, Penninx BW, Boomsma DI, Deloukas P, Spector TD, Frayling TM, Ferrucci L, Kong A, Thorsteinsdottir U, Stefansson K, van Duijn CM, Aulchenko YS, Cao A, Scuteri A, Schlessinger D, Uda M, Ruokonen A, Jarvelin MR, Waterworth DM, Vollenweider P, Peltonen L, Mooser V, Abecasis GR, Wareham NJ, Sladek R, Froguel P, Watanabe RM, Meigs JB, Groop L, Boehnke M, McCarthy MI, Florez JC, Barroso I. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42:105–116. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Florez JC, Jablonski KA, McAteer JB, Franks PW, Mason CC, Mather K, Horton E, Goldberg R, Dabelea D, Kahn SE, Arakaki RF, Shuldiner AR, Knowler WC. Effects of genetic variants previously associated with fasting glucose and insulin in the diabetes prevention program. PLoS ONE. 2012;7:e44424. doi: 10.1371/journal.pone.0044424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fujita H, Hara K, Shojima N, Horikoshi M, Iwata M, Hirota Y, Tobe K, Seino S, Kadowaki T. Variations with modest effects have an important role in the genetic background of type 2 diabetes and diabetes-related traits. J Hum Genet. 2012;57:776–779. doi: 10.1038/jhg.2012.110. [DOI] [PubMed] [Google Scholar]
- 61.Hong KW, Chung M, Cho SB. Meta-analysis of genome-wide association study of homeostasis model assessment beta cell function and insulin resistance in an East Asian population and the European results. Mol Genet Genomics. 2014;289:1247–1255. doi: 10.1007/s00438-014-0885-6. [DOI] [PubMed] [Google Scholar]
- 62.Hu C, Zhang R, Wang C, Wang J, Ma X, Hou X, Lu J, Yu W, Jiang F, Bao Y, Xiang K, Jia W. Variants from GIPR, TCF7L2, DGKB, MADD, CRY2, GLIS3, PROX1, SLC30A8 and IGF1 are associated with glucose metabolism in the Chinese. PLoS ONE. 2010;5:e15542. doi: 10.1371/journal.pone.0015542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kiani AK, John P, Bhatti A, Zia A, Shahid G, Akhtar P, Wang X, Demirci FY, Kamboh MI. Association of 32 type 1 diabetes risk loci in Pakistani patients. Diabetes Res Clin Pract. 2015;108:137–142. doi: 10.1016/j.diabres.2015.01.022. [DOI] [PubMed] [Google Scholar]
- 64.Li H, Gan W, Lu L, Dong X, Han X, Hu C, Yang Z, Sun L, Bao W, Li P, He M, Sun L, Wang Y, Zhu J, Ning Q, Tang Y, Zhang R, Wen J, Wang D, Zhu X, Guo K, Zuo X, Guo X, Yang H, Zhou X, Zhang X, Qi L, Loos RJ, Hu FB, Wu T, Liu Y, Liu L, Yang Z, Hu R, Jia W, Ji L, Li Y, Lin X. A genome-wide association study identifies GRK5 and RASGRP1 as type 2 diabetes loci in Chinese Hans. Diabetes. 2013;62:291–298. doi: 10.2337/db12-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu C, Li H, Qi L, Loos RJ, Qi Q, Lu L, Gan W, Lin X. Variants in GLIS3 and CRY2 are associated with type 2 diabetes and impaired fasting glucose in Chinese Hans. PLoS One. 2011;6:e21464. doi: 10.1371/journal.pone.0021464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mahajan A, Go MJ, Zhang W, Below JE, Gaulton KJ, Ferreira T, Horikoshi M, Johnson AD, Ng MC, Prokopenko I, Saleheen D, Wang X, Zeggini E, Abecasis GR, Adair LS, Almgren P, Atalay M, Aung T, Baldassarre D, Balkau B, Bao Y, Barnett AH, Barroso I, Basit A, Been LF, Beilby J, Bell GI, Benediktsson R, Bergman RN, Boehm BO, Boerwinkle E, Bonnycastle LL, Burtt N, Cai Q, Campbell H, Carey J, Cauchi S, Caulfield M, Chan JC, Chang LC, Chang TJ, Chang YC, Charpentier G, Chen CH, Chen H, Chen YT, Chia KS, Chidambaram M, Chines PS, Cho NH, Cho YM, Chuang LM, Collins FS, Cornelis MC, Couper DJ, Crenshaw AT, van Dam RM, Danesh J, Das D, de Faire U, Dedoussis G, Deloukas P, Dimas AS, Dina C, Doney AS, Donnelly PJ, Dorkhan M, van Duijn C, Dupuis J, Edkins S, Elliott P, Emilsson V, Erbel R, Eriksson JG, Escobedo J, Esko T, Eury E, Florez JC, Fontanillas P, Forouhi NG, Forsen T, Fox C, Fraser RM, Frayling TM, Froguel P, Frossard P, Gao Y, Gertow K, Gieger C, Gigante B, Grallert H, Grant GB, Grrop LC, Groves CJ, Grundberg E, Guiducci C, Hamsten A, Han BG, Hara K, Hassanali N, Hattersley AT, Hayward C, Hedman AK, Herder C, Hofman A, Holmen OL, Hovingh K, Hreidarsson AB, Hu C, Hu FB, Hui J, Humphries SE, Hunt SE, Hunter DJ, Hveem K, Hydrie ZI, Ikegami H, Illig T, Ingelsson E, Islam M, Isomaa B, Jackson AU, Jafar T, James A, Jia W, Jockel KH, Jonsson A, Jowett JB, Kadowaki T, Kang HM, Kanoni S, Kao WH, Kathiresan S, Kato N, Katulanda P, Keinanen-Kiukaanniemi KM, Kelly AM, Khan H, Khaw KT, Khor CC, Kim HL, Kim S, Kim YJ, Kinnunen L, Klopp N, Kong A, Korpi-Hyovalti E, Kowlessur S, Kraft P, Kravic J, Kristensen MM, Krithika S, Kumar A, Kumate J, Kuusisto J, Kwak SH, Laakso M, Lagou V, Lakka TA, Langenberg C, Langford C, Lawrence R, Leander K, Lee JM, Lee NR, Li M, Li X, Li Y, Liang J, Liju S, Lim WY, Lind L, Lindgren CM, Lindholm E, Liu CT, Liu JJ, Lobbens S, Long J, Loos RJ, Lu W, Luan J, Lyssenko V, Ma RC, Maeda S, Magi R, Mannisto S, Matthews DR, Meigs JB, Melander O, Metspalu A, Meyer J, Mirza G, Mihailov E, Moebus S, Mohan V, Mohlke KL, Morris AD, Muhleisen TW, Muller-Nurasyid M, Musk B, Nakamura J, Nakashima E, Navarro P, Ng PK, Nica AC, Nilsson PM, Njolstad I, Nothen MM, Ohnaka K, Ong TH, Owen KR, Palmer CN, Pankow JS, Park KS, Parkin M, Pechlivanis S, Pedersen NL, Peltonen L, Perry JR, Peters A, Pinidiyapathirage JM, Platou CG, Potter S, Price JF, Qi L, Radha V, Rallidis L, Rasheed A, Rathman W, Rauramaa R, Raychaudhuri S, Rayner NW, Rees SD, Rehnberg E, Ripatti S, Robertson N, Roden M, Rossin EJ, Rudan I, Rybin D, Saaristo TE, Salomaa V, Saltevo J, Samuel M, Sanghera DK, Saramies J, Scott J, Scott LJ, Scott RA, Segre AV, Sehmi J, Sennblad B, Shah N, Shah S, Shera AS, Shu XO, Shuldiner AR, Sigurdsson G, Sijbrands E, Silveira A, Sim X, Sivapalaratnam S, Small KS, So WY, Stancakova A, Stefansson K, Steinbach G, Steinthorsdottir V, Stirrups K, Strawbridge RJ, Stringham HM, Sun Q, Suo C, Syvanen AC, Takayanagi R, Takeuchi F, Tay WT, Teslovich TM, Thorand B, Thorleifsson G, Thorsteinsdottir U, Tikkanen E, Trakalo J, Tremoli E, Trip MD, Tsai FJ, Tuomi T, Tuomilehto J, Uitterlinden AG, Valladares-Salgado A, Vedantam S, Veglia F, Voight BF, Wang C, Wareham NJ, Wennauer R, Wickremasinghe AR, Wilsgaard T, Wilson JF, Wiltshire S, Winckler W, Wong TY, Wood AR, Wu JY, Wu Y, Yamamoto K, Yamauchi T, Yang M, Yengo L, Yokota M, Young R, Zabaneh D, Zhang F, Zhang R, Zheng W, Zimmet PZ, Altshuler D, Bowden DW, Cho YS, Cox NJ, Cruz M, Hanis CL, Kooner J, Lee JY, Seielstad M, Teo YY, Boehnke M, Parra EJ, Chambers JC, Tai ES, McCarthy MI, Morris AP. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet. 2014;46:234–244. doi: 10.1038/ng.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morris AP, Voight BF, Teslovich TM, Ferreira T, Segre AV, Steinthorsdottir V, Strawbridge RJ, Khan H, Grallert H, Mahajan A, Prokopenko I, Kang HM, Dina C, Esko T, Fraser RM, Kanoni S, Kumar A, Lagou V, Langenberg C, Luan J, Lindgren CM, Muller-Nurasyid M, Pechlivanis S, Rayner NW, Scott LJ, Wiltshire S, Yengo L, Kinnunen L, Rossin EJ, Raychaudhuri S, Johnson AD, Dimas AS, Loos RJ, Vedantam S, Chen H, Florez JC, Fox C, Liu CT, Rybin D, Couper DJ, Kao WH, Li M, Cornelis MC, Kraft P, Sun Q, van Dam RM, Stringham HM, Chines PS, Fischer K, Fontanillas P, Holmen OL, Hunt SE, Jackson AU, Kong A, Lawrence R, Meyer J, Perry JR, Platou CG, Potter S, Rehnberg E, Robertson N, Sivapalaratnam S, Stancakova A, Stirrups K, Thorleifsson G, Tikkanen E, Wood AR, Almgren P, Atalay M, Benediktsson R, Bonnycastle LL, Burtt N, Carey J, Charpentier G, Crenshaw AT, Doney AS, Dorkhan M, Edkins S, Emilsson V, Eury E, Forsen T, Gertow K, Gigante B, Grant GB, Groves CJ, Guiducci C, Herder C, Hreidarsson AB, Hui J, James A, Jonsson A, Rathmann W, Klopp N, Kravic J, Krjutskov K, Langford C, Leander K, Lindholm E, Lobbens S, Mannisto S, Mirza G, Muhleisen TW, Musk B, Parkin M, Rallidis L, Saramies J, Sennblad B, Shah S, Sigurethsson G, Silveira A, Steinbach G, Thorand B, Trakalo J, Veglia F, Wennauer R, Winckler W, Zabaneh D, Campbell H, van Duijn C, Uitterlinden AG, Hofman A, Sijbrands E, Abecasis GR, Owen KR, Zeggini E, Trip MD, Forouhi NG, Syvanen AC, Eriksson JG, Peltonen L, Nothen MM, Balkau B, Palmer CN, Lyssenko V, Tuomi T, Isomaa B, Hunter DJ, Qi L, Shuldiner AR, Roden M, Barroso I, Wilsgaard T, Beilby J, Hovingh K, Price JF, Wilson JF, Rauramaa R, Lakka TA, Lind L, Dedoussis G, Njolstad I, Pedersen NL, Khaw KT, Wareham NJ, Keinanen-Kiukaanniemi SM, Saaristo TE, Korpi-Hyovalti E, Saltevo J, Laakso M, Kuusisto J, Metspalu A, Collins FS, Mohlke KL, Bergman RN, Tuomilehto J, Boehm BO, Gieger C, Hveem K, Cauchi S, Froguel P, Baldassarre D, Tremoli E, Humphries SE, Saleheen D, Danesh J, Ingelsson E, Ripatti S, Salomaa V, Erbel R, Jockel KH, Moebus S, Peters A, Illig T, de Faire U, Hamsten A, Morris AD, Donnelly PJ, Frayling TM, Hattersley AT, Boerwinkle E, Melander O, Kathiresan S, Nilsson PM, Deloukas P, Thorsteinsdottir U, Groop LC, Stefansson K, Hu F, Pankow JS, Dupuis J, Meigs JB, Altshuler D, Boehnke M, McCarthy MI. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981–990. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Muller YL, Piaggi P, Chen P, Wiessner G, Okani C, Kobes S, Knowler WC, Bogardus C, Hanson RL, Baier LJ. Assessing variation across 8 established East Asian loci for type 2 diabetes mellitus in American Indians: suggestive evidence for new sex-specific diabetes signals in GLIS3 and ZFAND3. Diabetes Metab Res Rev. 2017;33:e2869. doi: 10.1002/dmrr.2869. [DOI] [PubMed] [Google Scholar]
- 69.Rees SD, Hydrie MZ, O’Hare JP, Kumar S, Shera AS, Basit A, Barnett AH, Kelly MA. Effects of 16 genetic variants on fasting glucose and type 2 diabetes in South Asians: ADCY5 and GLIS3 variants may predispose to type 2 diabetes. PLoS One. 2011;6:e24710. doi: 10.1371/journal.pone.0024710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sakai K, Imamura M, Tanaka Y, Iwata M, Hirose H, Kaku K, Maegawa H, Watada H, Tobe K, Kashiwagi A, Kawamori R, Maeda S. Replication study for the association of 9 East Asian GWAS-derived loci with susceptibility to type 2 diabetes in a Japanese population. PLoS One. 2013;8:e76317. doi: 10.1371/journal.pone.0076317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wagner R, Dudziak K, Herzberg-Schafer SA, Machicao F, Stefan N, Staiger H, Haring HU, Fritsche A. Glucose-raising genetic variants in MADD and ADCY5 impair conversion of proinsulin to insulin. PLoS One. 2011;6:e23639. doi: 10.1371/journal.pone.0023639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Goodarzi MO, Guo X, Cui J, Jones MR, Haritunians T, Xiang AH, Chen YD, Taylor KD, Buchanan TA, Hsueh WA, Raffel LJ, Rotter JI. Systematic evaluation of validated type 2 diabetes and glycaemic trait loci for association with insulin clearance. Diabetologia. 2013;56:1282–1290. doi: 10.1007/s00125-013-2880-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Winkler C, Krumsiek J, Buettner F, Angermuller C, Giannopoulou EZ, Theis FJ, Ziegler AG, Bonifacio E. Feature ranking of type 1 diabetes susceptibility genes improves prediction of type 1 diabetes. Diabetologia. 2014;57:2521–2529. doi: 10.1007/s00125-014-3362-1. [DOI] [PubMed] [Google Scholar]
- 74.Steck AK, Dong F, Wong R, Fouts A, Liu E, Romanos J, Wijmenga C, Norris JM, Rewers MJ. Improving prediction of type 1 diabetes by testing non-HLA genetic variants in addition to HLA markers. Pediatr Diabetes. 2014;15:355–362. doi: 10.1111/pedi.12092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grant SF, Qu HQ, Bradfield JP, Marchand L, Kim CE, Glessner JT, Grabs R, Taback SP, Frackelton EC, Eckert AW, Annaiah K, Lawson ML, Otieno FG, Santa E, Shaner JL, Smith RM, Skraban R, Imielinski M, Chiavacci RM, Grundmeier RW, Stanley CA, Kirsch SE, Waggott D, Paterson AD, Monos DS, Group DER. Polychronakos C, Hakonarson H. Follow-up analysis of genome-wide association data identifies novel loci for type 1 diabetes. Diabetes. 2009;58:290–295. doi: 10.2337/db08-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fortune MD, Guo H, Burren O, Schofield E, Walker NM, Ban M, Sawcer SJ, Bowes J, Worthington J, Barton A, Eyre S, Todd JA, Wallace C. Statistical colocalization of genetic risk variants for related autoimmune diseases in the context of common controls. Nat Genet. 2015;47:839–846. doi: 10.1038/ng.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Duarte GCK, Assmann TS, Dieter C, de Souza BM, Crispim D. GLIS3 rs7020673 and rs10758593 polymorphisms interact in the susceptibility for type 1 diabetes mellitus. Acta Diabetol. 2017;54:813–821. doi: 10.1007/s00592-017-1009-7. [DOI] [PubMed] [Google Scholar]
- 78.Awata T, Yamashita H, Kurihara S, Morita-Ohkubo T, Miyashita Y, Katayama S, Kawasaki E, Tanaka S, Ikegami H, Maruyama T, Shimada A, Takahashi K, Kawabata Y, Kobayashi T, Nishida N, Mawatari Y. A low-frequency GLIS3 variant associated with resistance to Japanese type 1 diabetes. Biochem Biophys Res Commun. 2013;437:521–525. doi: 10.1016/j.bbrc.2013.06.102. [DOI] [PubMed] [Google Scholar]
- 79.Kiani AK, Jahangir S, John P, Bhatti A, Zia A, Wang X, Demirci FY, Kamboh MI. Genetic link of type 1 diabetes susceptibility loci with rheumatoid arthritis in Pakistani patients. Immunogenetics. 2015;67:277–282. doi: 10.1007/s00251-015-0839-0. [DOI] [PubMed] [Google Scholar]
- 80.Casalone E, Tachmazidou I, Zengini E, Hatzikotoulas K, Hackinger S, Suveges D, Steinberg J, Rayner NW, Consortium A. Wilkinson JM, Panoutsopoulou K, Zeggini E. A novel variant in GLIS3 is associated with osteoarthritis. Ann Rheum Dis. 2018;77:620–623. doi: 10.1136/annrheumdis-2017-211848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Khor CC, Do T, Jia H, Nakano M, George R, Abu-Amero K, Duvesh R, Chen LJ, Li Z, Nongpiur ME, Perera SA, Qiao C, Wong HT, Sakai H, Barbosa de Melo M, Lee MC, Chan AS, Azhany Y, Dao TL, Ikeda Y, Perez-Grossmann RA, Zarnowski T, Day AC, Jonas JB, Tam PO, Tran TA, Ayub H, Akhtar F, Micheal S, Chew PT, Aljasim LA, Dada T, Luu TT, Awadalla MS, Kitnarong N, Wanichwecharungruang B, Aung YY, Mohamed-Noor J, Vijayan S, Sarangapani S, Husain R, Jap A, Baskaran M, Goh D, Su DH, Wang H, Yong VK, Yip LW, Trinh TB, Makornwattana M, Nguyen TT, Leuenberger EU, Park KH, Wiyogo WA, Kumar RS, Tello C, Kurimoto Y, Thapa SS, Pathanapitoon K, Salmon JF, Sohn YH, Fea A, Ozaki M, Lai JS, Tantisevi V, Khaing CC, Mizoguchi T, Nakano S, Kim CY, Tang G, Fan S, Wu R, Meng H, Nguyen TT, Tran TD, Ueno M, Martinez JM, Ramli N, Aung YM, Reyes RD, Vernon SA, Fang SK, Xie Z, Chen XY, Foo JN, Sim KS, Wong TT, Quek DT, Venkatesh R, Kavitha S, Krishnadas SR, Soumittra N, Shantha B, Lim BA, Ogle J, de Vasconcellos JP, Costa VP, Abe RY, de Souza BB, Sng CC, Aquino MC, Kosior-Jarecka E, Fong GB, Tamanaja VC, Fujita R, Jiang Y, Waseem N, Low S, Pham HN, Al-Shahwan S, Craven ER, Khan MI, Dada R, Mohanty K, Faiq MA, Hewitt AW, Burdon KP, Gan EH, Prutthipongsit A, Patthanathamrongkasem T, Catacutan MA, Felarca IR, Liao CS, Rusmayani E, Istiantoro VW, Consolandi G, Pignata G, Lavia C, Rojanapongpun P, Mangkornkanokpong L, Chansangpetch S, Chan JC, Choy BN, Shum JW, Than HM, Oo KT, Han AT, Yong VH, Ng XY, Goh SR, Chong YF, Hibberd ML, Seielstad M, Png E, Dunstan SJ, Chau NV, Bei J, Zeng YX, Karkey A, Basnyat B, Pasutto F, Paoli D, Frezzotti P, Wang JJ, Mitchell P, Fingert JH, Allingham RR, Hauser MA, Lim ST, Chew SH, Ebstein RP, Sakuntabhai A, Park KH, Ahn J, Boland G, Snippe H, Stead R, Quino R, Zaw SN, Lukasik U, Shetty R, Zahari M, Bae HW, Oo NL, Kubota T, Manassakorn A, Ho WL, Dallorto L, Hwang YH, Kiire CA, Kuroda M, Djamal ZE, Peregrino JI, Ghosh A, Jeoung JW, Hoan TS, Srisamran N, Sandragasu T, Set SH, Doan VH, Bhattacharya SS, Ho CL, Tan DT, Sihota R, Loon SC, Mori K, Kinoshita S, Hollander AI, Qamar R, Wang YX, Teo YY, Tai ES, Hartleben-Matkin C, Lozano-Giral D, Saw SM, Cheng CY, Zenteno JC, Pang CP, Bui HT, Hee O, Craig JE, Edward DP, Yonahara M, Neto JM, Guevara-Fujita ML, Xu L, Ritch R, Liza-Sharmini AT, Wong TY, Al-Obeidan S, Do NH, Sundaresan P, Tham CC, Foster PJ, Vijaya L, Tashiro K, Vithana EN, Wang N, Aung T. Genome-wide association study identifies five new susceptibility loci for primary angle closure glaucoma. Nat Genet. 2016;48:556–562. doi: 10.1038/ng.3540. [DOI] [PubMed] [Google Scholar]
- 82.Nongpiur ME, Cheng CY, Duvesh R, Vijayan S, Baskaran M, Khor CC, Allen J, Kavitha S, Venkatesh R, Goh D, Husain R, Boey PY, Quek D, Ho CL, Wong TT, Perera S, Wong TY, Krishnadas SR, Sundaresan P, Aung T, Vithana EN. Evaluation of primary angle closure glaucoma susceptibility loci in patients with early stages of angle closure disease. Ophthalmology. 2018;125:664–670. doi: 10.1016/j.ophtha.2017.11.016. [DOI] [PubMed] [Google Scholar]
- 83.Sanchez-Castro M, Eldjouzi H, Charpentier E, Busson PF, Hauet Q, Lindenbaum P, Delasalle-Guyomarch B, Baudry A, Pichon O, Pascal C, Lefort B, Bajolle F, Pezard P, Schott JJ, Dina C, Redon R, Gournay V, Bonnet D, Le Caignec C. Search for rare copy-number variants in congenital heart defects identifies novel candidate genes and a potential role for FOXC1 in patients with coarctation of the aorta. Circ Cardiovasc Genet. 2016;9:86–94. doi: 10.1161/CIRCGENETICS.115.001213. [DOI] [PubMed] [Google Scholar]
- 84.Wakil SM, Ram R, Muiya NP, Andres E, Mazhar N, Hagos S, Alshahid M, Meyer BF, Morahan G, Dzimiri N. A common variant association study reveals novel susceptibility loci for low HDL-cholesterol levels in ethnic Arabs. Clin Genet. 2016;90:518–525. doi: 10.1111/cge.12761. [DOI] [PubMed] [Google Scholar]
- 85.Cruchaga C, Kauwe JS, Harari O, Jin SC, Cai Y, Karch CM, Benitez BA, Jeng AT, Skorupa T, Carrell D, Bertelsen S, Bailey M, McKean D, Shulman JM, De Jager PL, Chibnik L, Bennett DA, Arnold SE, Harold D, Sims R, Gerrish A, Williams J, Van Deerlin VM, Lee VM, Shaw LM, Trojanowski JQ, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD, Peskind ER, Galasko D, Fagan AM, Holtzman DM, Morris JC, Goate AM. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron. 2013;78:256–268. doi: 10.1016/j.neuron.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Deming Y, Li Z, Kapoor M, Harari O, Del-Aguila JL, Black K, Carrell D, Cai Y, Fernandez MV, Budde J, Ma S, Saef B, Howells B, Huang KL, Bertelsen S, Fagan AM, Holtzman DM, Morris JC, Kim S, Saykin AJ, De Jager PL, Albert M, Moghekar A, O’Brien R, Riemenschneider M, Petersen RC, Blennow K, Zetterberg H, Minthon L, Van Deerlin VM, Lee VM, Shaw LM, Trojanowski JQ, Schellenberg G, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Peskind ER, Li G, Di Narzo AF, Alzheimer’s Disease Neuroimaging I, Alzheimer Disease Genetic C, Kauwe JS, Goate AM, Cruchaga C. Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol. 2017;133:839–856. doi: 10.1007/s00401-017-1685-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chung SJ, Kim MJ, Kim J, Ryu HS, Kim YJ, Kim SY, Lee JH. Association of type 2 diabetes GWAS loci and the risk of Parkinson’s and Alzheimer’s diseases. Parkinsonism Relat Disord. 2015;21:1435–1440. doi: 10.1016/j.parkreldis.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 88.Attanasio M, Uhlenhaut NH, Sousa VH, O’Toole JF, Otto E, Anlag K, Klugmann C, Treier AC, Helou J, Sayer JA, Seelow D, Nurnberg G, Becker C, Chudley AE, Nurnberg P, Hildebrandt F, Treier M. Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat Genet. 2007;39:1018–1024. doi: 10.1038/ng2072. [DOI] [PubMed] [Google Scholar]
- 89.Ramachandran H, Yakulov TA, Engel C, Muller B, Walz G. The C175R mutation alters nuclear localization and transcriptional activity of the nephronophthisis NPHP7 gene product. Eur J Hum Genet. 2016;24:774–778. doi: 10.1038/ejhg.2015.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Otto EA, Ramaswami G, Janssen S, Chaki M, Allen SJ, Zhou W, Airik R, Hurd TW, Ghosh AK, Wolf MT, Hoppe B, Neuhaus TJ, Bockenhauer D, Milford DV, Soliman NA, Antignac C, Saunier S, Johnson CA, Hildebrandt F. Mutation analysis of 18 nephronophthisis associated ciliopathy disease genes using a DNA pooling and next generation sequencing strategy. J Med Genet. 2011;48:105–116. doi: 10.1136/jmg.2010.082552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Halbritter J, Porath JD, Diaz KA, Braun DA, Kohl S, Chaki M, Allen SJ, Soliman NA, Hildebrandt F, Otto EA, Group GPNS Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum Genet. 2013;132:865–884. doi: 10.1007/s00439-013-1297-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kuo PH, Chuang LC, Su MH, Chen CH, Chen CH, Wu JY, Yen CJ, Wu YY, Liu SK, Chou MC, Chou WJ, Chiu YN, Tsai WC, Gau SS. Genome-wide association study for autism spectrum disorder in Taiwanese Han population. PLoS One. 2015;10:e0138695. doi: 10.1371/journal.pone.0138695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Song W, Chen YP, Huang R, Chen K, Pan PL, Li J, Yang Y, Shang HF. Glis1 rs797906: an increased risk factor for late-onset Parkinson’s disease in the Han Chinese population. Eur Neurol. 2012;68:89–92. doi: 10.1159/000337955. [DOI] [PubMed] [Google Scholar]
- 94.Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Kruger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao JJ, Huang XM, Wood NW, Lorenz D, Deuschl G, Chen HL, Riess O, Hardy JA, Singleton AB, Gasser T. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pankratz N, Wilk JB, Latourelle JC, DeStefano AL, Halter C, Pugh EW, Doheny KF, Gusella JF, Nichols WC, Foroud T, Myers RH, Investigators PP, Investigators G. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet. 2009;124:593–605. doi: 10.1007/s00439-008-0582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Haugarvoll K, Toft M, Skipper L, Heckman MG, Crook JE, Soto A, Ross OA, Hulihan MM, Kachergus JM, Sando SB, White LR, Lynch T, Gibson JM, Uitti RJ, Wszolek ZK, Aasly JO, Farrer MJ. Fine-mapping and candidate gene investigation within the PARK10 locus. Eur J Hum Genet. 2009;17:336–343. doi: 10.1038/ejhg.2008.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Maraganore DM, de Andrade M, Lesnick TG, Strain KJ, Farrer MJ, Rocca WA, Pant PVK, Frazer KA, Cox DR, Ballinger DG. High-resolution whole-genome association study of Parkinson disease. Am J Hum Genet. 2005;77:685–693. doi: 10.1086/496902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Divers J, Palmer ND, Lu L, Register TC, Carr JJ, Hicks PJ, Hightower RC, Smith SC, Xu J, Cox AJ, Hruska KA, Bowden DW, Lewis CE, Heiss G, Province MA, Borecki IB, Kerr KF, Chen YD, Palmas W, Rotter JI, Wassel CL, Bertoni AG, Herrington DM, Wagenknecht LE, Langefeld CD, Freedman BI. Admixture mapping of coronary artery calcified plaque in African Americans with type 2 diabetes mellitus. Circ Cardiovasc Genet. 2013;6:97–105. doi: 10.1161/CIRCGENETICS.112.964114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yamanaka S, Blau HM. Nuclear reprogramming to a pluripotent state by three approaches. Nature. 2010;465:704–712. doi: 10.1038/nature09229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Maekawa M, Yamaguchi K, Nakamura T, Shibukawa R, Kodanaka I, Ichisaka T, Kawamura Y, Mochizuki H, Goshima N, Yamanaka S. Direct reprogramming of somatic cells is promoted by maternal transcription factor GLIS1. Nature. 2011;474:225–229. doi: 10.1038/nature10106. [DOI] [PubMed] [Google Scholar]
- 101.Maekawa M, Yamanaka S. Glis1, a unique pro-reprogramming factor, may facilitate clinical applications of iPSC technology. Cell Cycle. 2011;10:3613–3614. doi: 10.4161/cc.10.21.17834. [DOI] [PubMed] [Google Scholar]
- 102.Yoshioka N, Gros E, Li HR, Kumar S, Deacon DC, Maron C, Muotri AR, Chi NC, Fu XD, Yu BD, Dowdy SF. Efficient generation of human iPSCs by a synthetic self-replicative RNA. Cell Stem Cell. 2013;13:246–254. doi: 10.1016/j.stem.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yoshioka N, Dowdy SF. Enhanced generation of iPSCs from older adult human cells by a synthetic five-factor self-replicative RNA. PLoS One. 2017;12:e0182018. doi: 10.1371/journal.pone.0182018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang L, Chen Y, Guan C, Zhao Z, Li Q, Yang J, Mo J, Wang B, Wu W, Yang X, Song L, Li J. Using low-risk factors to generate non-integrated human induced pluripotent stem cells from urine-derived cells. Stem Cell Res Ther. 2017;8:245. doi: 10.1186/s13287-017-0698-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu J, Han Q, Peng T, Peng M, Wei B, Li D, Wang X, Yu S, Yang J, Cao S, Huang K, Hutchins AP, Liu H, Kuang J, Zhou Z, Chen J, Wu H, Guo L, Chen Y, Chen Y, Li X, Wu H, Liao B, He W, Song H, Yao H, Pan G, Chen J, Pei D. The oncogene c-Jun impedes somatic cell reprogramming. Nat Cell Biol. 2015;17:856–867. doi: 10.1038/ncb3193. [DOI] [PubMed] [Google Scholar]
- 106.Lee SY, Noh HB, Kim HT, Lee KI, Hwang DY. Glis family proteins are differentially implicated in the cellular reprogramming of human somatic cells. Oncotarget. 2017;8:77041–77049. doi: 10.18632/oncotarget.20334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pells S, Koutsouraki E, Morfopoulou S, Valencia-Cadavid S, Tomlinson SR, Kalathur R, Futschik ME, De Sousa PA. Novel human embryonic stem cell regulators identified by conserved and distinct CPG island methylation state. PLoS One. 2015;10:e0131102. doi: 10.1371/journal.pone.0131102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.D’Alessio AC, Fan ZP, Wert KJ, Baranov P, Cohen MA, Saini JS, Cohick E, Charniga C, Dadon D, Hannett NM, Young MJ, Temple S, Jaenisch R, Lee TI, Young RA. A systematic approach to identify candidate transcription factors that control cell identity. Stem Cell Rep. 2015;5:763–775. doi: 10.1016/j.stemcr.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Habeb AM, Al-Magamsi MS, Eid IM, Ali MI, Hattersley AT, Hussain K, Ellard S. Incidence, genetics, and clinical phenotype of permanent neonatal diabetes mellitus in Northwest Saudi Arabia. Pediatr Diabetes. 2012;13:499–505. doi: 10.1111/j.1399-5448.2011.00828.x. [DOI] [PubMed] [Google Scholar]
- 110.Schoenmakers N, Chatterjee VK. Thyroid gland: TSHR mutations and subclinical congenital hypothyroidism. Nat Rev Endocrinol. 2015;11:258–259. doi: 10.1038/nrendo.2015.27. [DOI] [PubMed] [Google Scholar]
- 111.Grasberger H, Refetoff S. Genetic causes of congenital hypothyroidism due to dyshormonogenesis. Curr Opin Pediatr. 2011;23:421–428. doi: 10.1097/MOP.0b013e32834726a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zoeller RT, Tan SW, Tyl RW. General background on the hypothalamic-pituitary-thyroid (HPT) axis. Crit Rev Toxicol. 2007;37:11–53. doi: 10.1080/10408440601123446. [DOI] [PubMed] [Google Scholar]
- 113.Spitzweg C, Morris JC. The sodium iodide symporter: its pathophysiological and therapeutic implications. Clin Endocrinol (Oxf) 2002;57:559–574. doi: 10.1046/j.1365-2265.2002.01640.x. [DOI] [PubMed] [Google Scholar]
- 114.Kuhnen P, Turan S, Frohler S, Guran T, Abali S, Biebermann H, Bereket A, Gruters A, Chen W, Krude H. Identification of PENDRIN (SLC26A4) mutations in patients with congenital hypothyroidism and “apparent” thyroid dysgenesis. J Clin Endocrinol Metab. 2014;99:E169–E176. doi: 10.1210/jc.2013-2619. [DOI] [PubMed] [Google Scholar]
- 115.Postiglione MP, Parlato R, Rodriguez-Mallon A, Rosica A, Mithbaokar P, Maresca M, Marians RC, Davies TF, Zannini MS, De Felice M, Di Lauro R. Role of the thyroid-stimulating hormone receptor signaling in development and differentiation of the thyroid gland. Proc Natl Acad Sci USA. 2002;99:15462–15467. doi: 10.1073/pnas.242328999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Brewer C, Yeager N, Di Cristofano A. Thyroid-stimulating hormone initiated proliferative signals converge in vivo on the mTOR kinase without activating AKT. Cancer Res. 2007;67:8002–8006. doi: 10.1158/0008-5472.CAN-07-2471. [DOI] [PubMed] [Google Scholar]
- 117.Murtaugh LC, Melton DA. Genes, signals, and lineages in pancreas development. Annu Rev Cell Dev Biol. 2003;19:71–89. doi: 10.1146/annurev.cellbio.19.111301.144752. [DOI] [PubMed] [Google Scholar]
- 118.Pan FC, Wright C. Pancreas organogenesis: from bud to plexus to gland. Dev Dyn. 2011;240:530–565. doi: 10.1002/dvdy.22584. [DOI] [PubMed] [Google Scholar]
- 119.Kang HS, Takeda Y, Jeon K, Jetten AM. The spatiotemporal pattern of Glis3 expression indicates a regulatory function in bipotent and endocrine progenitors during early pancreatic development and in beta, PP and ductal cells. PLoS One. 2016;11:e0157138. doi: 10.1371/journal.pone.0157138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gradwohl G, Dierich A, LeMeur M, Guillemot F. Neurogenin 3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci USA. 2000;97:1607–1611. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.De Vas MG, Kopp JL, Heliot C, Sander M, Cereghini S, Haumaitre C. Hnf1b controls pancreas morphogenesis and the generation of Ngn3+ endocrine progenitors. Development. 2015;142:871–882. doi: 10.1242/dev.110759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Oliver-Krasinski JM, Stoffers DA. On the origin of the beta cell. Genes Dev. 2008;22:1998–2021. doi: 10.1101/gad.1670808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hang Y, Stein R. MafA and MafB activity in pancreatic beta cells. Trends Endocrinol Metab. 2011;22:364–373. doi: 10.1016/j.tem.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.van der Meulen T, Huising MO. Maturation of stem cell-derived beta-cells guided by the expression of urocortin 3. Rev Diabet Stud. 2014;11:115–132. doi: 10.1900/RDS.2014.11.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yang Y, Bush SP, Wen X, Cao W, Chan L. Differential gene dosage effects of diabetes-associated gene Glis3 in pancreatic beta cell differentiation and function. Endocrinology. 2017;158:9–20. doi: 10.1210/en.2016-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yang Y, Chang BH, Chan L. Sustained expression of the transcription factor Glis3 is required for normal beta cell function in adults. EMBO Mol Med. 2013;5:92–104. doi: 10.1002/emmm.201201398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dooley J, Tian L, Schonefeldt S, Delghingaro-Augusto V, Garcia-Perez JE, Pasciuto E, Di Marino D, Carr EJ, Oskolkov N, Lyssenko V, Franckaert D, Lagou V, Overbergh L, Vandenbussche J, Allemeersch J, Chabot-Roy G, Dahlstrom JE, Laybutt DR, Petrovsky N, Socha L, Gevaert K, Jetten AM, Lambrechts D, Linterman MA, Goodnow CC, Nolan CJ, Lesage S, Schlenner SM, Liston A. Genetic predisposition for beta cell fragility underlies type 1 and type 2 diabetes. Nat Genet. 2016;48:519–527. doi: 10.1038/ng.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nogueira TC, Paula FM, Villate O, Colli ML, Moura RF, Cunha DA, Marselli L, Marchetti P, Cnop M, Julier C, Eizirik DL. GLIS3, a susceptibility gene for type 1 and type 2 diabetes, modulates pancreatic beta cell apoptosis via regulation of a splice variant of the BH3-only protein BIM. PLoS Genet. 2013;9:e1003532. doi: 10.1371/journal.pgen.1003532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Andrali SS, Sampley ML, Vanderford NL, Ozcan S. Glucose regulation of insulin gene expression in pancreatic beta-cells. Biochem J. 2008;415:1–10. doi: 10.1042/BJ20081029. [DOI] [PubMed] [Google Scholar]
- 130.Kang HS, Chen LY, Lichti-Kaiser K, Liao G, Gerrish K, Bortner CD, Yao HH, Eddy EM, Jetten AM. Transcription factor Glis3: a new and critical regulator of postnatal stages of mouse spermatogenesis. Stem Cells. 2016;34:2772–2783. doi: 10.1002/stem.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kanatsu-Shinohara M, Shinohara T. Spermatogonial stem cell self-renewal and development. Annu Rev Cell Dev Biol. 2013;29:163–187. doi: 10.1146/annurev-cellbio-101512-122353. [DOI] [PubMed] [Google Scholar]
- 132.Nagano MC, Yeh JR. The identity and fate decision control of spermatogonial stem cells: where is the point of no return? Curr Top Dev Biol. 2013;102:61–95. doi: 10.1016/B978-0-12-416024-8.00003-9. [DOI] [PubMed] [Google Scholar]
- 133.Oatley JM, Brinster RL. The germline stem cell niche unit in mammalian testes. Physiol Rev. 2012;92:577–595. doi: 10.1152/physrev.00025.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Griswold MD. Spermatogenesis: the commitment to meiosis. Physiol Rev. 2016;96:1–17. doi: 10.1152/physrev.00013.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sada A, Hasegawa K, Pin PH, Saga Y. NANOS2 acts downstream of glial cell line-derived neurotrophic factor signaling to suppress differentiation of spermatogonial stem cells. Stem Cells. 2012;30:280–291. doi: 10.1002/stem.790. [DOI] [PubMed] [Google Scholar]
- 136.Chakraborty P, Buaas FW, Sharma M, Snyder E, de Rooij DG, Braun RE. LIN28A marks the spermatogonial progenitor population and regulates its cyclic expansion. Stem Cells. 2014;32:860–873. doi: 10.1002/stem.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bak CW, Yoon TK, Choi Y. Functions of PIWI proteins in spermatogenesis. Clin Exp Reprod Med. 2011;38:61–67. doi: 10.5653/cerm.2011.38.2.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Goertz MJ, Wu Z, Gallardo TD, Hamra FK, Castrillon DH. Foxo1 is required in mouse spermatogonial stem cells for their maintenance and the initiation of spermatogenesis. J Clin Invest. 2011;121:3456–3466. doi: 10.1172/JCI57984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Takahashi K, Sakurai N, Emura N, Hashizume T, Sawai K. Effects of downregulating GLIS1 transcript on preimplantation development and gene expression of bovine embryos. J Reprod Dev. 2015;61:369–374. doi: 10.1262/jrd.2015-029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tosic M, Allen A, Willmann D, Lepper C, Kim J, Duteil D, Schule R. Lsd1 regulates skeletal muscle regeneration and directs the fate of satellite cells. Nat Commun. 2018;9:366. doi: 10.1038/s41467-017-02740-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lu D, Rauhauser A, Li B, Ren C, McEnery K, Zhu J, Chaki M, Vadnagara K, Elhadi S, Jetten AM, Igarashi P, Attanasio M. Loss of Glis2/NPHP7 causes kidney epithelial cell senescence and suppresses cyst growth in the Kif3a mouse model of cystic kidney disease. Kidney Int. 2016;89:1307–1323. doi: 10.1016/j.kint.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LS, Somlo S, Igarashi P. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci USA. 2003;100:5286–5291. doi: 10.1073/pnas.0836980100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rauhauser AA, Ren C, Lu D, Li B, Zhu J, McEnery K, Vadnagara K, Zepeda-Orozco D, Zhou XJ, Lin F, Jetten AM, Attanasio M. Hedgehog signaling indirectly affects tubular cell survival after obstructive kidney injury. Am J Physiol Renal Physiol. 2015;309:F770–F778. doi: 10.1152/ajprenal.00232.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rasouly HM, Kumar S, Chan S, Pisarek-Horowitz A, Sharma R, Xi QJ, Nishizaki Y, Higashi Y, Salant DJ, Maas RL, Lu W. Loss of Zeb2 in mesenchyme-derived nephrons causes primary glomerulocystic disease. Kidney Int. 2016;90:1262–1273. doi: 10.1016/j.kint.2016.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Haumaitre C, Fabre M, Cormier S, Baumann C, Delezoide AL, Cereghini S. Severe pancreas hypoplasia and multicystic renal dysplasia in two human fetuses carrying novel HNF1beta/MODY5 mutations. Hum Mol Genet. 2006;15:2363–2375. doi: 10.1093/hmg/ddl161. [DOI] [PubMed] [Google Scholar]
- 146.Kolb RJ, Nauli SM. Ciliary dysfunction in polycystic kidney disease: an emerging model with polarizing potential. Front Biosci. 2008;13:4451–4466. doi: 10.2741/3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ma M, Gallagher AR, Somlo S. Ciliary mechanisms of cyst formation in polycystic kidney disease. Cold Spring Harb Perspect Biol. 2017;9:a028209. doi: 10.1101/cshperspect.a028209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Torres VE, Harris PC. Mechanisms of disease: autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol. 2006;2:40–55. doi: 10.1038/ncpneph0070. [DOI] [PubMed] [Google Scholar]
- 149.Yoder BK. Role of primary cilia in the pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2007;18:1381–1388. doi: 10.1681/ASN.2006111215. [DOI] [PubMed] [Google Scholar]
- 150.Reiter JF, Leroux MR. Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol. 2017;18:533–547. doi: 10.1038/nrm.2017.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Song CJ, Zimmerman KA, Henke SJ, Yoder BK. Inflammation and fibrosis in polycystic kidney disease. Results Probl Cell Differ. 2017;60:323–344. doi: 10.1007/978-3-319-51436-9_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hilgendorf KI, Johnson CT, Jackson PK. The primary cilium as a cellular receiver: organizing ciliary GPCR signaling. Curr Opin Cell Biol. 2016;39:84–92. doi: 10.1016/j.ceb.2016.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Braun DA, Hildebrandt F. Ciliopathies. Cold Spring Harb Perspect Biol. 2017;9:a028209. doi: 10.1101/cshperspect.a028191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11:331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bangs F, Anderson KV. Primary cilia and mammalian Hedgehog signaling. Cold Spring Harb Perspect Biol. 2016;9:a028175. doi: 10.1101/cshperspect.a028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Schou KB, Pedersen LB, Christensen ST. Ins and outs of GPCR signaling in primary cilia. EMBO Rep. 2015;16:1099–1113. doi: 10.15252/embr.201540530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mykytyn K, Askwith C. G-protein-coupled receptor signaling in cilia. Cold Spring Harb Perspect Biol. 2017;9:1028183. doi: 10.1101/cshperspect.a028183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ruppersburg CC, Hartzell HC. The Ca2+-activated Cl− channel ANO1/TMEM16A regulates primary ciliogenesis. Mol Biol Cell. 2014;25:1793–1807. doi: 10.1091/mbc.e13-10-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Flannery RJ, Kleene NK, Kleene SJ. A TRPM4-dependent current in murine renal primary cilia. Am J Physiol Renal Physiol. 2015;309:F697–F707. doi: 10.1152/ajprenal.00294.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Siroky BJ, Kleene NK, Kleene SJ, Varnell CD, Jr, Comer RG, Liu J, Lu L, Pachciarz NW, Bissler JJ, Dixon BP. Primary cilia regulate the osmotic stress response of renal epithelial cells through TRPM3. Am J Physiol Renal Physiol. 2017;312:F791–F805. doi: 10.1152/ajprenal.00465.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Gerdes JM, Davis EE, Katsanis N. The vertebrate primary cilium in development, homeostasis, and disease. Cell. 2009;137:32–45. doi: 10.1016/j.cell.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Praetorius HA, Spring KR. A physiological view of the primary cilium. Annu Rev Physiol. 2005;67:515–529. doi: 10.1146/annurev.physiol.67.040403.101353. [DOI] [PubMed] [Google Scholar]
- 163.Orhon I, Dupont N, Pampliega O, Cuervo AM, Codogno P. Autophagy and regulation of cilia function and assembly. Cell Death Differ. 2015;22:389–397. doi: 10.1038/cdd.2014.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Michaud EJ, Yoder BK. The primary cilium in cell signaling and cancer. Cancer Res. 2006;66:6463–6467. doi: 10.1158/0008-5472.CAN-06-0462. [DOI] [PubMed] [Google Scholar]
- 165.Paul BM, Vanden Heuvel GB. Kidney: polycystic kidney disease. Wiley Interdiscip Rev Dev Biol. 2014;3:465–487. doi: 10.1002/wdev.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Mick DU, Rodrigues RB, Leib RD, Adams CM, Chien AS, Gygi SP, Nachury MV. Proteomics of primary cilia by proximity labeling. Dev Cell. 2015;35:497–512. doi: 10.1016/j.devcel.2015.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Han Y, Xiong Y, Shi X, Wu J, Zhao Y, Jiang J. Regulation of gli ciliary localization and Hedgehog signaling by the PY-NLS/karyopherin-beta2 nuclear import system. PLoS Biol. 2017;15:e2002063. doi: 10.1371/journal.pbio.2002063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mukhopadhyay S, Rohatgi R. G-protein-coupled receptors, Hedgehog signaling and primary cilia. Semin Cell Dev Biol. 2014;33:63–72. doi: 10.1016/j.semcdb.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]