

Abstract
BACKGROUND
Neurodegenerative diseases involving protein aggregation often accompany psychiatric symptoms. Frontotemporal lobar degeneration (FTLD) associated with TAR DNA-binding protein 43 kDa (TDP-43) aggregation is characterized by progressive neuronal atrophy in frontal and temporal lobes of cerebral cortex. Furthermore, patients with FTLD display mental dysfunction in multiple behavioral dimensions. Nevertheless, their molecular origin for psychiatric symptoms remains unclear.
METHODS
In FTLD neurons and mouse models with TDP-43 aggregates, we examined co-aggregation between TDP-43 and disrupted in schizophrenia 1 (DISC1), a key player in the pathology of mental conditions and its effects on local translation in dendrites and psychiatric behaviors. The protein co-aggregation and the expression level of synaptic proteins were also investigated with postmortem brains from FTLD patients (n=6).
RESULTS
We found cytosolic TDP-43/DISC1 co-aggregates in brains of both FTLD mouse model and patients. At the mechanistic levels, the TDP-43/DISC1 co-aggregates disrupted the activity-dependent dendritic local translation through impairment of translation initiation and, in turn, reduced synaptic protein expression. Behavioral deficits detected in FTLD model mice were ameliorated by exogenous DISC1 expression.
CONCLUSIONS
Our findings reveal a novel role of the aggregate-prone TDP-43-DISC1 protein complex in regulating local translation, which affects aberrant behaviors relevant to multiple psychiatric dimensions.
Keywords: FTLD, co-aggregation, TDP-43, DISC1, local translation, psychiatric behaviors
INTRODUCTION
Neurodegenerative disorders display multiple types of functional abnormalities, such as motor, cognitive, and psychiatric deficits (1,2). Patients of frontotemporal lobar degeneration (FTLD), characterized by atrophy in the frontal and temporal lobes of the brain, frequently show psychiatric dysfunction in multiple behavioral dimensions (3,4).
In FTLD patient brain, TAR DNA-binding protein 43 (TDP-43) is a major component of ubiquitin-positive hyper-phosphorylated inclusions (5), suggesting that its aggregation is implicated in FTLD pathogenesis (6,7). TDP-43 is a heterogeneous ribonucleoprotein containing two highly conserved RNA recognition motifs that regulate RNA processing, including exon splicing, mRNA transport and microRNA biogenesis (8). In neurons, TDP-43 is also a constituent of RNA granules in dendrites (8,9) where it interacts with RNA binding proteins including fragile X mental-retardation protein (FMRP) and Staufen1, and regulates mRNA transport and local translation (9), which are critical for synaptic plasticity (10–13). Considerable effort has been made to delineate the molecular mechanisms associated with neurodegeneration and motor dysfunction caused by TDP-43 aggregation (14). However, it remains unclear how TDP-43 aggregation might mediate abnormal behaviors associated with FTLD (3,4) and the molecular mechanisms therein.
Causative proteins for neurodegenerative diseases, including TDP-43, often form co-aggregates with their binding partners (15–18). The functional influences of such co-aggregation have been investigated mostly in the context of cellular dysfunction and neuronal death (17,19,20). Yet, it remains elusive how functional effects of co-aggregation may underlie higher brain functions. Importantly, co-aggregation of disease-associated proteins is specific (20–23). Such selective co-aggregation suggests that each type of co-aggregation may impact different signaling pathways, leading to distinct dimensions of behaviors. DISC1, an important molecule in the pathology of a wide range of brain conditions (24,25), interacts with 14-3-3 protein, RACK1 and hnRNP-U (26,27), all of which are also reported to interact with TDP-43 (28–30), shows an aggregation-prone nature and plays critical roles in mental conditions (24,31,32). Therefore, we focused on DISC1 as a possible binding and co-aggregation protein with TDP-43. Here, we investigate the novel hypothesis that, in FTLD, the co-aggregation of DISC1 with TDP-43 may trigger neuronal dysfunction in dendrites and psychiatric manifestations including social deficits.
METHODS AND MATERIALS
See the supplement for further detailed Methods and Materials.
Animals and Brain Samples
All animals used in this study were housed at a maximum of 5 per cage and in a temperature controlled room under a 12-h light/12-h dark cycle with free access to food and water. All of the experiments were performed in compliance with relevant laws and guidelines issued by RIKEN.
Postmortem brain samples of FTLD and controls were obtained from Tokyo Metropolitan Institute of Gerontology and Fukushimura Brain Bank under a protocol approved by the local research ethics committee of RIKEN. Informed consent was obtained for all human tissue samples by the Tokyo Metropolitan Institute of Gerontology and Fukushimura Brain Bank and all the samples were anonymized and cannot be tracked back to individual patients. All autopsy cases underwent neuropathological evaluation of FTLD and all patients with FTLD met the clinical diagnostic criteria of FTLD. Subjects that did not carry a diagnosis of FTLD were used as controls.
Plasmids and Antibodies
pCSII-CMV, pCAG-HIVgp, and pCAG-VSV-G plasmids were obtained from RIKEN Bioresource center. An N-terminal truncated (220-414) form (TDP-220C) of the mouse TDP43 cDNA was subcloned into the pCSII-CMV vector with Venus (K206A) at the N-terminus. Full-length mouse DISC1 cDNA with a HA tag at the C-terminus or mouse DISC1 (RNAi resistant) tagged with HA at the N-terminus and with Flagx3 at the C-terminus was subcloned into the pCSII-CMV-IRES-mCherry plasmid or CSII-CMV vector, respectively. The Gaussian luciferase coding region was amplified by PCR using a pCMV-Gluc control plasmid (New England Biosciences). FUGW-mouse DISC1-Scramble RNAi-IRES-GFP, FUGW-mouse DISC1RNAi-IRES-GFP and FUGW-mouse DISC1RNAi-IRES-HA-full-length mouse DISC1 (RNAi resistant) were reported previously (33). DISC1 rabbit polyclonal antibodies, m317C, m595C, h316C and h598C were raised against the recombinant DISC1 protein fragments; residues 317-852 and 595-852 in mouse DISC1 and 316-854 and 598-854 in human DISC1, respectively (Research Resources Center in RIKEN Brain Science Institute). DISC1 mouse monoclonal antibodies HM6 and M49 were raised against the recombinant mouse DISC1 protein fragment (residues 317-852) (MBL). The monoclonal DISC1 antibody 2B3 and polyclonal DISC1 antibodies m317C and h316C are described previously (23,34,35). The antibody that recognizes the N-terminal domain of TDP-43 was raised against recombinant full-length mouse TDP-43 (MBL). In this study, we used the antibodies against TDP-43 (mouse monoclonal FL4, a kind gift from Dr. Cleveland), TDP-43 (mouse monoclonal, Abnova, H00023435-M01), TDP-43 (rabbit polyclonal, Proteintech, 10782-2-AP), phosphorylated-Ser409/410 of TDP43 (rabbit polyclonal, CosmoBio, CAC-TIP-PTD-M01), FMRP (mouse monoclonal, DSHB, 2F5-1-S), FUS/TLS (mouse monoclonal, SantaCruz, sc-47711), GFP (mouse monoclonal, Nacalai Tesque, 04363-24), GFP (rabbit polyclonal, MLB, 598), puromycin (mouse monoclonal, Millipore, MABE343),, ubiquitin (rabbit polyclonal, DAKO, z045801), NR2B (mouse monoclonal, NeuroMab, 75-028), PSD95 (mouse monoclonal, NeuroMab, 75-028), NR1 (mouse monoclonal, NeuroMab, 75-272), GluR2 (mouse monoclonal, NeuroMab, 75-002), mGluR1/5 (mouse monoclonal, NeuroMab, 75-116), CaMKIIα (rabbit polyclonal, Sigma), SV2 (mouse monoclonal, DSHB, SV2) MAP2a/b (mouse monoclonal, NeoMarker), α–synuclein (mouse monoclonal, Covance, S1G-39720-200) and Tau (mouse monoclonal, Millipore, MAB361).
Statistical Analysis
The statistical significance of the data was examined by t test for analyses of two groups (n ≥ 3 for cell biology data unless otherwise indicated) or by one-way ANOVA with a Bonferroni’s multiple comparison test for analyses of three or more groups unless otherwise indicated. The p values of <0.05 (*), <0.01 (**) and <0.001 (***) were considered to be statistically significant.
RESULTS
DISC1 forms co-aggregates with TDP-43
First, we examined co-immunoprecipitation of DISC1 and TDP-43 in vivo. DISC1 formed a complex with TDP-43 in both mouse and human brain (Figure 1A,B and Supplemental Figure S1A,B) and the binding was higher in cerebral cortex than in cerebellum (Figure 1C). Furthermore, we found that DISC1 also bound to fused in sarcoma/translated in liposarcoma (FUS/TLS) and FMRP (Figure 1D), both of which interact with TDP-43 and are the RNA-binding proteins associated with dendritic local translation (6,11,36). Therefore, we asked whether RNA is critical for the interaction. We treated mouse brain homogenates with or without RNase, followed by co-immunoprecipitation. RNase treatment strikingly abolished the binding of DISC1 to these RNA-binding proteins (Figure 1D), indicating the RNA-dependent DISC1 interaction. In sporadic FTLD patients, aggregation of a C-terminal fragment of TDP-43 in cytosol is a hallmark of FTLD neurons (5). Therefore, we examined whether DISC1 forms co-aggregates with the C-terminal fragment of TDP-43. First, we established cultured cortical neurons that expressed an N-terminally Venus tagged C-terminal fragment (residues 220-414) of TDP-43, termed TDP-220C, using a lentivirus system. In both cytosol and neurites, TDP-220C formed, ubiquitinated and hyper-phosphorylated aggregates, which are observed in FTLD patient brains (8) (Figure 1E and Supplemental Figure S2A,B). Thus, the TDP-220C expressed system used in this study recapitulates the characteristics of human FTLD. TDP-220C aggregates sequestered endogenous DISC1 (Figure 1E and Supplemental Figure S2C) while the expression of full-length TDP-43 did not induce self-aggregation nor aggregation of DISC1 (Supplemental Figure S2D). In contrast to RNA-dependent binding of DISC1 to TDP-43, the interactions between DISC1 and TDP-220C aggregates were RNA-independent (Supplemental Figure S2C,E), indicating that this binding is mediated by protein-protein interactions. Furthermore, a filter trap assay showed that detergent-resistant insoluble DISC1 was increased in neurons expressing TDP-220C (Supplemental Figure S2F). These results are consistent with the aggregation-prone nature of DISC1 (31), although the physiological consequences of DISC1 aggregation are not fully understood.
Figure 1.
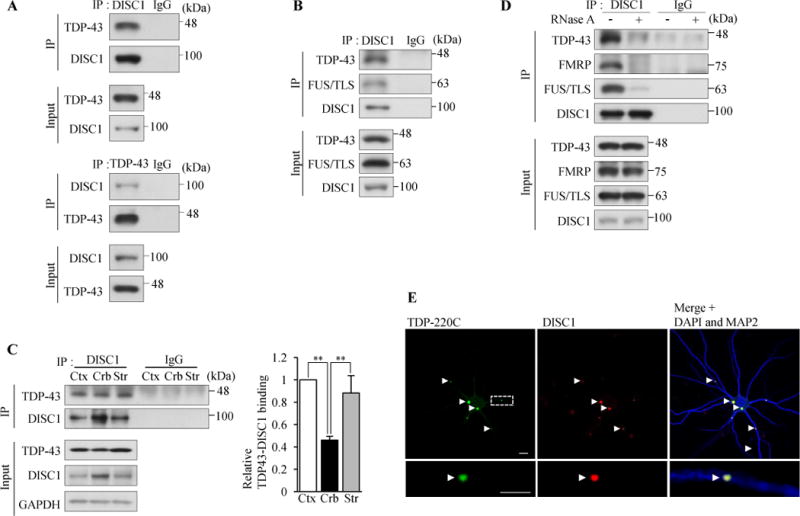
DISC1 binds to and forms co-aggregates with TDP-43 in neurons. (A) TDP-43 interacts with DISC1 in vivo. DISC1 (m317C) or TDP-43 was immunoprecipitated from homogenates of wild-type mouse cerebral cortex, followed by western blotting (2B3 for DISC1). (B) DISC1 was immunoprecipitated (h598C) from healthy human temporal lobes, followed by western blotting (HM6-5 for DISC1). (C) The binding of DISC1 to TDP-43 is more dominant in cerebral cortex and striatum than that in cerebellum. DISC1 was immunoprecipitated with an anti-DISC1 antibody (m595C) from homogenates of wild-type mouse cerebral cortex, striatum or cerebellum, followed by western blotting (M49 for DISC1) (Left). Co-immunoprecipitated TDP-43 was normalized to immunoprecipitated DISC1 and the binding between DISC1 and TDP-43 is expressed as the relative ratio to cerebral cortex (Right) (n=4, F(2,9)=16.87, P=0.0009, one-way ANOVA; **P<0.01, Bonferroni’s multiple comparison test post hoc). Error bars represent S.E.M. (D) DISC1 binds to TDP-43, FMRP and FUS/TLS in an RNA-dependent manner. Mouse brain (wild-type) homogenates were treated with or without 200 μg/ml RNase A, followed by immunoprecipitation with an anti-DISC1 antibody (m317C) and western blotting (2B3 for DISC1). (E) Cultured cortical neurons were infected with a lentivirus expression vector encoding Venus-TDP-220C (green) and endogenous DISC1 was immunostained with an anti-DISC1 antibody (HM6-5) (red). Nuclei were stained with DAPI (blue) and dendrites were stained with an anti-MAP2 antibody (blue). A representative image is shown for the whole neuron (Top) and dendrite (Bottom), respectively. 85% of neurons contained TDP-220C aggregates both in soma and dendrites, and 91% of TDP-220C aggregates were DISC1-positive (n=216). Scale bar represents 5 μm.
DISC1 mediates the initiation step of dendritic local translation
A previous study implicated a role of DISC1 in translation (37) although DISC1’s role in translation has never been investigated. Furthermore, TDP-43 and FMRP are reported to be associated with the translational machinery and interact with polyribosomes (38,39). Since we found that DISC1 can interact with both TDP-43 and FMRP, we examined whether DISC1 is also associated with the translational machinery by polysome gradient centrifugation analysis. DISC1 was detected in polyribosome fractions and co-migrated with ribosomal S6 (RS6), TDP-43 and FMRP after either EDTA or RNase treatment (38), demonstrating that DISC1, as well as TDP-43 and FMRP, can interact with polyribosomes (Figure 2A and Supplemental Figure S3A). Next we examined whether DISC1 interacts with stalled ribosomes and suppresses translation, as suggested for FMRP (40,41). When neurons were treated with hippuristanol, an inhibitor of eIF4A to induce ribosome run-off (41), a large population of FMRP remained in polyribosome fractions as previously observed (41). However, the amount of DISC1 in polyribosome fractions was significantly reduced (Supplemental Figure S3B), showing that DISC1 is not associated with stalled ribosomes and hence DISC1 is not a negative regulator of translation.
Figure 2.
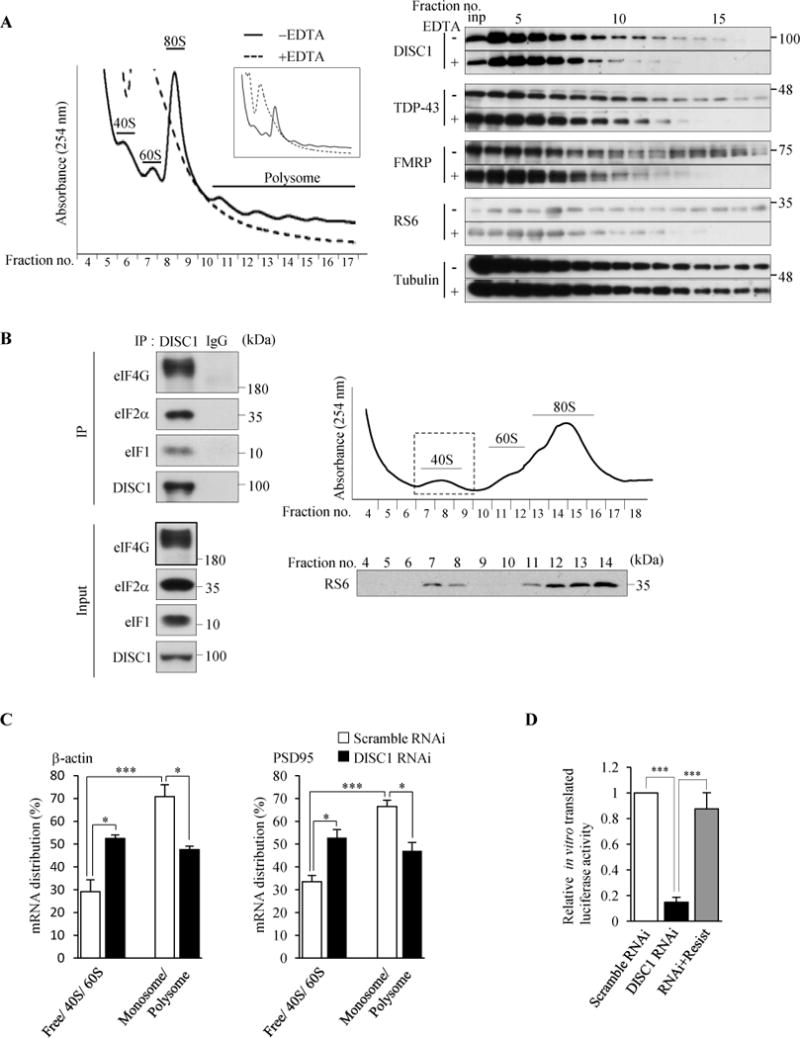
DISC1 binds to initiation factors and regulates translation initiation. (A) The polysome gradient analysis using lysates of cultured cortical neurons that were treated with or without 30 mM EDTA prior to centrifugation. A representative absorption profile of sucrose gradient at 254 nm is shown (left). The inset shows the overall view of the absorption profile. Fractions were collected and analyzed by western blotting with indicated antibodies (M49 for DISC1) (right). DISC1 showed the co-migration with polysomes both before and after the EDTA treatment. “inp” denotes input. (B) DISC1 binds to eIF4G, eIF2α and eIF1 in the 40S ribosome fraction. DISC1 was immunoprecipitated (m595C) from 40S ribosome fractions purified from wild-type mouse cerebral cortex, followed by western blotting. The right top panel shows the absorption profile of sucrose gradient at 254 nm and the collected fractions were analyzed by western blotting with an anti-RS6 antibody. (C) Ribosome-free (Free), 40S, 60S, 80S monosome and polysome fractions were obtained from N2a cell lysates by sucrose gradient centrifugation. mRNA levels of β-actin and PSD95 in each fraction were analyzed by RT-qPCR. The percentages of mRNA distribution in Free + 40S + 80S and monosome + polysome fractions are shown. (n=3, β-actin: F(3,8)=19.85, P=0.0005; PSD95: F(3,8)=18.68, P=0.0006, one-way ANOVA, *P<0.05, ***P<0.001 Bonferroni’s multiple comparison test post hoc). (D) In vitro translation using N2a cell lysates. In vitro transcribed mRNA encoding Gaussian luciferase (Gluc) driven by the m7GpppG cap structure was added to cell lysates and incubated for 5 hours followed by measurement of luciferase activity. The luciferase activity of in vitro translated Gluc is expressed as the relative ratio to control cells (n=4, F(2,9)=36.46, P<0.0001, one-way ANOVA; *P<0.05, ***P<0.001, Bonferroni’s multiple comparison test post hoc). Throughout the figures, error bars represent S.E.M.
In addition to the repression of translation by stalling translating ribosomes, FMRP is known to impair translation initiation by inhibiting binding of eIF4G to eIF4E on 40S ribosome during initiation (42). Since a substantial amount of DISC1 co-sediments with the 40S ribosome fraction as observed for FMRP (Figure 2A), we next examined whether DISC1 also plays a role in translation initiation. We first isolated 40S ribosome subunit fraction and examined whether DISC1 interacts with initiation factors in the 40S ribosome subunit. DISC1 interacted with the initiation factors eIF4G, eIF2α and eIF1 in the 40S ribosome subunit fraction (Figure 2B), suggesting that DISC1 plays a critical role in translation initiation. Furthermore, we examined whether DISC1 and TDP-43 forms a complex in the 40S ribosome subunit fraction and found that TDP-43 also resides within this fraction and interacts with DISC1 and eIF4G, suggesting that TDP-43 may also play a role in translation initiation together with DISC1 (Supplemental Figure S3C).
To further investigate whether DISC1 is functionally involved in translation initiation, we next examined how the distribution of mRNAs was altered by DISC1 knockdown. After fractionating the lysates of control and DISC1-knockdown mouse neuroblastoma (N2a) cells by sucrose gradient experiments, we quantified the amounts of β-actin and PSD95 mRNAs, both of which are known to be localized in dendrites (43,44), in each fraction by RT-qPCR. Inhibition of translation initiation was expected to shift the position of the target mRNAs from monosome and polysome fractions to those of ribosome-free or 40S subunit fractions whereas inhibition of peptide chain elongation would increase an amount of the target mRNAs in polysome fractions (45,46). We found that the target mRNA levels were reduced in monosome and polysome fractions while increased in ribosome-free, 40S and 60S subunit fractions by depletion of DISC1 (Figure 2C), suggesting that DISC1 plays a functional role in translation initiation.
To further confirm that DISC1 is functionally involved in translation initiation, we performed an in vitro translation assay using N2a cells whose translation activity is detectable compared to neurons. When an equal amount of in vitro transcribed reporter mRNA bearing a 5′-end cap structure, whose translation requires formation of translation initiation complex, was added to the lysates of control and DISC1-knockdown cells, more than 80% of in vitro translation products were reduced by DISC1 depletion and the compromised translation was restored by co-expression of the RNAi-resistant form of DISC1 (Figure 2D). Together, these results directly show that DISC1 indeed plays a role in translation and promotes the initiation step.
To gain further insights into the possible roles of DISC1 in local translation in dendrites, we examined whether DISC1 regulates translation in neurons by Surface Sensing of Translation (SUnSET). This method monitors global protein synthesis by the incorporation of puromycin into newly synthesized polypeptides (47). Neuronal stimulation induces local protein synthesis in dendrites (48,49). Therefore, we knocked down endogenous DISC1 in cultured cortical neurons by lentiviral infection of DISC1 RNAi (Supplemental Figure S4A) to examine the effects of DISC1 depletion on protein synthesis induced by neuronal stimulation. Stimulation with depolarizing KCl (9,50) increased the amounts of DISC1 and phosphorylated RS6 in ribosome-enriched fractions (Supplemental Figure S4B), suggesting the involvement of DISC1 in translation activated by neuronal stimulation. We then evaluated the amount of newly synthesized proteins in the isolated synaptosomal fraction (Supplemental Figure S4C) with or without neuronal stimulation with KCl. In basal culture conditions without stimulation (5 mM KCl), the amount of newly synthesized proteins was similar between control and DISC1-knockdown neurons (Figure 3A). Neuronal stimulation with 55 mM KCl in control neurons enhanced new protein synthesis, which did not depend on transcription but translation activity (Supplemental Figure S4D,E), but this increased new protein synthesis was not observed in DISC1-knockdown neurons (Figure 3A). To avoid any off-target effects of RNAi (25), we co-expressed an RNAi-resistant form of DISC1 (RNAi+Resist), which restored the suppressed new protein synthesis by DISC1 depletion (Figure 3A). Furthermore, contrary to the effect of DISC1 depletion in the synaptosomal fraction, the DISC1 depletion had no significant effect on new protein synthesis in the total fraction upon neuronal stimulation (Supplemental Figure S5A). These results indicate that DISC1 plays a role in translation in dendrites when translation rates are enhanced by neuronal stimulation.
Figure 3.
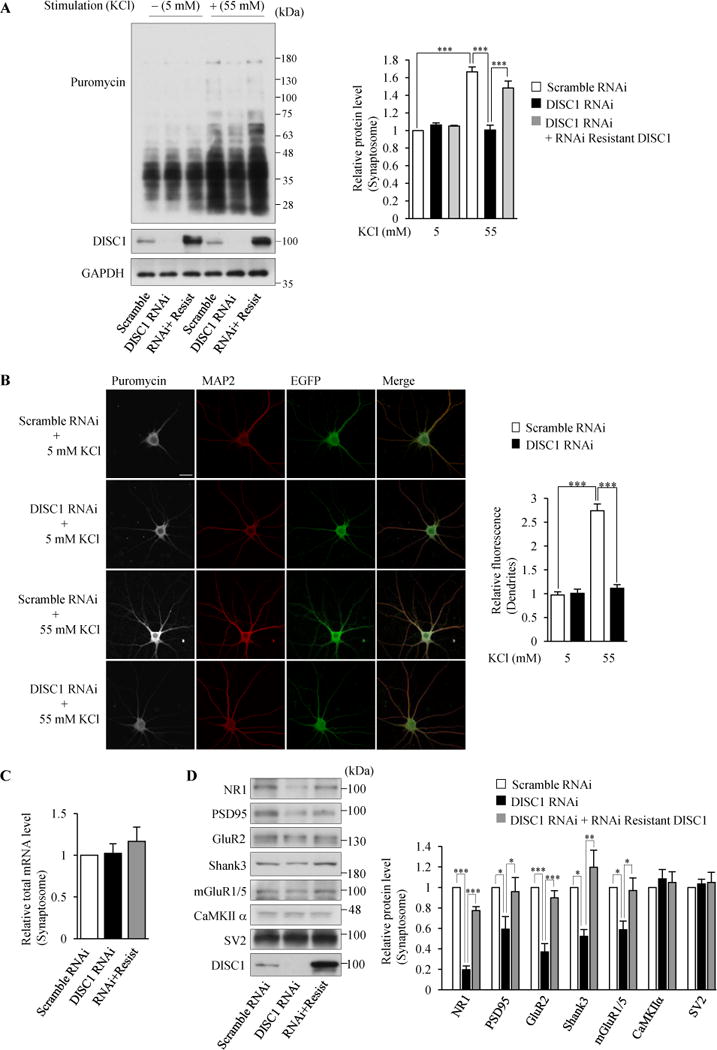
Local translation in dendrites is regulated by DISC1 in neurons. (A) The isolated synaptosomal fraction was subjected to western blotting with an anti-puromycin antibody for detection of puromycin-incorporated newly synthesized proteins. The signal intensities of puromycin-labeled polypeptides were normalized to those of GAPDH and then expressed as the relative ratio of control neurons (right) (n=4, F(5,18)=28.06, P<0.0001, one-way ANOVA; ***P<0.001, Bonferroni’s multiple comparison test post hoc). (B) Neurons were immunostained with anti-puromycin (gray) and anti-MAP2 (red) antibodies. The fluorescent intensities in MAP2-labeled dendrites were measured and normalized by those in 5 mM KCl treated-control neurons (right). (n=19,18,25,21 for Scramble RNAi + 5 mM KCl, DISC1 RNAi + 5 mM KCl, Scamble RNAi + 55 mM KCl and DISC1 RNAi + 55 mM KCl respectively. F(3,79)=74.17, P<0.0001, one-way ANOVA; ***P<0.001, Bonferroni’s multiple comparison test post hoc). Scale bar represents 25 μM. (C) Total mRNA levels in the synaptosomal fraction were not affected by DISC1 knockdown. A synaptosomal fraction was isolated from neurons in which indicated lentivirus was infected. Total mRNA levels in the synaptosomal fraction were measured by a fluorometer and normalized to total RNA levels. The levels of total mRNA relative to those in control neurons are shown (n=3, F(2,6)=0.5678, P=0.5945, one-way ANOVA; Bonferroni’s multiple comparison test post hoc). (D) The protein levels in the synaptosomal fraction were examined by the knockdown of DISC1. The levels of synaptic proteins relative to those in control (Scramble RNAi) neurons are shown (right) (n=4 for CaMKIIα, n=3 for Shank3 and n=6 for rest of genes, NR1: F(2,15)=176.7, P<0.0001, PSD95: F(2,15)=5.386 P=0.0173, GluR2: F(2,15)=30.71 P<0.0001, Shank3: F(2,6)=11.22 P=0.0094, mGluR1/5: F(2,15)=7.155, P=0.0066, one-way ANOVA; *P<0.05, ***P<0.001, Bonferroni’s multiple comparison test post hoc). Throughout the figures, error bars represent S.E.M.
To confirm the involvement of DISC1 in local translation in dendrites, we visualized puromycin-conjugated nascent polypeptides. We found the DISC1 depletion reduced new protein synthesis in dendrites under stimulation (Figure 3B), consistent with the observation in Figure 3A. Furthermore, the stimulation with brain derived neurotrophic factor (BDNF) also enhanced new protein synthesis in control neurons, which was impaired by DISC1 depletion (Supplemental Figure S5B). These results indicate a novel function of DISC1 in regulating dendrite translation that is dependent on neuronal activity. Total mRNA levels in the synaptosomal fraction were not altered by the loss of DISC1 (Figure 3C). Thus, it is unlikely that the impaired global new protein synthesis in stimulated DISC1-knockdown neurons (Figure 3A,B) was simply due to the reduced mRNA levels in dendrites. Rather, the decrease in translation activity by DISC1 depletion (Figure 2D) was associated with reduced global new protein synthesis in N2a cells (Supplemental Figure S5C). These results suggest that the decreased translation activity is primarily responsible for the reduced SUnSET signals in dendrites of DISC1-depleted neurons (Figure 3A,B). Taken together, DISC1 plays a major novel role in regulating translational activity in neurons.
Next, since various synaptic genes are translated locally in dendrites (10,11), we examined whether the decreased translation activity by DISC1 depletion causes a reduction of synaptic protein levels. The knockdown of DISC1 significantly decreased protein levels of NR1, PSD95, GluR2, NR2B, PSD95 and Shank3 in the synaptosomal fraction (Figure 3D). In contrast, the protein levels of CaMKIIα whose mRNA is reported to be localized in dendrites (44) and presynaptic SV2 were not affected by the knockdown of DISC1. Moreover, we observed that the DISC1 depletion reduced the protein levels of surface-localized neuronal receptors (Supplemental Figure S6). Together, these results indicate that DISC1 positively regulates local translation in dendrites through promotion of translation initiation.
DISC1-TDP43 co-aggregation impairs dendritic local translation
Since endogenous DISC1 is readily sequestered into TDP-220C aggregates (Figure 1E), we hypothesized that the co-aggregation of DISC1 with TDP-220C might have pathological effects similar to DISC1 depletion, including impaired dendritic local translation (Supplemental Figure S7). As expected, the TDP-220C aggregation in neurons (Supplemental Figure S8A) caused a reduction of protein levels of synaptic genes and surface-localized neuronal receptors (Figure 4A and Supplemental Figure S8B). Importantly, the co-expression of DISC1 normalized the reduction of synaptic protein levels and surface-localized neuronal receptors (Figure 4A and Supplemental Figure S8B). Moreover, the TDP-220C aggregation in neurons decreased the induction of new protein synthesis in the synaptosomal fraction by neuronal stimulation, which could be restored by co-expression of DISC1 (Figure 4B). This rescue effect was not caused by the decrease in TDP-220C protein level due to the co-expression of DISC1 (Supplemental Figure S8C). Our results revealed that the loss of functional DISC1 mediates the local translation defects caused by the TDP-220C aggregation. Taken together, the findings support that co-aggregation of DISC1 with TDP-220C impaired the role of DISC1 in dendritic local translation.
Figure 4.
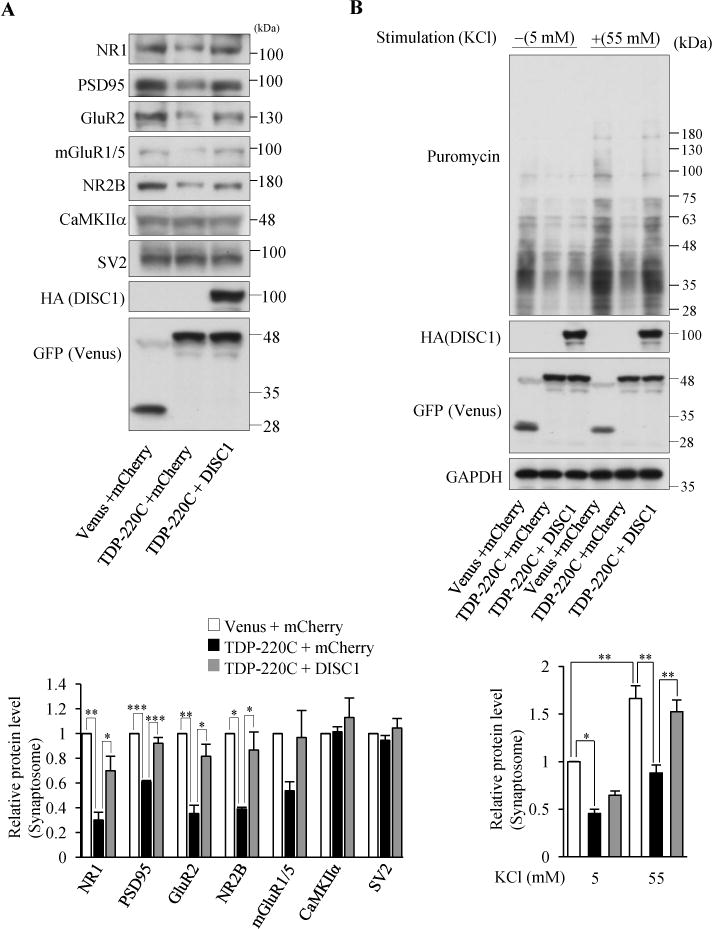
Co-aggregation of TDP43 and DISC1 impairs DISC1-mediated local translation. (A) Indicated synaptic proteins in the synaptosomal fraction of cultured cortical neurons infected with indicated lentivirus were detected by western blotting (top). The levels of synaptic proteins relative to those in control neurons are shown (bottom), (n=3 for NR2B n=4 for rest of genes, NR1: F(2,9)=15.60, P=0.0012; PSD95: F(2,9)=41.39, P<0.0001; GluR2: F(2,9)=16.17, P=0.0010; NR2B: F(2,9)=10.50, P=0.0110, one-way ANOVA; *P<0.05, **P<0.01, ***P<0.001, Bonferroni’s multiple comparison test post hoc). (B) Co-expression of DISC1 restored the decrease in neural stimulation-dependent protein synthesis by TDP-220C aggregation. Cultured cortical neurons were stimulated with 55 mM KCl, followed by the treatment with 10 μg/ml puromycin. The puromycin-labeled proteins in the isolated synaptosomal fraction were detected by western blotting with an anti-puromycin antibody. The signal intensities of puromycin-incorporated polypeptides were normalized to those of GAPDH and then expressed as the relative ratio of control neurons (right) (n=4, F(5,18)=18.99, P<0.0001, one-way ANOVA; *P<0.05, **P<0.01, Bonferroni’s multiple comparison test post hoc). Throughout the figures, error bars represent S.E.M.
DISC1 co-aggregation with TDP-43 in FTLD patient brain
Next, we explored whether neurons in human FTLD brain, which are known to contain TDP-43 aggregates, recapitulate the observation in cultured cortical neurons with TDP-220C aggregates. In control human brain, TDP-43 was localized mainly in nucleoli whereas DISC1 was localized both in the nucleus and cytosol. By contrast, in FTLD forebrain, TDP-43 was depleted from nucleoli and TDP-43 aggregates were observed in cytosol, which is a hallmark of FTLD molecular pathology (5). DISC1 was colocalized with TDP-43 aggregates in FTLD patient brains (Figure 5A and Supplemental Figure S9). Furthermore, detergent-resistant insolubility of both DISC1 and phosphorylated TDP-43 was detected while a soluble DISC1 level was reduced in FTLD patient brains (Figure 5B–D), indicating that functional DISC1 was significantly lost by co-aggregation with TDP-43. Finally, we examined whether protein levels of synaptic genes in FTLD brains were reduced or not, likewise in cultured cortical neurons with TDP-220C aggregates. Protein levels of several synaptic genes showed a significant decrease in FTLD patient brains compared with control brains (Figure 5E). Together, these results from human FTLD brain are consistent with the observations in cultured cortical neurons with TDP-220C aggregates.
Figure 5.
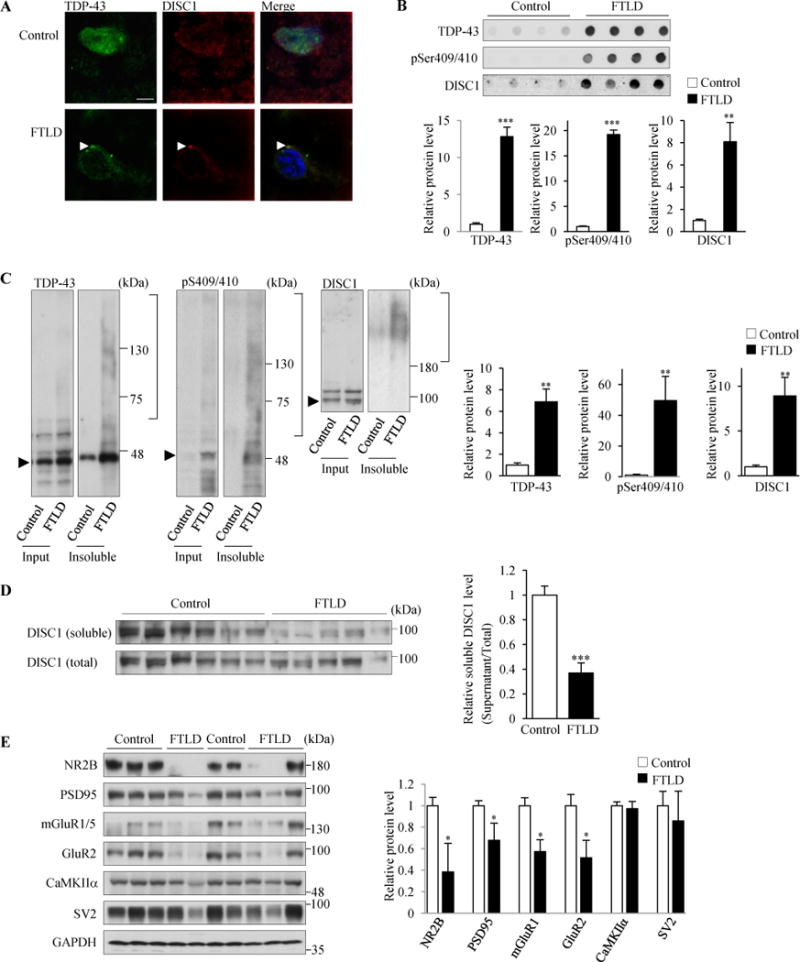
TDP-43 aggregates sequestered endogenous DISC1 in FTLD patient brain. (A) DISC1 and TDP-43 are mislocalized and co-aggregated in the cytosol in FTLD patient brains. DISC1 and TDP-43 were immunostained using anti-DISC1 antibody (h598C) and anti-TDP43 antibodies, respectively, in control (Control, top panels) and FTLD patient brains (FTLD, bottom panels). Brain samples from three different FTLD patients were stained. In neurons containing TDP-43 aggregates, 89% of TDP-43 aggregates were DISC1-positive (n=27). Other images immunostained with another anti-DISC1 antibody (HM6-5) are shown in Supplemental Figure S8D. Scale bar represents 5 μm. (B–D) Insoluble DISC1 levels were increased while soluble DISC1 levels were decreased in FTLD patient brains. (B) 125 μg of the homogenates of temporal lobes from control (n=5) and FTLD (n=5) subjects were incubated with 2% sarkosyl and amounts of insoluble proteins were analyzed by the filter-trap assay, followed by immunoblotting with indicated antibodies (h598C for DISC1). The levels of each genes relative to those in control are also shown (below) (TDP43: t=7.833, ***P<0.0001; pSer409/410: t=12.928, ***P<0.0001; DISC1: t=4.051, **P=0.0037, unpaired two-tailed t-tes). (C) The homogenates of temporal lobe from control (n=6) and FTLD (n=5) subjects were incubated with 2% sarkosyl and then centrifuged at 100,000×g. The resulting sarkosyl-insoluble pellets and the input were analyzed by western blotting with indicated antibodies (h598C for DISC1). Arrowheads indicate the band position of each monomeric protein. The levels of each genes relative to those in control are shown (right) (TDP43: t=3.246, **P=0.0098; pSer409/410: t=3.415, **P=0.0077; DISC1: t=3.253, **P=0.0099, unpaired two-tailed t-test). (D) The homogenates of temporal lobe from control (n=6) and FTLD (n=5) subjects were incubated with 2% sarkosyl and then centrifuged at 100,000×g. The resulting sarkosyl-soluble supernatans and the input were analyzed by western blotting with an anti DISC1 antibody (h598). The signal intensities of DISC1 in supernatants were once normalized to those in the input and then expressed as the relative ratio of control brains (right). (t=5.798, ***P=0.0003, unpaired two-tailed t-test). (E) Homogenates of temporal lobes from control (n=5) and FTLD patient brains (n=5) were subjected to western blotting with indicated antibodies (left). GAPDH was used as a loading control. The signal intensities of each gene relative to those in control brains are shown (right) (NR2B: t=2.689, *P=0.0275; PSD95: t=2.404, *P=0.0429; mGlulR1: t=2.369, *P=0.0453; GluR2: t=2.964, *P=0.0181, unpaired two-tailed t-test). Throughout the figures, error bars represent S.E.M.
Amelioration of behavioral deficits in multiple psychiatric dimensions by DISC1 co-expression in TDP-220C mice
We examined in vivo consequences of DISC1/TDP-220C co-aggregation. We stereotaxically injected adeno-associated virus (AAV) encoding N-terminally Venus-tagged TDP-220C into the frontal cortex of mice (hereafter termed TDP-220C mice) at 6 weeks old. The AAV infection induced the formation of highly phosphorylated TDP-220C aggregates in neurons in frontal cortex (Supplemental Figure S10A–D). Endogenous DISC1 and full-length TDP-43 were sequestered into TDP-220C aggregates (Supplemental Figure S10C,D). Protein levels of postsynaptic genes showed a reduction in TDP-220C mice (Figure 6A), consistent with the results from cultured neurons (Figure 4A). Importantly, the reduced protein levels in the synaptosomal fraction of TDP-220C mice were normalized by co-expression of DISC1 without any effect on the expression level of TDP-220C (Figure 6A and Supplemental Figure S10E). These results show that a loss of DISC1 function mediates the reduced abundance of postsynaptic proteins in TDP-220C mice.
Figure 6.
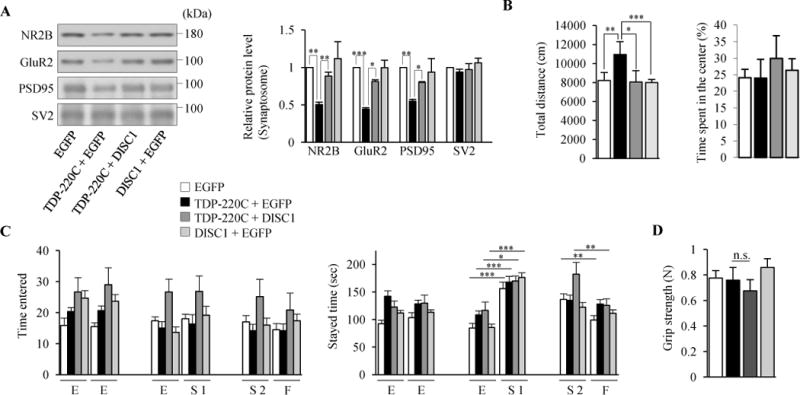
Psychiatric symptoms in TDP-220C mice are rescued by exogenous DISC1 expression. (A to D) AAV encoding indicated genes was stereotaxically injected into mouse prefrontal cortex. (A) Indicated synaptic proteins in the synaptosomal fraction were detected by western blotting (left). The levels of synaptic proteins relative to those in control neurons are shown (right) (n=4, NR2B: F(3,12)=5.231, P=0.0154; GluR2: F(3,12)=25.59, P<0.0001; PSD95: F(3,12)=4.819, P=0.0199; SV2: F(3,12)=0.9337, P=0.4545, one-way ANOVA; *P<0.05, **P<0.01, ***P<0.001, Bonferroni’s multiple comparison test post hoc). (B) In the open field test, TDP-220C mice showed hyperactivity, which was restored by the co-expression of DISC1 (left). The time spent in a center region was not altered between all mice groups (right). n=17, 17, 17, 18 for EGFP (white), TDP-220C+EGFP (black), TDP-220C+DISC1 (dark gray), DISC1+EGFP (light gray) mice, respectively (F(3,65)=6.371, P=0.0007, one-way ANOVA, *P<0.05, **P<0.01, ***P<0.001, Bonferroni’s multiple comparison test post hoc). (C) In the social interaction test, TDP-220C mice were less interested in novel stranger mice, which was rescued by the co-expression of DISC1. During the first encounter, all mice group spent more time in the area with first stranger mice (S1) compared to empty cage (E). During the second encounter, EGFP control mice spent more time with novel, unfamiliar mice (S2) than with familiar mice (F) whereas TDP-220C mice spend equal time with unfamiliar and familiar mice. This behavior was normalized by co-expression of DISC1. n=23, 23, 21, 24 for EGFP (white), TDP-220C+EGFP (black), TDP 220C+DISC1 (dark gray), DISC1+EGFP (light gray) mice, respectively (EGFP: F(5,132)=9.048, P<0.0001; TDP-220C+EGFP: F(5,132)=4.875, P=0.0004; TDP 220C+DISC1: F(5,120)=3.666, P=0.004; DISC1+EGFP: F(5,138)=19.95, P<0.0001, one-way ANOVA, *P<0.05, **P<0.01, ***P<0.001, Bonferroni’s multiple comparison test post hoc). (D) Measurement of forearm grip strength. Grip strength was comparable between all mice groups. n=, 12, 11, 12, 12 for EGFP (white), TDP-220C+EGFP (black), TDP 220C+DISC1 (dark gray), DISC1+EGFP (light gray) mice, respectively. No statistical differences were detected (F(3,39)=2.796, P=0.0528, one-way ANOVA). Throughout the figures, error bars represent S.E.M.
Next, we examined a selected behavioral repertoire of TDP-220C mice two weeks after AAV injection. TDP-220C mice showed significantly increased locomotor activity (hyperactivity) but not in the time spent in a center region in the open field test (Figure 6B). Next we investigated social interactions with the three-chamber test. Control mice interacted more with an unfamiliar stranger mouse than a familiar mouse, but TDP-220C mice did not show significant differences (Figure 6C, right). The time spent in each area of the two resident mice was comparable for control and TDP-220C mice, indicating that the social interaction defect of TDP-220C mice was not caused by their hyperactive phenotypes (Figure 6C, left). Importantly, the hyperactivity and social deficits were rescued by DISC1 co-expression (Figure 6B,C), indicating that restoration of DISC1 function was critical for behavior in these dimensions. In contrast, in the Morris water maze test, TDP-220C mice showed a learning impairment but co-expression of DISC1 did not normalize the learning, indicating that the defect in the hippocampal-dependent learning task in TDP-220C mice is independent of DISC1 dysfunction (Supplemental Figure S10F). As an additional control, the grip strength test confirmed that defects of neuromuscular function which might affect the behavioral tests had not yet been observed in TDP-220C mice (Figure 6D). Collectively, the hyperactivity and disturbed sociability in TDP-220C mice are selectively mediated by the loss of DISC1 function, providing experimental support for the role of TDP-43-DISC1 interactions in FTLD-related behavioral symptoms in FTLD mice.
DISCUSSION
In this study, we found that TDP-43, a causal factor in FTLD, forms co-aggregates with DISC1, which lead to impaired activity-dependent dendritic local translation and mental deficits in FTLD models (Supplemental Figure S7). Importantly, abnormal behaviors in some dimensions in FTLD mice were normalized by exogenous expression of DISC1 in frontal cortex, demonstrating that the loss of soluble functional DISC1 due to co-aggregation is causally responsible for the behaviors. In this study, we established both cultured cortical neurons and mice that express TDP-220C as FTLD models, which faithfully recapitulate the formation of hyper-phosphorylated, ubiquitinated and cytoplasmic aggregates that are the main hallmarks of pathophysiological FTLD (6). Future studies with a full-length TDP-43 model from the endogenous locus will address the influence of TDP-43-DISC1 co-aggregation on broader aspects of TDP-43 proteinopathy.
The impaired activity-dependent local translation in dendrites could be caused by defects of two distinct functions of DISC1, transport of mRNAs, a previously reported function of DISC1 (27) and translation activity, a novel function reported here. Our SUnSET experiments using neurons showed a complete deficiency of neuronal stimulation-dependent translation by DISC1 depletion (Figure 3A,B) whereas DISC1-depleted cells showed an approximately 80% decrease in cap-dependent translation activity in the in vitro translation assay (Figure 2D). These results suggest that the reduced translation activity contributes to about 80% of the overall decrease in dendritic local translation in DISC1-kncokdown neurons (Figure 3A,B). Importantly, the in vitro translation assay showed that the loss of DISC1 significantly impaired translation initiation. Since we found that DISC1 interacts with eIF1, eIF2α and eIF4G on a 40S ribosome subunit (Figure 2B) but does not have significant effects on eIF4F complex formation (Supplemental Figure S11), DISC1 is likely to participate in scanning on mRNA to initiate translation. Taken together, although further studies will decipher how DISC1 mediates local translation of a specific cohort of synaptic genes, DISC1 plays critical role in translation activity through the initiation step, compared to the transport of mRNAs (27), in dendrites of translationally active neurons.
Our results indicate that DISC1 regulates translation of critical synaptic genes, some of which are associated with psychiatric disorders (Figure 3D). Among these, Shank3 is a scaffold protein that organizes macromolecular complexes at the postsynapse (51) and a mutation in SHANK3 is strongly linked to psychiatric disorders including schizophrenia and autism spectrum disorders (52,53). Likewise, TDP-43 aggregation might also disturb DISC1-FMRP interactions, resulting in the misregulation of various FMRP-target mRNAs in FTLD. Recent genome wide association studies of schizophrenia highlighted FMRP-target genes that play critical roles in postsynaptric functions (54,55).
Several lines of DISC1 mutant mice with diverse behavioral phenotypes have been previously reported (56). Among these DISC1 mutant mice, dominant negative transgenic mice (57), point mutant mice (58) and exon2/3-deleted mice (59) showed some phenotypic overlap with TDP-220C mice, including increased locomotion and social impairment. Notably, TDP-220C mice are not phenotypically the same as the previously reported DISC1 mutant mice. Such phenotypic differences may be caused by the gain-of-function of DISC1 and other proteins by their co-aggregation with TDP-220C. DISC1 is proposed to regulate synaptic plasticity and cognitive behavior (27,33,56), but its underlying molecular mechanisms have not fully elucidated yet. Given that regulation of mRNA transport and dendritic local translation are suggested to be critical for synaptic function and plasticity as well as cognitive function, our findings provide a candidate molecular basis for impaired synaptic plasticity and behavioral deficits relevant to psychiatric conditions observed upon functional modulation of DISC1 in neurons and mice (33,56,59,60).
Given that dendritic local translation is a critical process for higher brain functions (12,61), our co-aggregation model suggests a resolution to the long-standing puzzle of why mental disorders are frequently observed in many neurodegenerative disorders. The co-aggregation model might also help to explain the complex heterogeneous nature in neuropsychiatric diseases including FTLD (3,4) because co-aggregates could sequester a range of aggregation-prone proteins that might also include other risk factors for psychiatric diseases such as DISC1 but likely involving other disease candidate molecules. The species and amounts of aggregation-prone proteins sequestered into insoluble or soluble aggregates may differ between patients and these inter-individual differences may partly explain the intrinsic heterogeneity of phenotypic penetrance of psychiatric symptoms.
Supplementary Material
Acknowledgments
We thank Koko Ishizuka, Miles Houslay and Charles Yokoyama for helpful discussion, Hiroshi Matsukawa for help with visualization of newly synthesized peptides in dendrites, Miwa Higaki for help with primary culture preparation and virus production, Hiroyuki Hatsuta for preparation of human brains, Naomi Takahashi for help with subcloning, Dr. Cleveland (University of California) for providing antibodies, Dr. Miyawaki (RIKEN) for his help with microplate luminometer. DNA sequencing and production of polyclonal antibodies were performed at RIKEN Brain Science Institute Research Resources Center facility. Animals were maintained by RIKEN Brain Science Institute animal facility. This work was supported by USPHS grants MH-105660 (A.S.), MH-094268 Silvo O. Conte center (A.S.),, MH-092443 (A.S.), grants from Stanley (A.S.), RUSK (A.S.), S-R foundations (A.S.), NARSAD (A.S.), and Maryland Stem Cell Research Fund (A.S.), the Next program LS129 (M.T.), Grant in Aid for Scientific Research on Innovative Areas (26116004, M.T.), Young Scientists (B) 23700398 (R.E.), from MEXT, Japan, The Uehara Memorial Foundation (M.T.), Daiichi Sankyo Foundation of Life Science (M.T.), Takeda Science Foundation (M.T.) and Suzuken Memorial Foundation (M.T).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
The authors report no biomedical financial interests or potential conflicts of interest.
Supplemental information includes 11 figures, figure legends, methods and references.
References
- 1.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 2.Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;6:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Josephs KA, Hodges JR, Snowden JS, Mackenzie IR, Neumann M, Mann DM, et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol. 2011;122:137–153. doi: 10.1007/s00401-011-0839-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Irish M, Piguet O, Hodges JR. Self-projection and the default network in frontotemporal dementia. Nat Rev Neurol. 2012;8:152–161. doi: 10.1038/nrneurol.2012.11. [DOI] [PubMed] [Google Scholar]
- 5.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 6.Cohen TJ, Lee VM, Trojanowski JQ. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol Med. 2011;11:659–667. doi: 10.1016/j.molmed.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;3:416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee EB, Lee VM, Trojanowski JQ. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci. 2012;13:38–50. doi: 10.1038/nrn3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang IF, Wu LS, Shen CK. TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J Neurochem. 2008;105:797–806. doi: 10.1111/j.1471-4159.2007.05190.x. [DOI] [PubMed] [Google Scholar]
- 10.Bramham CR, Wells DG. Dendritic mRNA: transport, translation and function. Nat Rev Neurosci. 2007;8:776–789. doi: 10.1038/nrn2150. [DOI] [PubMed] [Google Scholar]
- 11.Holt CE, Schuman EM. The central dogma decentralized: new perspectives on RNA function and local translation in neurons. Neuron. 2013;80:648–657. doi: 10.1016/j.neuron.2013.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buffington SA, Huang W, Costa-Mattioli M. Translation control in synaptic plasticity and cognitive dysfunction. Annu Rev Neurosci. 2014;37:17–38. doi: 10.1146/annurev-neuro-071013-014100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doyle M, Kiebler MA. Mechanisms of dendritic mRNA transport and its role in synaptic tagging. EMBO J. 2011;30:3540–3552. doi: 10.1038/emboj.2011.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baralle M, Buratti E, Baralle FE. The role of TDP-43 in the pathogenesis of ALS and FTLD. Biochem Soc Trans. 2013;41:1536–1540. doi: 10.1042/BST20130186. [DOI] [PubMed] [Google Scholar]
- 15.Charles V, Mezey E, Reddy PH, Dehejia A, Young TA, Polymeropoulos MH, et al. Alpha-synuclein immunoreactivity of huntingtin polyglutamine aggregates in striatum and cortex of Huntington’s disease patients and transgenic mouse models. Neurosci Lett. 2000;289:29–32. doi: 10.1016/s0304-3940(00)01247-7. [DOI] [PubMed] [Google Scholar]
- 16.Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, et al. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science. 2003;300:636–640. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- 17.Blum D, Herrera F, Francelle L, Mendens T, Basquin M, Obriot H, et al. Mutant huntingtin alters Tau phosphorylation and subcellular distribution. Hum Mol Genet. 2015;24:76–85. doi: 10.1093/hmg/ddu421. [DOI] [PubMed] [Google Scholar]
- 18.Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–1075. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 20.Pocas GM, Branco-Santos J, Herrera F, Outeiro TF, Domingos PM. α-Synuclein modifies mutant huntingtin aggregation and neurotoxicity in Drosophila. Hum Mol Genet. 2015;24:1898–1907. doi: 10.1093/hmg/ddu606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bachhuber T, Katzmarski N, McCarter JF, Loreth D, Tahirovic S, Kamp F, et al. Inhibition of amyloid-β plaque formation by α-synuclein. Nat Med. 2015;21:802–807. doi: 10.1038/nm.3885. [DOI] [PubMed] [Google Scholar]
- 22.Trossbach SV, Bader V, Hecher L, Pum ME, Masoud ST, Prikulis I, et al. Misassembly of full-length Disrupted-in-Schizophrenia 1 protein is linked to altered dopamine homeostasis and behavioral deficits. Mol Psychiatry. 2016;11:1561–1572. doi: 10.1038/mp.2015.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanaka M, Ishizuka K, Nekooki-Machida Y, Endo R, Takashima N, Sasaki H, et al. Aggregation of scaffolding protein DISC1 dysregulates phophodiesterase 4 in Huntington’s disease. J Clin Invest. 2017;127:1438–1450. doi: 10.1172/JCI85594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brandon NJ, Sawa A. Linking neurodevelopmental and synaptic theories of mental illness through DISC1. Nat Rev Neurosci. 2011;12:707–722. doi: 10.1038/nrn3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niwa M, Cash-Padgett T, Kubo KI, Saito A, Ishii K, Sumitomo A, et al. DISC1 a key molecular lead in psychiatry and neurodevelopment: No-More Disrupted-in-Schizophrenia. Mol Psychiatry. 2016;21:1488–1489. doi: 10.1038/mp.2016.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taya S, Shinoda T, Tsuboi D, Asaki J, Nagai K, Hikita T, et al. DISC1 regulates the transport of the NUDEL/LIS1/14-3-3epsilon complex through kinesin-1. J Neurosci. 2007;27:15–26. doi: 10.1523/JNEUROSCI.3826-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsuboi D, Kuroda K, Tanaka M, Namba T, Iizuka Y, Taya S, et al. Disrupted-in-schizophrenia 1 regulates transport of ITPR1 mRNA for synaptic plasticity. Nat Neurosci. 2015;18:698–707. doi: 10.1038/nn.3984. [DOI] [PubMed] [Google Scholar]
- 28.Freibaum BD, Chitta RK, High AA, Taylor JP. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res. 2010;9:1104–1120. doi: 10.1021/pr901076y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Russo A, Scardigli R, La Regina F, Murray ME, Romano N, Dickson DW, et al. Increased cytoplasmic TDP-43 reduces global protein synthesis by interacting with RACK1 on polyribosomes. Hum Mol Genet. 2017;26:1407–1418. doi: 10.1093/hmg/ddx035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suzuki H, Shibagaki Y, Hattori S, Matsuoka M. Nuclear TDP-43 causes neuronal toxicity by escaping from the inhibitory regulation by hnRNPs. Hum Mol Genet. 2015;24:1513–1527. doi: 10.1093/hmg/ddu563. [DOI] [PubMed] [Google Scholar]
- 31.Leliveld SR, Bader V, Hendriks P, Prikulis I, Sajnani G, Requena JR, et al. Insolubility of disrupted-in-schizophrenia 1 disrupts oligomer-dependent interactions with nuclear distribution element 1 and is associated with sporadic mental disease. J Neurosci. 2008;28:3839–3845. doi: 10.1523/JNEUROSCI.5389-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Porteous DJ, Millar JK, Brandon NJ, Sawa A. Disc1 at 10: connecting psychiatric genetics and neuroscience. Trends Mol Med. 2011;12:699–706. doi: 10.1016/j.molmed.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hayashi-Takagi A, Takaki M, Graziane N, Seshadri S, Murdoch H, Dunlop AJ, et al. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci. 2011;13:327–332. doi: 10.1038/nn.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shahani N, Seshadri S, Jaaro-Peled H, Ishizuka K, Hirota-Tsuyada Y, Wang Q, et al. DISC1 regulates trafficking and processing of APP and Aβ generation. Mol Psychiatry. 2015;20:874–879. doi: 10.1038/mp.2014.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seshadri S, Faust T, Ishizuka K, Delevich K, Chung Y, Kim SH, et al. Interneuronal DISC1 regulates NRG1-ErbB4 signalling and excitatory-inhibitory synapse formation in the mature cortex. Nat Commun. 2015;6 doi: 10.1038/ncomms10118. ncomm10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sama RR, Ward CL, Bosco DA. Functions of FUS/TLS from DNA repair to stress response: Implications for ALS. ASN Neuro. 2014;6 doi: 10.1177/1759091414544472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Camargo LM, Collura V, Rain JC, Mizuguchi K, Hermjakob H, Kerrien S, et al. Disrupted in Schizophrenia 1 Interactome: evidence for the close connectivity of risk genes and a potential synaptic basis for schizophrenia. Mol Psychiatry. 2007;12:74–86. doi: 10.1038/sj.mp.4001880. [DOI] [PubMed] [Google Scholar]
- 38.Stefani G, Fraser CE, Darnell JC, Darnell RB. Fragile X mental retardation protein is associated with translating polyribosomes in neuronal cells. J Neurosci. 2004;24:7272–7276. doi: 10.1523/JNEUROSCI.2306-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coyne AN, Siddegowda BB. Futch/MAP1B mRNA is a translational target of TDP-43 and is neuroprotective in a Drosophila model of amyotrophic lateral sclerosis. J Neurosci. 2014;34:15962–15974. doi: 10.1523/JNEUROSCI.2526-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ceman S, O’Donnell WT, Reed M, Patton S, Pohl J, Warren ST. Phosphorylation influences the translation state of FMRP-associated polyribosomes. Hum Mol Genet. 2003;12:3295–3305. doi: 10.1093/hmg/ddg350. [DOI] [PubMed] [Google Scholar]
- 41.Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Napoli I, Mercaldo V, Boyl PP, Eleuteri B, Zalfa F, De Rubeis S, et al. The fragile X syndrome protein represses activity-dependent translation through CYFIP1, a new 4E-BP. Cell. 2008;134:1042–54. doi: 10.1016/j.cell.2008.07.031. [DOI] [PubMed] [Google Scholar]
- 43.Knowles RB, Sabry JH, Martone ME, Deerinck TJ, Ellisman MH, Bassell GJ, et al. Translocation of RNA granules in living neurons. J Neurosci. 1996;16:7812–7820. doi: 10.1523/JNEUROSCI.16-24-07812.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cajigas IJ, Tushev G, Will TJ, Dieck ST, Fuerst N, Schuman EM. The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron. 2012;74:453–466. doi: 10.1016/j.neuron.2012.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, Gao W, Svitkin YV, Chen AP, Cheng YC. DCB-3503, a Tylophorine Analog, Inhibits Protein Synthesis through a Novel Mechanism. PLoS One. 2010;5:e11607. doi: 10.1371/journal.pone.0011607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Friday AJ, Henderson MA, Morrison JK, Hoffman JL, Keiper BD. Spatial and temporal translational control of germ cell mRNAs mediated by the eIF4E isoform IFE-1. J Cell Sci. 2015;128:4487–4498. doi: 10.1242/jcs.172684. [DOI] [PubMed] [Google Scholar]
- 47.Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6:275–277. doi: 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- 48.Ramocki MB, Zoghbi HY. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature. 2008;455:912–918. doi: 10.1038/nature07457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ebert DH, Greenberg ME. Activity-dependent neuronal signalling and autism spectrum disorder. Nature. 2013;493:327–337. doi: 10.1038/nature11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- 51.Jiang YH, Ehlers MD. Modeling autism by SHANK gene mutations in mice. Neuron. 2013;78:8–27. doi: 10.1016/j.neuron.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilson HL, Wong AC, Shaw SR, Tse WY, Stapleton GA, Phelan MC, et al. Molecular characterization of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J Med Genet. 2003;40:575–584. doi: 10.1136/jmg.40.8.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gauthier J, Champagne N, Lafreniere RG, Xiong L, Spiegelman D, Brustein E, et al. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc Natl Acad Sci USA. 2010;107:7863–7868. doi: 10.1073/pnas.0906232107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tomoda T, Sumitomo A, Jaaro-Peled H, Sawa A. Utility and validity of DISC1 mouse models in biological psychiatry. Neuroscience. 2016;321:99–107. doi: 10.1016/j.neuroscience.2015.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hikida T, Jaaro-Peled H, Seshadri S, Oishi K, Hookway C, Kong S, et al. Dominant negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc Natl Acad Sci USA. 2007;104:14501–14506. doi: 10.1073/pnas.0704774104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Clapcote SJ, Lipina TV, Millar JK, Mackie S, Christie S, Ogawa F, et al. Behavioral Phenotypes of Disc1 Missense Mutations in Mice. Neuron. 2007;54:387–402. doi: 10.1016/j.neuron.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 59.Kuroda K, Yamada S, Tanaka M, Iizuka M, Yano H, Mori D, et al. Behavioral alterations associated with targeted disruption of exon 2 and 3 of the Disc1 gene in the mouse. Hum Mol Genet. 2011;20:4666–4683. doi: 10.1093/hmg/ddr400. [DOI] [PubMed] [Google Scholar]
- 60.Kvajo M, McKellar H, Drew LJ, Lepagnol-Bestel AM, Xiao L, Levy RJ, et al. Altered axonal targeting and short-term plasticity in the hippocampus of Disc1 mutant mice. Proc Natl Acad Sci USA. 2011;108:1349–1358. doi: 10.1073/pnas.1114113108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu-Yesucevitz L, Bassell GJ, Gitler AD, Hart AC, Klann E, Richter JD, et al. Local RNA translation at the synapse and in disease. J Neurosci. 2011;31:16086–16093. doi: 10.1523/JNEUROSCI.4105-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.