

Abstract
RAS is the most frequently mutated gene across human cancers, but developing inhibitors of mutant RAS has proven to be challenging. Given the difficulties of targeting RAS directly, drugs that impact the other components of pathways where mutant RAS operates may potentially be effective. However, the system-level features, including different localizations of RAS isoforms, competition between downstream effectors, and interlocking feedback and feed-forward loops, must be understood to fully grasp the opportunities and limitations of inhibiting specific targets. Mathematical modeling can help us discern the system-level impacts of these features in normal and cancer cells. New technologies enable the acquisition of experimental data that will facilitate development of realistic models of oncogenic RAS behavior. In light of the wealth of empirical data accumulated over decades of study and the advancement of experimental methods for gathering new data, modelers now have the opportunity to advance progress toward realization of targeted treatment for mutant RAS-driven cancers.
Keywords: RAS, ERK cascade, mechanistic modeling, mathematical modeling, systems biology
1. Introduction
Following the discovery that mutant RAS genes can cause oncogenic transformation (reviewed by Barbacid in 1987 [1]), RAS signaling has been in the limelight of scientific interest for over three decades [2]. RAS proteins are GTPases and key transducers of receptor tyrosine kinase (RTK) signaling, which is central for cell proliferation and survival. Although more than 170 RAS-related proteins have been identified in humans, three RAS family members are recognized as important oncogenes: HRAS, NRAS, and KRAS (with splice isoforms KRAS4A and KRAS4B) [3]. Variants in these three RAS genes are the most prevalent mutations across human cancers, appearing in more than 30% of all cases, yet therapies to selectively inhibit mutant RAS are only recently emerging [4,5].
RAS receives activating inputs from guanine nucleotide exchange factors (GEFs) and deactivating inputs from GTPase activating proteins (GAPs) (Fig. 1). GEFs (e.g., SOS1-2, RASGRP1-4, RASGRF1-2) facilitate RAS conformational change from the inactive GDP-bound form to the active GTP-bound form by promoting nucleotide release. As intracellular GTP/GDP ratios are estimated to be greater than 10 under nutrient replete conditions [6,7], a free RAS G domain is more likely to bind GTP than GDP. GAPs (e.g., RASA1, NF1, SYNGAP1, RASAL1, RASA3-4) prompt the reverse transition by accelerating GTP hydrolysis [8-13]. Thus, RAS GAPs have the potential to function as tumor suppressors, and loss-of-function mutations affecting RAS GAPs have the potential to be oncogenic. Indeed, suppression of NF1 is found in a variety of cancers including glioblastoma, non-small cell lung cancer, and melanoma, and mutations in NF1 are found in neurofibromatosis type 1 [14]. Similarly, inactivation of RASA1 can promote melanoma tumorigenesis via RAS activation, and lower expression of RASA1 is associated with decreased survival in melanoma patients harboring BRAF mutations [15]. RAS has similar affinities for GTP and GDP, but because of intrinsic GTPase activity and a low GDP/GTP exchange rate in the absence of GEF activity, inactive RAS-GDP is the prevailing form in unstimulated cells [16].
Fig. 1.
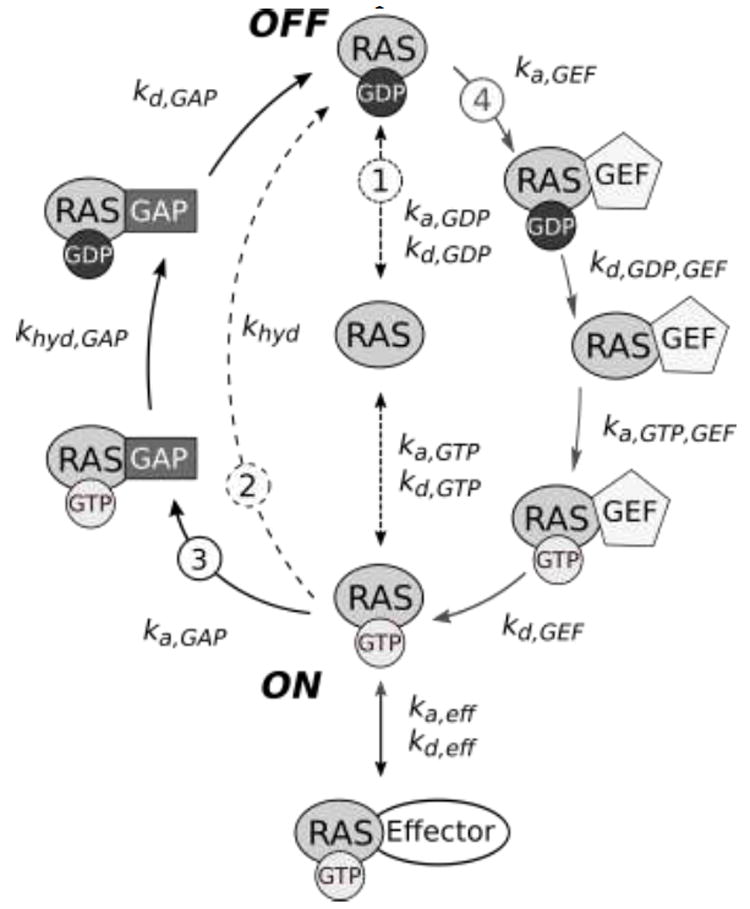
The RAS activation cycle. RAS can bind either GTP or GDP, and is active when bound to GTP. In the active configuration, it is able to interact with downstream effectors. RAS activation/deactivation can occur through multiple processes. Processes of interest are labeled with circled numbers: 1 shows free nucleotide exchange, 2 describes RAS-catalyzed hydrolysis of GTP to GDP, 3 depicts GTP hydrolysis stimulated by GAP, and 4 is GEF-induced GDP release from RAS to facilitate GTP binding. Rate constants for each step are labeled; variable names correspond to those in Table 1.
RAS interacts with a number of downstream effectors, which become activated when recruited to the GTP-bound form of RAS. These effectors include RAF kinase family proteins (ARAF, BRAF, and CRAF), phosphoinositide 3-kinase (PI3K), RAS-like (RAL) small GTPases (RALGDS, RGL, RGL2, RAGL3), phospholipase C epsilon (PLCε), the RNA-silencing effector Argonaute 2 (AGO2), and RAS association domain family (RASSF) proteins (NORE1, RASSF1A) [11,17,18]. The pathways emanating from RAS effectors influence a wide variety of cellular processes, including mitogenic, survival, pro-apoptotic, inflammation, DNA-damage repair, and senescence pathways [19] (Fig. 2). Furthermore, GTP-bound KRAS4B proteins may form RAS-RAS dimers that promote dimerization and activation of RAF kinases while hiding other RAS effector binding regions [20].
Fig. 2.
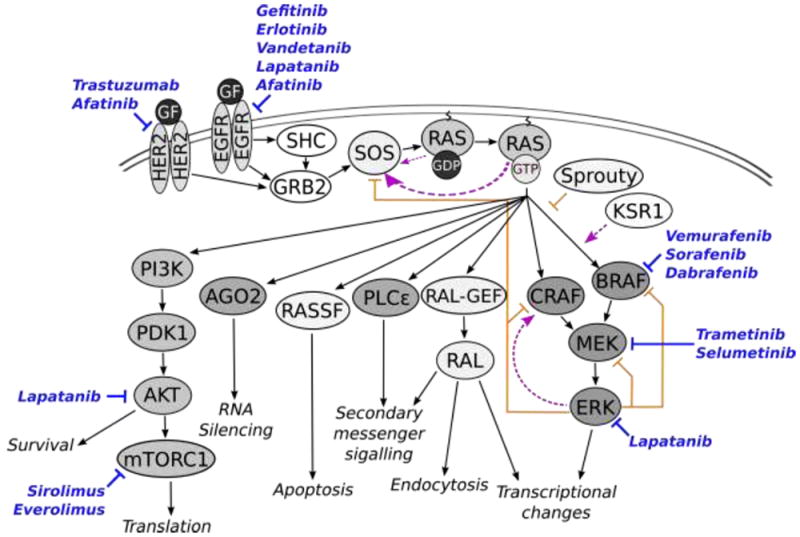
Molecularly targeted drugs impacting networks related to RAS signaling. RAS receives activating inputs from EGFR and HER2 triggered by growth factors. Signals propagate from RAS to downstream effectors including RASSF, RAF, RAL-GEF, PLCε, and PI3K, resulting in varied impacts on cellular phenotype, shown with gray text and arrows. Positive feedbacks are indicated with dashed lines and arrows in magenta. Note that the positive feedback from RAS-GTP to SOS is stronger than that of RAS-GDP, as indicated by the thickness of the line. Negative feedbacks are shown with orange lines with blunt ends. Inhibitory drugs are shown in blue with their target indicated by a blue line with a blunt end.
Most oncogenic RAS mutations disrupt the GTPase activity of RAS isoforms, locking RAS in the GTP-bound state and resulting in constitutive activation of downstream cell signaling pathways. Over 99% of all oncogenic RAS mutations occur in codons 12, 13, and 61 [21]. Codons 12 and 13 are located in one of four primary sequence regions critical for GTP-binding. Codon 61 falls in a region that is important for both GTP-binding and GEF-binding (the Switch II region) [22]. Although codons 12, 13, and 61 are in regions that are identical for all RAS isoforms, the distribution of oncogenic mutations differs between these isoforms [21]. Constitutive activation of NRAS by mutation at codon 61 is more common in melanoma [23], whereas mutations in KRAS codons 12 and 13 are common in colorectal, lung, and pancreatic cancers [24]. Interestingly, 80% of oncogenic KRAS mutations occur in codon 12 [21].
The prevalence of RAS mutations in cancers, availability of empirical data accumulated over decades of study, and the complexity of RAS signaling networks render RAS a promising candidate for investigation via mathematical modeling. Models have proven useful in simulating both the RAS activation cycle as well as the larger network surrounding RAS, including the extracellular signal-related (ERK) cascade [25]. In 2000, Brightman and Fell published an an ordinary differential equation (ODE) model describing regulation of ERK that considered RAS activation and GEF/GAP activity [26]. This model revealed the importance of feedback regulation in achieving either sustained or transient activation of RAS, MEK, and ERK. In 2002, Schoeberl et al. [27] produced an ODE model of the ERK pathway, consisting of 101 reactions and 94 species, many of which were included in Kholodenko et al.’s [28] 1999 model of signal transduction from the epidermal growth factor receptor (EGFR) through SOS. This model was applied to predict how dynamics of growth factor binding impact ERK activation. However, it lacked GAP regulation and considered GAP activity as a constant factor (reviewed by Orton et al. [29]). In 2004, Markevich et al. described an early mechanistic model focused on RAS activation by RTKs [16]. This model captured the regulation of wildtype RAS by GEFs and GAPs as well as the consequences of changes in RAS intrinsic nucleotide exchange activity and GTPase activity. Importantly, the model demonstrated that RAS activation patterns can be explained by delays between the activation of GEFs and GAPs by RTKs, resulting in transient RAS activation in response to epidermal growth factor (EGF) treatment. In 2007, mechanistic models began to be used to study the impact of mutations on RAS signaling, with the model of Stites et al. [30] comparing wildtype and oncogenic mutant RAS to infer strategies for selectively inhibiting the oncogenic network. In 2009, Orton et. al. modeled the ERK pathway to predict the result of EGFR overexpression or mutations in RAS, BRAF, and EGFR [31]. In 2015, the model of Stites et al. (2007) was expanded to simulate random mutagenesis throughout the network, leading to the conclusion that mutations in the tumor suppressor gene NF1 work in concert with mutations in RAS signaling to drive cancer [32].
Mathematical modeling promises to help us understand distinct RAS signaling patterns in the context of different adaptive topologies of the RAS network and diverse cellular backgrounds [33]. Yet, existing models have mostly focused on RAS activation within a single RTK pathway, neglecting to consider the impacts of intricate feedback and feedforward interactions between multiple RAS effector pathways. Furthermore, there is an unmet need for modeling studies that evaluate the phenotypic consequences of the broad spectrum of RAS mutations and that consider differential localization of RAS isoforms. In this review, we describe several new technologies that can generate the data needed to develop more sophisticated models of RAS signaling. We summarize complex and nonlinear phenomena involved in RAS signaling, which provide novel opportunities for mathematical modeling studies. In light of these developments, the future application of improved mathematical models of RAS signaling could enable prediction of clinical responses to drugs and their combinations and to eventually aid in the rational design of cancer therapies.
2. New technologies enable development of improved mathematical models
2.1 Measuring equilibrium and rate constants for mutant forms of RAS
RAS mutations associated with cancer, such as mutations at codons 12, 13, and 61, result in impaired RAS GTPase activity and a decrease in the binding and/or catalytic activities of GAPs [2,34,35]. Integrating experimental data from cells harboring oncogenic RAS into mathematical models provides insight into the aberrant signaling underlying RAS-driven cancers.
One technique commonly used to measure rate constants for RAS activation involves N-methylanthraniloyl derivatives of GTP/GDP that can be tracked with fluorescence spectroscopy [36,37]. Using this methodology, rate constants for tens of variants of mutant RAS have been reported [38-41] and incorporated into mechanistic models [30,42-44]. Obtaining rate constants for RAS variants as well as for other mutant enzymes involved in RAS signaling will continue to be important for understanding RAS-driven diseases. Going forward, the application of advanced techniques leveraging nuclear magnetic resonance (NMR) spectroscopy may well provide new information on signaling pathways, with NMR being capable of simultaneous measurement of many relevant processes, including binding, catalysis, post-translational modification, and conformational change [45-47]. NMR has been used to study hydrolysis and nucleotide exchange rates of oncogenic RAS mutants in real-time [48]. It may also be possible to approximate rate constants in cases where a specific variant has not been measured, as there is good correlation between structure-derived energy values and rate constants across libraries of mutant RAS [44]. Table 1 lists many of the available experimentally determined parameters for wildtype and oncogenic mutant RAS.
Table 1.
Parameters relevant for modeling the RAS activation cycle, for wildtype (WT) RAS and 15 oncogenic RAS variants. Rate constants correspond to those in Fig. 1. Variables have units s-1 for first-order reactions and M-1s-1 for second-order reactions. Values are formatted for the RAS isoform for which the measurement was taken. Values for KRAS are in bold, values for HRAS are italic, and values for NRAS are underlineds
| RAS Mutation | ka, GTP (1/M/s) | kd,GTP (1/s) | ka,GDP (1/M/s) | kd,GDP(1/s) | khyd (1/s) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| None (WT) | 2.05E-4 | [37] | 6.33E-5 | [48] | 1.47E-4 | [48] | ||||
| 1.00E-4 | [174] | 4.20E-4 | [174] | 3.40E-4 | [174] | |||||
| 2.00E-3 | [175] | 2.00E-3 | [175] | 6.80E-4 | [175] | |||||
| 1.40E8 | [174] | 9.00E-5 | [176] | 5.10E7 | [174] | 1.20E-4 | [176] | 2.10E-4 | [176] | |
| 2.20E6 | [173] | 2.50E-4 | [177] | 2.30E6 | [173] | 1.08E-4 | [177] | 3.49E-4 | [177] | |
| 2.33E-4 | [180] | 3.00E-5 | [38] | 9.30E-3 | [38] | |||||
| 2.17E-4 | [180] | 2.17E-4 | [180] | |||||||
| 1.60E-5 | [173] | |||||||||
| 0.013 | [181] | |||||||||
|
| ||||||||||
| G12A | 200E-3 | [175] | 2.00E-3 | [175] | 1.30E-5 | [175] | ||||
| 9.00E-5 | [176] | 5.00E-5 | [176] | 5.00E-5 | [176] | |||||
|
| ||||||||||
| G12C | 2.00E-3 | [175] | 2.00E-3 | [175] | 4.90E-4 | [175] | ||||
| 6.00E-5 | [176] | 2.20E-4 | [176] | 1.80E-4 | [176] | |||||
|
| ||||||||||
| G12D | 5.00E-4 | [174] | 2.00E-4 | [174] | 1.50E-4 | [174] | ||||
| 2.00E-3 | [175] | 2.00E-3 | [175] | 1.90E-4 | [175] | |||||
| 4.80E8 | [174] | 9.50E-4 | [176] | 7.00E7 | [174] | 1.60E-4 | [176] | 1.40E-4 | [176] | |
| 7.54E6 | [30] | 1.25E-3 | [30] | 3.16E6 | [30] | 5.16E-5 | [30] | 1.40E-4 | [30] | |
| 6.00E-4 | [180] | 1.67E-4 | [180] | 9.33E-5 | [180] | |||||
|
| ||||||||||
| G12E | 4.90E-4 | [176] | 1.50E-4 | [176] | 1.60E-4 | [176] | ||||
|
| ||||||||||
| G12R | 2.00E-3 | [175] | 2.00E-3 | [175] | 1.80E-5 | [175] | ||||
| 6.17E-3 | [181] | |||||||||
|
| ||||||||||
| G12S | 2.00E-5 | [176] | 4.80E-4 | [176] | 1.20E-4 | [176] | ||||
|
| ||||||||||
| G12V | 8.00E-5 | [174] | 3.33E-5 | [48] | 1.48E-5 | [48] | ||||
| 5.80E8 | [174] | 2.00E-3 | [175] | 1.30E-4 | [174] | 4.00E-5 | [174] | |||
| 9.11E6 | [30] | 9.00E-5 | [176] | 1.16E8 | [174] | 2.00E-3 | [175] | 4.20E-5 | [175] | |
| 2.00E-4 | [30] | 5.23E6 | [30] | 2.00E-5 | [176] | 5.00E-6 | [176] | |||
| 2.00E-5 | [38] | 1.50E-3 | [38] | |||||||
| 3.35E-5 | [30] | 5.23E-5 | [30] | |||||||
| 3.83E-3 | [181] | |||||||||
|
| ||||||||||
| G13C | 8.00E-05 | [176] | 2.50E-4 | [176] | 1.10E-4 | [176] | ||||
|
| ||||||||||
| G13D | 0.018 | [175] | 9.45E-4 | [48] | 5.17E-5 | [48] | ||||
| 6.30E-04 | [176] | 0.027 | [175] | 9.60E-5 | [175] | |||||
| 1.60E-4 | [176] | 1.90E-4 | [176] | |||||||
|
| ||||||||||
| G13S | 7.00E-5 | [176] | 3.60E-4 | [176] | 3.20E-4 | [176] | ||||
|
| ||||||||||
| G13V | 1.20E-4 | [176] | 3.40E-4 | [176] | 2.00E-4 | [176] | ||||
|
| ||||||||||
| Q61H | 2.00E-3 | [175] | 2.00E-3 | [175] | 1.30E-5 | [175] | ||||
| 1.53E-4 | [177] | 1.60E-4 | [177] | 3.05E-5 | [177] | |||||
| 0.012 | [181] | |||||||||
|
| ||||||||||
| Q61K | 1.50E-4 | [177] | 1.52E-4 | [177] | 7.07E-4 | [177] | ||||
|
| ||||||||||
| Q61L | 2.00E-3 | [175] | 1.50E-4 | [48] | 1.83E-6 | [48] | ||||
| 2.67E-4 | [177] | 2.00E-3 | [175] | 8.00E-6 | [175] | |||||
| 2.67E-4 | [177] | 7.58E-6 | [177] | |||||||
|
| ||||||||||
| Q61P | 3.17E-4 | [177] | 1.13E-4 | [177] | 2.72E-5 | [177] | ||||
|
| ||||||||||
| Q61R | 1.12E-4 | [177] | 1.23E-4 | [177] | 6.10E-6 | [177] | ||||
|
| ||||||||||
| Q61W | 2.00E-4 | [177] | 1.08E-4 | [177] | 2.53E-5 | [177] | ||||
|
| ||||||||||
| 10G11 | 5.50E-4 | [180] | 1.18E-3 | [180] | 1.83E-5 | [180] | ||||
GGEF is RASGRF1. SGEF is SOS1. RGAP is RASA1. NGAP is NF1. *calculated from Lenzen et al. [173] using nucleotide dissociation rate as kr = 3.9 s-1 = kd,GDP,GEF and Kd (Kd = Kd4), such that kf = ka,GTP,GEF = kr/Kd. **calculated from Lenzen et al. [173], kr = Kd3kf3 = (0.6e-3)(3.4e4), assuming the same dissociation rate of GEF from RAS-GTP and RAS-GDP.
With recent technological advances, binding affinities can be measured in high-throughput fashion, which will assist in the development of more accurate and comprehensive mechanistic models. Techniques using protein domain microarrays and fluorescence-polarization (FP) allow for the measurement of thousands of equilibrium dissociation constants [49,50]. Of particular importance when the goal is to model cell signaling networks, these techniques measure binding affinities in a domain-specific manner. Other high-throughput techniques for quantifying binding affinities are based on chromatography or deep sequencing [51-53]. Affinities measured by high-throughput proteomics are already contributing to the development of new mechanistic models of cell signaling [54]. For RAS-related models, effort is still needed to quantify affinities for the various RAS-effector interactions. A summary of available equilibrium dissociation constants for RAS isoforms and 13 effectors is presented in Table 2.
Table 2.
Equilibrium dissociation constants (KD) for RAS effectors. KD = kd,eff/ka,eff as in Fig. 1. Values have units of μM. A dash indicates that no value for the isoform was reported in the cited study
Finally, techniques enabling real-time activity quantification for single molecules allow for insight into the degree of intracellular variation in reaction rates. For instance, Iversen et al. [55] measured nucleotide exchange rates on single SOS molecules with a novel assay involving a synthetic membrane partitioned into micrometer-scale compartments. Interestingly, they observed a very wide distribution of catalytic activities, with a small proportion of SOS molecules transiently entering a state of very high catalytic activity. From a modeling perspective, this observation suggests that ODE-based models may not appropriately capture some aspects of RAS signaling. Stochastic modeling approaches are applicable when molecular species with low copy numbers, such as rare highly active SOS molecules, contribute significantly to system behavior.
2.2 Protein copy number measurements
Protein copy number measurements are key data necessary for the development of realistic mechanistic models. Techniques for measuring the proteome are becoming increasingly complete, fast, and cost-effective [56]. Mass-spectrometry (MS)-based approaches have demonstrated the ability to quantify abundances of thousands of mammalian proteins in parallel [57-60]. New proteomics methods have been introduced that eliminate the need for spiking-in reference peptides, counting cells, or protein concentration measurements, which simplifies protocols for large-scale proteomics studies [61] (see the excellent recent review on MS-based proteomic technologies by Aebersold and Mann [62]).
Improvements in targeted proteomics techniques expedite quantification of specific subsets of proteins across varying conditions, such as across cells types, or in response to external stimuli [54,63]. In contrast to shotgun proteomics, targeted proteomics techniques are capable of detecting proteins expressed at nanomolar levels (corresponding to a detection threshold of approximately 1000 cytosolic molecules per cell) [64]. A recent study used ultra-sensitive targeted proteomics to measure abundances of 26 proteins in the ERK pathway in normal human mammary epithelial cells (HMECs), breast cancer cell lines, and normal human fibroblasts [65]. This analysis revealed that the majority of proteins in the pathway have similar expression levels across cell types, and helped to quantify relative abundances between members of the pathway. Proteomics raises awareness of previous modeling assumptions that may not be valid across cell types, thereby improving the accuracy of future models. For instance, the assumption that the EGFR adapter molecules SHC1, GRB2, and GAB1 are present in excess [28] would be incorrect in many cell lines, as these proteins have been found to have per-cell quantities ranging from 3,000 to 55,000, whereas EGFR was found to have a median per-cell copy number of 210,000 across seven normal and cancer cell lines [65].
Protein phosphorylation plays an important role in many cellular regulatory processes, including signaling downstream of RAS. Thus, new and developing phosphoproteomic techniques for quantitatively monitoring changes in protein phosphorylation can potentially be applied to gain insights into RAS signaling dynamics and to support modeling studies of RAS-related pathways. Sudhir et al. [66] identified phosphorylation events in human bronchial epithelial cells (HBECs) with and without expression of the oncogenic mutant KRAS G12V. A total of 52 proteins were differentially phosphorylated upon transformation with mutant KRAS. This analysis allowed for identification of new pathways by which oncogenic and wildtype RAS activation influence human cells. Others have used phosphoproteomics to study alterations in phosphorylation arising from the presence of oncogenic KRAS G12V, activated CDC42 G12V, or knockout of the downstream effector PAK4 [67]. Phosphoproteomics was also applied to study serine-threonine family kinases, identifying proteins with decreased phosphorylation following addition of EGFR, PI3K, mTOR, and MEK inhibitors [68].
2.3 Genetically engineered cell lines to study effects of RAS network mutations
The ability to genetically alter human cell lines is an exciting avenue by which to study the biological basis for disease. The introduction and maturation of CRISPR/Cas9 and related CRISPR technologies has resulted in significant improvements in genome engineering efficiency with reduced off-target effects [69-72]. CRISPR/Cas9 is exceptionally versatile and can provide the means for controlled studies of heterozygous and homozygous mutations, as well as gene amplification or overexpression, gene deletion or repression, and isoform effects. To our knowledge, CRISPR-Cas9 has not yet been used to directly support mechanistic RAS modeling, but findings from the application of this technology are already offering intriguing results that can be further analyzed computationally. Related to RAS signaling, CRISPR-libraries have been used to engineer cells for identifying mechanisms of resistance to the BRAF inhibitor vemurafenib [69,73]. Vemurafenib resistance was observed with CRISPR-induced overexpression of BCAR3, EGFR, and a number of G protein-coupled receptors (GPR35, LPAR1, LPAR5, and P2RY8), and following deletion of NF1, NF2, and MED12.
Genome-wide screening with CRISPR can be used to identify important pathway components that may have been neglected in prior modeling efforts. For instance, Wang et al. [74] used CRISPR to engineer acute myeloid leukemia (AML) cell lines to examine gene essentiality. One of the genes determined to be essential for the proliferation of RAS-driven AML was SHOC2. A complex of SHOC2, protein phosphatase 1 (PP1), the scaffold protein SCRIB, and the RAS-family member MRAS is capable of promoting ERK activation by dephosphorylating the inhibitory S259 site on CRAF [75,76]. However, SCRIB has also been noted to have inhibitory effects on ERK activation through interactions with MEK and ERK [76]. No mathematical model has yet accounted for the cooperative role of MRAS in RAS signaling. The presence of both positive and negative effects on ERK by proteins involved in MRAS regulation could contribute to observed temporal pulses in ERK [33], which is certainly worthy of model-guided investigation.
2.4 Real-time monitoring and control
Recent advances in real-time cellular monitoring enable direct observation of how input signals propagate through RAS signaling networks and contribute to resulting cellular phenotypes, providing mechanistic insights and empirical data useful for defining and parameterizing mechanistic models. For instance, Harvey et al. [77] examined neuronal RAS signaling by applying a Förster (or fluorescence) resonance energy transfer (FRET)-based sensor. HRAS was tagged with a green fluorescent protein and the RAS-binding domain of RAF was tagged with two red fluorescent proteins. Two-photon fluorescence lifetime imaging (2pFLIM) was used to generate spatial profiles of RAS activation by tracking FRET arising from RAS/RAF interaction. RAS activation was observed to be reduced with inhibition of calcium/calmodulin dependent protein kinase II (CaMKII), PI3K, or protein kinase C (PKC). The spatial data was used to parameterize a model accounting for one-dimensional diffusion and the rate of RAS deactivation, leading to the conclusion that rapid diffusion of active RAS was the primary mechanism for the observed spreading of RAS activation along dendrites and to neighboring dendritic spines. However, these data [77] might call for an alternative explanation. Diffusion distance is proportional to the square root of time, and diffusion is a notoriously slow mechanism for propagating signals over large distances [78]. A positive feedback loop in the RAS pathway induced by RAS-mediated activation of SOS (see Section 3.1.2) can endow excitability and/or bistability features to the spatial propagation along dendrites [79]. The wave-like propagation of RAS activation can be expected to transmit RAS signals more effectively than diffusion [80].
Numerous other studies providing real-time data have not yet received attention from modelers. Verissimo et al. [81] applied real-time confocal imaging to scrutinize differences in response to afatinib (an EGFR/HER2 inhibitor) and selumetinib (a MEK1/MEK2 inhibitor) treatment between patient-derived colorectal cancer (CRC) organoids with or without mutations in KRAS. KRAS mutant organoids entered a state of cell-cycle arrest upon treatment, whereas the CRC organoids with wildtype KRAS exhibited cell death. A similar FRET sensor was applied to study ERK activation and localization dynamics [82,83] and different cell-fate decisions resulting from distinct dynamics, as well as to quantify cell-to-cell variability in ERK activation within a population [84]. Improved FRET-based ERK activity sensors with larger dynamic range have been described recently [85], as was a method using single fluorescent-proteins to measure kinase activity in live single cells [86]. Time-course data generated from these and related technologies will be useful for developing accurate models of decision-making networks.
Related to recording observations in real-time is the ability to control cellular networks and measure the response to varying input patterns. Toettcher et al. [87] designed a system that allows for light-activated RAS signaling based on the plant Phy-PIF system. The Phy-PIF module is recruited to the membrane when red light is detected [88]. Because the SOS catalytic domain is fused to PIF, PIF-SOS localizes to the membrane and activates RAS when cells are exposed to a specific wavelength of light. Studies using this technique helped elucidate how the RAS signaling network can modulate a range of cellular behaviors, because differing sets of downstream factors are activated in response to changes in the duration of RAS activation [87]. This intriguing experimental result has not yet been recapitulated through mechanistic modeling.
2.5 Molecularly targeted drugs
Drugs that target critical signaling enzymes, thereby inhibiting specific pathways driving disease manifestation, might improve treatment efficacy and reduce negative side effects. A number of these agents, deemed “molecularly targeted” drugs, are now applied to treat various cancers. The tyrosine kinase inhibitor imatinib, FDA-approved in 2001, was a revolutionary molecularly targeted drug for treatment of chronic myeloid leukemia (CML) [89,90]. Imatinib blocks ATP binding to the kinases ABL, BCR-ABL, PDGFRA, and c-KIT, thereby inhibiting activation and preventing signaling to the downstream pathways controlling leukemogenesis. The success of imatinib paved the way for additional drugs targeting products of mutated cell signaling genes, including those involved in RAS signaling (Fig. 2). Vemurafenib, FDA-approved in 2011, is applied to treat BRAF V600E metastatic melanoma [91]. Approximately half of subcutaneous and cutaneous melanomas have a mutation in BRAF [92,93]. Sorafenib and dabrafenib are other examples of FDA-approved RAF inhibitors. Gefitinib, erlotinib, and afatinib are tyrosine kinase inhibitors targeting EGFR, approved for use in treating advanced non-small cell lung cancer [94-96]. Vandetanib and lapatanib also target EGFR [97], with lapatanib additionally capable of inhibiting ERK and AKT [98,99]. Trametinib inhibits MEK, and was approved for treating BRAF V600E melanoma when used in combination with dabrafenib [100]. Selumetinib is another MEK inhibitor for treatment of differentiated thyroid cancer [101]. Trastuzumab targets the epidermal growth factor receptor HER2 (aka ErbB2) and is approved for treating breast, gastric, and gastro-oesophageal junction cancers [102,103]. Other agents are approved for targeting related pathways, such as the macrolides sirolimus and everolimus, which inhibit mTOR [104].
Recently, methods to inhibit RAS, once thought of as an “undruggable” target, are showing promise [105]. Molecules have been developed that prevent oncogenic RAS signaling by selectively binding KRAS G12C [106,107], interfering with KRAS plasma membrane localization by inhibiting PDEδ [108,109], disrupting palmitoylation-driven localization of HRAS by binding APT1 [110], or disrupting KRAS4B membrane organization by preventing calmodulin binding to the C-terminal hypervariable region [111].
Clinical and experimental observations show that resistance to molecularly targeted cancer drugs occurs frequently [112,113]. One means of resistance is the generation of secondary mutations affecting the drug target site, as is observed in EGFR following gefitinib or erlotinib treatment [114]. Resistance can also emerge through re-activation of inhibited pathways, either bypassing the targeted enzyme or through activation of parallel, alternate pathways. For instance, resistance to BRAF inhibitors can be conveyed by re-activation of ERK or activation of the parallel PI3K/AKT/mTOR pathway [93,115]. ERK re-activation can be achieved via drug-induced allosteric activation of BRAF/CRAF heterodimers, or by the kinase COT, which activates MEK1/2 [116-118]. Mechanistic models of cell signaling can be used to further characterize mechanisms of resistance to molecularly targeted therapeutics. For instance, Iadevaia et al. [119] developed an ODE model of the insulin-like growth factor 1 receptor (IGF1R) signaling network using mass action kinetics parameterized with particle swarm optimization against experimental time-course data. This model was able to predict resistance pathways in a breast cancer cell line after treatment with inhibitors targeting molecules in the network. A model of RAS signaling revealed a basis for differences in EGFR inhibitor efficacy in cells encoding different oncogenic RAS mutants [43].
3. Complex features of RAS signaling continue to necessitate application of mathematical modeling
3.1 Feedback in RAS-associated signaling pathways
Serum starved cells expressing wildtype EGFR and RAS usually respond to EGF stimulation by transient RAS activation lasting 10 30 minutes [120]. Temporal activation patterns of RAS are influenced not only by delays in activation of RAS-GEFs and RAS-GAPs [16], but also by positive and negative feedbacks from RAS downstream effectors to GEFs and GAPs of RAS [121,122]. In the following sections, we discuss feedback loops impacting RAS signaling.
3.1.1 Negative Feedback
Within the ERK cascade, negative feedback loops from ERK to SOS, RAF, and MEK have been discovered and characterized (Fig. 2). A unifying mechanism for these feedback loops is phosphorylation: ERK phosphorylates MEK1 as well as CRAF, BRAF and SOS. MEK1 possesses a threonine residue at position 292 in the protein kinase domain that is phosphorylated by ERK; this residue is not present in MEK2 [123,124]. SOS1 is phosphorylated by ERK at four serine residues (S1132, S1167, S1178, and S1193), yet SOS2 is only phosphorylated at one location, which may indicate that negative feedback from ERK impairs the activities of these isoforms to varying degrees [125]. Phosphorylated SOS is recruited to the membrane less efficiently due to impaired interaction with GRB2 [126,127]. CRAF kinase activity is attenuated upon phosphorylation at six sites, five of which are phosphorylated by ERK (S29, S296, S301, and S642) [128]. ERK phosphorylates BRAF on four sites (S151, T401, S750, and T753), which inhibits BRAF/CRAF dimerization and BRAF binding to RAS-GTP [129].
Negative regulators of RAS signaling can enable the preferential activation of downstream effector pathways. For instance, consider the Sprouty family of proteins, which are versatile regulators of ERK signaling [130-132]. Upon growth factor stimulation, Sprouty localizes to the plasma membrane and suppresses signaling from RAS to ERK, promoting instead RAS signaling via PI3K [130,131]. Decreased Sprouty expression, and therefore decreased ability to reduce ERK activation, has been linked to several cancer types, including melanoma, breast, liver, lung, metastatic prostate, and other cancers [132]. Interestingly, Sprouty’s mechanism of action does not appear to be consistent across isoforms or across cell types; Sprouty proteins have been shown to interact with many partners in the ERK cascade, including GRB2, GAP1, and BRAF [133-136].
3.1.2 Positive Feedback
Intriguingly, in addition to negative feedback loops, the RAS/ERK pathway also contains positive feedback loops (Fig. 2), including positive feedback from RAS to its activator SOS [37]. Both structural and kinetic data suggest that RAS is able to bind to SOS at two distinct sites, such that RAS may act not only as a substrate, but also as an allosteric activator of SOS (Fig. 3) [37,137]. When RAS is bound to the SOS allosteric site, the dissociation rate of GDP from RAS bound to the SOS GEF domain is increased by up to 75-fold [138]. The enhancement of SOS GEF activity by binding the SOS allosteric site is more pronounced for RAS-GTP than for RAS-GDP [138,139]. In addition to allosteric activation, binding of RAS contributes to recruitment of SOS to the plasma membrane, which is known to increase SOS GEF activity by co-localizing SOS and RAS molecules [55, 140].
Fig. 3.
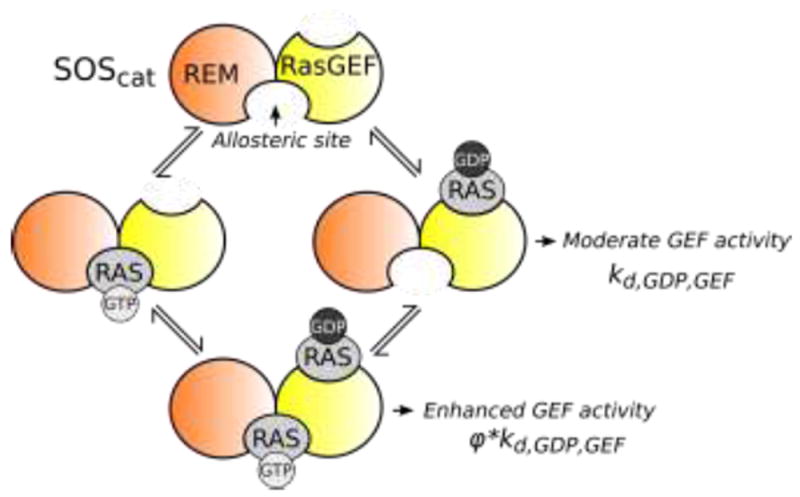
Positive feedback from RAS to SOS is achieved via binding of RAS to the SOS allosteric site that bridges the RAS exchanger motif (REM) and RAS-GEF domains, which together are responsible for SOS GEF activity. When the allosteric site is empty, SOS has moderate GEF activity, facilitating release of GDP from RAS-GDP bound at the RAS-GEF site with a rate constant kd,GDP,GEF. When RAS is bound to the REM domain, GEF activity increases to a rate characterized by the rate constant φ*kd,GDP,GEF , where φ > 1.
Positive feedback from active RAS to its GEF SOS may result in bistability in RAS-GTP regulation and hysteresis in RAS-GTP levels in response to stimuli. Das et al. [141] developed a model of bistable RAS behavior suggesting that positive RAS-SOS feedback might account for a digital all-or-nothing response of RAS-GTP to external stimuli. Following transfection of Jurkat cells with the SOS catalytic domain, which has higher catalytic activity than the full-length SOS, the authors observed bimodal expression of the cell-surface marker CD69 (e.g., CD69 expression was correlated with ERK activity). Other indirect evidence of bistability came from observations that populations of HEK293 cells split into subpopulations with either high or low levels of active ERK (ppERK) after transfection with oncogenic KRAS G12V [142] (although the emergence of a bimodal cell population response can be alternatively explained, see [143-145]).
Whereas ERK-mediated phosphorylation of SOS and CRAF/BRAF generates negative feedback, ERK- phosphorylation of the RAF kinase inhibitor protein (RKIP) creates a positive feedback loop, because it results in enhanced CRAF activity [146]. This positive feedback loop was not considered in a recent modeling effort focused on feedback in the ERK cascade, although the negative feedback from ERK to RAF was included [147]. Historically, ERK pathway models have not analyzed cases where both positive and negative feedback loops have been identified between two signaling proteins. This situation may occur for several enzyme pairs in the ERK pathway where so-called hidden negative feedback is brought about by the sequestration of an upstream kinase by a downstream kinase within a protein-protein complex [148]. Such feedback combinations can generate intricate dynamic behaviors, calling for use of computational models to aid in reasoning about these behaviors [147,149].
Transcriptional feedback loops in the RAS network include not only negative regulators, such as Sprouty [130,133,134], but also positive transcriptional regulators. In response to growth factor stimulation, the scaffold protein, KSR1, localizes to the plasma membrane and facilitates activation of the RAS-to-ERK pathway [150,151]. In quiescent cells, KSR1 is phosphorylated on S392 and localized in the cytoplasm. The RhoGEF GEF-H1 is necessary for recruitment of the phosphatase PP2A to KSR1 and dephosphorylation of KSR1 on S392 [151]. Because ERK signaling markedly enhances the GEF-H1 level, its expression creates a positive transcriptional feedback loop in the ERK pathway.
3.1.3 Interlocked negative and positive feedback loops
Mathematical models have not yet considered all negative and possible feedbacks involved in RAS signaling [65,152], and it remains an open challenge to discern the feedbacks that are most significant for clinical applications. However, models have begun to address the implications of interlocking sets of feedback and feedforward connections between signaling pathways emanating from RAS. For instance, RAS regulation works in concert with feedback connections from RAS effector pathways such as ERK. When both negative feedback from ERK to SOS as well as positive feedback from RAS to SOS are considered, models predict the occurrence of relaxation oscillations consistent with experimental observations [147]. Negative feedback between ERK and RAF and between ERK and MEK further determines the shape of the oscillating waveforms. Taking into account spatial heterogeneity, one might also consider the possibility of the formation of traveling phosphorylation waves [153,79].
Interlocked feedback loops originating from RAS downstream effector pathways influence RAS activation patterns. For instance, a mathematical model of the EGFR signaling network and validating experiments show that the RAS/PI3K/AKT and ERK signaling pathways interact through the scaffold protein GAB1 [154]. A positive feedback from PI3K to GAB1 strongly activates RAS in response to weak EGF stimuli in HEK293 cells and also accounts for the increase in RAS-to-ERK signaling by insulin in these cells [121]. Experiments on HeLa cells have shown that transient RAS activation in response to EGF stimulation is substantially prolonged if ERK is inhibited [122]. This effect is mainly explained by the ability of ERK or its downstream effectors to inactivate SOS (RAS-GEF) and activate NF1 (RAS-GAP).
3.2 Trafficking and co-localization
The amino acid sequences of the RAS isoforms KRAS4A, KRAS4B, HRAS, and NRAS are 80% the same, but these isoforms possess distinct, non-redundant functionalities [22]. All RAS isoforms are translated in the cytosol and then transported to membranes after post-translational modifications in the C-terminal hypervariable region (HVR) that differ for each isoform. Because all differences in sequence identity are found in the HVR rather than in the regions involved in binding GTP/GDP or effectors, RAS isoform localization may underlie isoform-specific activity [155]. RAS activation can occur at the plasma membrane (PM) and endomembranes of the endoplasmic reticulum (ER)/Golgi complex, mitochondria, and endosomes [155], although the localization for each isoform may differ between cell types. In neurons it has been shown that KRAS, but not HRAS, can reversibly transfer between the PM and intracellular membranes through an interaction with Ca2+ and calmodulin [156]. Localization can be specifically controlled to occur at certain regions of larger cellular membranes. For instance, NRAS was observed at both caveolin-positive and caveolin-negative regions of the PM, but KRAS was preferentially located at caveolin-positive domains [157].
Localization of RAS has implications for the associated signaling pathways by impacting which effectors are activated. One study found that HRAS localized to the ER or to lipid rafts in the PM activated ERK and AKT more readily than HRAS localized at the bulk membrane or the Golgi apparatus [158]. In contrast, HRAS located at the Golgi apparatus was more effective in activating RAL-GDS than HRAS located at the ER or lipid rafts in the PM. Mathematical models are only beginning to consider localization and trafficking of RAS isoforms. Schmick et al. [159] provided one such model, which used cellular automata to simulate KRAS trafficking. The model reproduced experimental observations, including a decrease in plasma-membrane bound KRAS upon inhibition of PDEδ or upon introduction of a phosphomimetic mutant KRAS, but it did not examine the effects of differential KRAS localization on downstream effector activation. Future models of RAS signaling that include isoform-specific RAS trafficking and localization will provide a more complete picture of RAS influence on cellular phenotype.
3.3 Competition between RAS effectors
Available structures of RAS in complex with GEFs, GAPs and other effectors show that binding interfaces overlap on the RAS proteins [160]. Therefore, despite having multiple direct interaction partners, a single RAS molecule can bind only one effector, GEF, or GAP at a time. Competition between multiple RAS binding partners implies that any change in the abundance or affinity of a single downstream effector not only impacts its binding to RAS, but can also affect the RAS-GTP level and interaction of RAS with other effectors. As RAS effector pathways crosstalk through feedback loops, inhibition of one pathway can activate or inhibit other signaling branches. In addition, oncogenic RAS variants also influence interactions with effectors; KRAS G12V and G12C preferentially activate RAL-GDS, but KRAS G12D preferentially activates RAF and PI3K [35].
Mechanistic models can provide insights into the dynamics of effector competition. Stites et al. [54] presented a rule-based model of EGFR signaling to identify important protein-protein interactions across 11 cell lines. This model simultaneously considered experimentally determined protein copy numbers and protein binding affinities and found potential cell-line specific differences in the rank order of recruited EGFR binding partners. Such an approach could also be applied to evaluate effector binding to each of the RAS isoforms and to make predictions regarding the abundances of different RAS-effector complexes in disparate cellular backgrounds.
4. Conclusions and outlook
Integration of mathematical modeling into the clinical setting has the potential to revolutionize treatments by informing therapeutic strategies in a patient- and tumor-specific manner [161]. While models specifically incorporating RAS-activation have not, to our knowledge, been directly used to inform clinical practice, there are many promising studies that indicate how mathematical modeling is already being applied to generate relevant predictions. For instance, Chmielecki et al. [162] suggested that the dosing of EGFR tyrosine kinase inhibitors (TKIs) for non-small cell lung cancers was not optimized for mutant EGFR. By incorporating data from engineered cell lines into evolutionary models of cancer, the authors were able to predict the frequency of resistant cells in a population treated with varying doses and schedules of TKI as well as the time necessary to reestablish drug sensitivity in treated populations. A dosing schedule found to reduce the emergence of resistance was validated in the human lung adenocarcinoma cell line PC-9 [162], but has not yet been applied in the clinic. Numerous other recent computational attempts simulate therapeutic effects or dosing schedules with the goal of improving efficacy and/or evading resistance [163-166]. Other computational studies examine the potential effects from combination therapies [118,167-169]. Future efforts toward this end will benefit from the availability of the NCI-ALMANAC database, which contains a systematic evaluation of interactions between 104 anticancer drugs across 60 human cancer cell lines [170]. Models have also been used to guide the design or predict the outcome of clinical trials [168,171].
Mathematical models of RAS signaling and the MAPK pathway include many potential targets for therapeutic agents, including drugs targeting upstream RTKs or downstream RAS effectors. Unfortunately, single drug therapies often fail because of molecular interactions that bypass the block, almost inevitably resulting in resistance [172]. Patients with cancers driven by RAS mutations are commonly excluded from treatment with RTK inhibitors, although combination therapy that suppresses both RTKs and RAS downstream effectors, such as RAF or MEK, might be more effective than RAF/MEK therapy alone [81]. However, selecting the best drug combinations for patients with diverse expression and mutation profiles remains an open challenge. The number of intuitively promising drug combinations applicable to treatment of RAS-driven cancer is large, and mathematical, mechanism-based modeling offers an avenue by which to predict effective combinatorial treatments that exhibit synergy, overcome/prevent resistance, or avoid toxicity. Inferring optimal treatment for RAS-driven cancers will be made possible through improvements in understanding of the biology underlying RAS signaling. Accurate predictive models should capture the complexities of the network described here, including the myriad positive and negative feedbacks described, or the possibility of effector competition, or localization specific differences between RAS isoforms, all of which have ramifications for downstream signaling. We foresee an improvement in the clinical relevance of mathematical models of RAS signaling as our comprehension of the mechanisms underlying disease is bolstered by technological advances in quantification and monitoring of biological systems.
Acknowledgments
This work was supported by the Joint Design of Advanced Computing Solutions for Cancer (JDACS4C) program established by the U.S. Department of Energy, by grants from the National Institutes of Health (NIH/NIGMS grant no. GM111510 and NIH/NCI grant no. CA197398), and by the European Union grants SmartNanoTox (Grant no. 686098), NanoCommons (Grant no. 731032) and MSCA-IF-2016 SAMNets grant no. 750688). Los Alamos National Laboratory is operated by the U.S. Department of Energy under contract DE-AC5206NA25396. We thank D. D. Von Hoff for carefully reading the manuscript.
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barbacid M. Ras Genes. Annu Rev Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- 2.Stephen AG, Esposito D, Bagni RG, McCormick F. Dragging Ras back in the ring. Cancer Cell. 2014;25:272–281. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 3.Colicelli J. Human RAS Superfamily Proteins and Related GTPases. Sci STKE. 2004;250:RE13. doi: 10.1126/stke.2502004re13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Novotny CJ, Hamilton GL, Mccormick F, Shokat KM. Farnesyltransferase-Mediated Delivery of a Covalent Inhibitor Overcomes Alternative Prenylation to Mislocalize K-Ras. ACS Chem Biol. 2017;12:1956–1962. doi: 10.1021/acschembio.7b00374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ho AL, Chau NG, Wong DJL, Cabanillas ME, Bauman JR, Bible KC, Brose MS, Calvo E, Boni V, Burrows F, Melvin CL, Scholz CR, Gualberto A. An open-label, phase II study of tipifarnib for the treatment of HRAS mutant solid tumors, including squamous cell carcinomas of the head and neck. J Clin Oncol. 2017;35 [Google Scholar]
- 6.Trahey M, Mccormick F. A Cytoplasmic Protein Stimulates Normal N-ras p21 GTPase, but Does not Affect Oncogenic Mutants. Science. 1987;238:542–545. doi: 10.1126/science.2821624. [DOI] [PubMed] [Google Scholar]
- 7.Pilz RB, Huvar I, Scheele JS, Van den Berghe G, Boss GR. A Decrease in the Intracellular Guanosine Concentration Is Necessary for Granulocytic Differentiation of HL-60 Cells , but Growth Cessation and Differentiation Are Not Associated with a Change in the Activation State of Ras, the Transforming Principle of. Cell Growth Differ. 1997;8:53–59. [PubMed] [Google Scholar]
- 8.Allin C, Ahmadian MR, Wittinghofer A, Gerwert K. Monitoring the GAP catalyzed H-Ras GTPase reaction at atomic resolution in real time. Proc Natl Acad Sci. 2001;98:7754–7759. doi: 10.1073/pnas.131549798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allin C, Gerwert K. Ras Catalyzes GTP Hydrolysis by Shifting Negative Charges from γ-toPhosphate As Revealed by Time-Resolved FTIR Difference Spectroscopy. Biochemistry. 2001;40:3037–3046. doi: 10.1021/bi0017024. [DOI] [PubMed] [Google Scholar]
- 10.Ahmadian MR, Stege P, Scheffzek K, Wittinghofer A. Confirmation of the arginine-finger hypothesis for the GAP-stimulated GTP-hydrolysis reaction of Ras. Nat Struct Mol Biol. 1997;4:686–689. doi: 10.1038/nsb0997-686. [DOI] [PubMed] [Google Scholar]
- 11.Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10:842–857. doi: 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denhardt DT. Signal-transducing protein phosphorylation cascades mediated by Ras/Rho proteins in the mammalian cell: the potential for multiplex signalling. Biochem J. 1996;318:729–747. doi: 10.1042/bj3180729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: Critical Elements in the Control of Small G Proteins. Cell. 2007;129:865–877. doi: 10.1016/j.cell.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 14.McLaughlin SK, Olsen SN, Dake B, De Raedt T, Lim E, Bronson RT, Beroukhim R, Polyak K, Brown M, Kuperwasser C, Cichowski K. The RasGAP Gene, RASAL2 , Is a Tumor and Metastasis Suppressor. Cancer Cell. 2013;24:365–378. doi: 10.1016/j.ccr.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sung H, Kanchi KL, Wang X, Hill KS, Jane L, Lee J, Kim Y, Dees ND, Ding L, Jamie K, Yang S, Sarnaik AA, Sondak VK, Mulé JJ, Wilson RK, Weber JS, Kim M. Inactivation of RASA1 promotes melanoma tumorigenesis via R-Ras activation. Oncotarget. 2016;7:23885. doi: 10.18632/oncotarget.8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Markevich NI, Moehren G, Demin OV, Kiyatkin A, Hoek JB, Kholodenko BN. Signal processing at the Ras circuit: what shapes Ras activation patterns? Syst Biol (Stevenage) 2004;1:104–113. doi: 10.1049/sb:20045003. [DOI] [PubMed] [Google Scholar]
- 17.Neel NF, Martin TD, Stratford JK, Zand TP, Reiner DJ, Der CJ. The RalGEF-Ral Effector Signaling Network: The Road Less Traveled for Anti-Ras Drug Discovery. Genes Cancer. 2011;2:275–287. doi: 10.1177/1947601911407329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shankar S, Pitchiaya S, Malik R, Kothari V, Hosono Y, Yocum AK, Gundlapalli H, White Y, Firestone A, Cao X, Dhanasekaran SM, Stuckey JA, Bollag G, Shannon K, Walter NG, Kumar-Sinha C, Chinnaiyan AM. KRAS Engages AGO2 to Enhance Cellular Transformation. Cell Rep. 2016;14:1448–1461. doi: 10.1016/j.celrep.2016.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Joneson T, Bar-Sagi D. Ras effectors and their role in mitogenesis and oncogenesis. J Mod Med. 1997;75:587–593. doi: 10.1007/s001090050143. [DOI] [PubMed] [Google Scholar]
- 20.Muratcioglu S, Chavan TS, Freed BC, Jang H, Khavrutskii L, Natasha Freed R, Dyba MA, Stefanisko K, Tarasov SG, Gursoy A, Keskin O, Tarasova NI, Gaponenko V, Nussinov R. GTP-Dependent K-Ras Dimerization. Structure. 2015;23:1325–1335. doi: 10.1016/j.str.2015.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prior IA, Lewis PD, Mattos C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012;72:2457–2468. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castellano E, Santos E. Functional Specificity of Ras Isoforms: So Similar but So Different. Genes Cancer. 2011;2:216–231. doi: 10.1177/1947601911408081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vu HL, Aplin AE. Targeting mutant NRAS signaling pathways in melanoma. Pharmacol Res. 2016;107:111–116. doi: 10.1016/j.phrs.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.T. Cancer Genome Atlas Research Network. Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stites EC, Ravichandran KS. Mechanistic modeling to investigate signaling by oncogenic Ras mutants, Wiley Interdiscip. Rev Syst Biol Med. 2012;4:117–127. doi: 10.1002/wsbm.156. [DOI] [PubMed] [Google Scholar]
- 26.Brightman FA, Fell DA. Differential feedback regulation of the MAPK cascade underlies the quantitative differences in EGF and NGF signalling in PC12 cells. FEBS Lett. 2000;482:169–174. doi: 10.1016/s0014-5793(00)02037-8. [DOI] [PubMed] [Google Scholar]
- 27.Schoeberl B, Eichler-Jonsson C, Gilles ED, Müller G. Computational modeling of the dynamics of the MAP kinase cascade activated by surface and internalized EGF receptors. Nat Biotechnol. 2002;20:370–375. doi: 10.1038/nbt0402-370. [DOI] [PubMed] [Google Scholar]
- 28.Kholodenko BN, Demin OV, Moehren G, Hoek JB. Quantification of Short Term Signaling by the Epidermal Growth Factor Receptor. J Biol Chem. 1999;274:30169–30181. doi: 10.1074/jbc.274.42.30169. [DOI] [PubMed] [Google Scholar]
- 29.Orton RJ, Sturm OE, Vyshemirsky V, Calder M, Gilbert DR, Kolch W. Computational modelling of the receptor-tyrosine-kinase-activated MAPK pathway. Biochem J. 2005;392:249–261. doi: 10.1042/BJ20050908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stites EC, Trampont PC, Ma Z, Ravichandran KS. Network Analysis of Oncogenic Ras Activation in Cancer. Science. 2007;318:463–468. doi: 10.1126/science.1144642. [DOI] [PubMed] [Google Scholar]
- 31.Orton RJ, Adriaens ME, Gormand A, Sturm OE, Kolch W, Gilbert DR. Computational modelling of cancerous mutations in the EGFR/ERK signalling pathway. BMC Syst Biol. 2009;3:100. doi: 10.1186/1752-0509-3-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stites EC, Trampont PC, Haney LB, Walk SF, Ravichandran KS. Cooperation between Noncanonical Ras Network Mutations. Cell Rep. 2015;10:307–316. doi: 10.1016/j.celrep.2014.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kholodenko BN, Hancock JF, Kolch W. Signalling ballet in space and time. Nat Rev Mol Cell Biol. 2010;11:414–426. doi: 10.1038/nrm2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buhrman G, Holzapfel G, Fetics S, Mattos C. Allosteric modulation of Ras positions Q61 for a direct role in catalysis. Proc Natl Acad Sci. 2010;107:4931–4936. doi: 10.1073/pnas.0912226107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ihle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, Blumenschein GR, Tsao A, Liu S, Larsen JE, Wang J, Diao L, Coombes KR, Chen L, Zhang S, Abdelmelek MF, Tang X, Papadimitrakopoulou V, Minna JD, Lippman SM, Hong WK, Herbst RS, Wistuba II, Heymach JV, Powis G. Effect of KRAS Oncogene Substitutions on Protein Behavior: Implications for Signaling and Clinical Outcome. J Natl Cancer Inst. 2012;104:228–239. doi: 10.1093/jnci/djr523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.John J, Sohmen R, Feuerstein J, Linke R, Wittinghofer A, Goody RS. Kinetics of Interaction of Nucleotides with Nucleotide-Free H-ras p21. Biochemistry. 1990;29:6058–6065. doi: 10.1021/bi00477a025. [DOI] [PubMed] [Google Scholar]
- 37.Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, Pirruccello M, Bar-Sagi D, Kuriyan J. Structural evidence for feedback activation by Ras·GTP of the Ras-specific nucleotide exchange factor SOS. Cell. 2003;112:685–695. doi: 10.1016/s0092-8674(03)00149-1. [DOI] [PubMed] [Google Scholar]
- 38.Gremer L, Merbitz-Zahradnik T, Dvorsky R, Cirstea IC, Kratz CP, Zenker M, Wittinghofer A, Ahmadian MR. Germline KRAS mutations cause aberrant biochemical and physical properties leading to developmental disorders. Hum Mutat. 2010;32:33–43. doi: 10.1002/humu.21377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gremer L, De Luca A, Merbitz-Zahradnik T, Dallapiccola B, Morlot S, Tartaglia M, Kutsche K, Ahmadian MR, Rosenberger G. Duplication of Glu 37 in the switch I region of HRAS impairs effector/GAP binding and underlies Costello syndrome by promoting enhanced growth factor-dependent MAPK and AKT activation. Hum Mol Genet. 2010;19:790–802. doi: 10.1093/hmg/ddp548. [DOI] [PubMed] [Google Scholar]
- 40.Cirstea IC, Gremer L, Dvorsky R, Zhang S, Piekorz RP, Zenker M, Ahmadian MR. Diverging gain-of-function mechanisms of two novel KRAS mutations associated with Noonan and cardio-facio-cutaneous syndromes. Hum Mol Genet. 2013;22:262–270. doi: 10.1093/hmg/dds426. [DOI] [PubMed] [Google Scholar]
- 41.Denayer E, Parret A, Chmara M, Schubbert S, Vogels A, Devriendt K, Frijns JP, Rybin V, de Ravel TJ, Shannon K, Cools J, Scheffzek K, Legius E. Mutation Analysis in Costello Syndrome: Functional and Structural Characterization of the HRAS p.Lys117Arg Mutation. Hum Mutat. 2008;29:232–239. doi: 10.1002/humu.20616. [DOI] [PubMed] [Google Scholar]
- 42.Kenney C, Stites EC. A computational panel of pathological RAS mutants with implications for personalized medicine and genetic medicine. bioRxiv. 2017 [Google Scholar]
- 43.Stites EC. Differences in sensitivity to EGFR inhibitors could be explained by described biochemical differences between oncogenic Ras mutants. bioRxiv. 2014 [Google Scholar]
- 44.Kiel C, Serrano L. Structure-energy-based predictions and network modelling of RASopathy and cancer missense mutations. Mol Syst Biol. 2014;10:727. doi: 10.1002/msb.20145092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baldwin AJ, Kay LE. NMR spectroscopy brings invisible protein states into focus. Nat Chem Biol. 2009;5:808–814. doi: 10.1038/nchembio.238. [DOI] [PubMed] [Google Scholar]
- 46.Mittermaier AK, Kay LE. Observing biological dynamics at atomic resolution using NMR. Trends Biochem Sci. 2009:601–611. doi: 10.1016/j.tibs.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 47.Theillet FX, Smet-Nocca C, Liokatis S, Thongwichian R, Kosten J, Yoon MK, Kriwacki RW, Landrieu I, Lippens G, Selenko P. Cell signaling, post-translational protein modifications and NMR spectroscopy. J Biomol NMR. 2012;54:217–236. doi: 10.1007/s10858-012-9674-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith MJ, Neel BG, Ikura M. NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc Natl Acad Sci. 2013;110 doi: 10.1073/pnas.1218173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hause RJ, Jr, Leung KK, Barkinge JL, Ciaccio MF, Chuu C, Jones RB. Comprehensive Binary Interaction Mapping of SH2 Domains via Fluorescence Polarization Reveals Novel Functional Diversification of ErbB Receptors. PLoS One. 2012;7:e44471. doi: 10.1371/journal.pone.0044471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaushansky A, Allen JE, Gordus A, Stiffler MA, Karp ES, Chang BH, MacBeath G. Quantifying protein-protein interactions in high throughput using protein domain microarrays. Nat Protoc. 2010;5:773–790. doi: 10.1038/nprot.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vincentelli R, Luck K, Poirson J, Polanowska J, Adbat J, Blémont M, Turchetto J, Iv F, Ricquier K, Straub M-L, Forster A, Cassonnet P, Borg J-P, Jacob Y, Masson M, Nominé Y, Reboul J, Wolff N, Charbonnier S, Travé G. Quantifying domain-ligand affinities and specificities by high-throughput holdup assay. Nat Methods. 2015;12:787–793. doi: 10.1038/nmeth.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adams RM, Mora T, Walczak AM, Kinney JB. Measuring the sequence-affinity landscape of antibodies with massively parallel titration curves. Elife. 2016:e23156. doi: 10.7554/eLife.23156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kowalsky CA, Whitehead TA. Determination of binding affinity upon mutation for type I dockerin-cohesion complexes from Clostridium thermocellum and Clostridium cellulolyticum using deep sequencing. Proteins Struct Funct Bioinforma. 2016;84:1914–1928. doi: 10.1002/prot.25175. [DOI] [PubMed] [Google Scholar]
- 54.Stites EC, Aziz M, Creamer MS, Von Hoff DD, Posner RG, Hlavacek WS. Use of Mechanistic Models to Integrate and Analyze Multiple Proteomic Datasets. Biophys J. 2015;108:1819–1829. doi: 10.1016/j.bpj.2015.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iversen L, Tu H, Lin W, Christensen SM, Abel SM, Iwig J, Wu H, Gureasko J, Rhodes C, Petit RS, Hansen SD, Thill P, Yu C, Stamou D. Ras activation by SOS: Allosteric regulation by altered fluctuation dynamics. Science. 2014;345:50–54. doi: 10.1126/science.1250373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mann M, Kulak NA, Nagaraj N, Cox J. The Coming Age of Complete, Accurate, and Ubiquitous Proteomes. Mol Cell. 2013;49:583–590. doi: 10.1016/j.molcel.2013.01.029. [DOI] [PubMed] [Google Scholar]
- 57.Geiger T, Wehner A, Schaab C, Cox J, Mann M. Comparative Proteomic Analysis of Eleven Common Cell Lines Reveals Ubiquitous but Varying Expression of Most Proteins. Mol Cell Proteomics. 2012;11 doi: 10.1074/mcp.M111.014050. M111.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Paabo S, Mann M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol. 2011;7:548. doi: 10.1038/msb.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 60.Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods. 2014;11:319–324. doi: 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- 61.Wiśniewski JR, Hein MY, Cox J, Mann M. A “Proteomic Ruler” for Protein Copy Number and Concentration Estimation without Spike-in Standards. Mol Cell Proteomics. 2014;13:3497–3506. doi: 10.1074/mcp.M113.037309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537:347–355. doi: 10.1038/nature19949. [DOI] [PubMed] [Google Scholar]
- 63.Kiel C, Ebhardt HA, Burnier J, Portugal C, Sabido E, Zimmermann T, Aebersold R, Serrano L. Quantification of ErbB network proteins in three cell types using complementary approaches identifies cell-general and cell-type-specific signaling proteins. J Proteome Res. 2014;13:300–313. doi: 10.1021/pr400878x. [DOI] [PubMed] [Google Scholar]
- 64.Shi T, Fillmore TL, Sun X, Zhao R, Schepmoes AA, Hossain M, Xie F, Wu S, Kim J-S, Jones N, Moore RJ, Pasa-Tolic L, Kagan J, Rodland KD, Liu T, Tang K, Camp DG, II, Smith RD, Qian W-J. Antibody-free, targeted mass-spectrometric approach for quantification of proteins at low picogram per milliliter levels in human plasma/serum. Proc Natl Acad Sci. 2012;109:15395–15400. doi: 10.1073/pnas.1204366109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shi T, Niepel M, Mcdermott JE, Gao Y, Nicora CD, Chrisler WB, Markillie LM, Petyuk VA, Smith RD, Rodland KD, Sorger PK, Qian WJ, Wiley HS. Conservation of protein abundance patterns reveals the regulatory architecture of the EGFR-MAPK pathway. Sci Signal. 2016;9 doi: 10.1126/scisignal.aaf0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sudhir PR, Hsu CL, Wang MJ, Wang YT, Chen YJ, Sung TY, Hsu WL, Yang UC, Chen JY. Phosphoproteomics identifies oncogenic Ras signaling targets and their involvement in lung adenocarcinomas. PLoS One. 2011;6 doi: 10.1371/journal.pone.0020199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gnad F, Young A, Zhou W, Lyle K, Ong CC, Stokes MP, Silva JC, Belvin M, Friedman LS, Koeppen H, Minden A, Hoeflich KP. Systems-wide Analysis of K-Ras, Cdc42, and PAK4 Signaling by Quantitative Phosphoproteomics. Mol Cell Proteomics. 2013;12:2070–2080. doi: 10.1074/mcp.M112.027052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moritz A, Li Y, Guo A, Villen J, Wang Y, MacNeill J, Kornhauser J, Sprott K, Zhou J, Possemato A, Ren JM, Hornbeck P, Cantley LC, Gygi SP, Rush J, Comb MJ. Akt-RSK-S6 Kinase Signaling Networks Activated by Oncogenic Receptor Tyrosine Kinases. Sci Signal. 2010;3:ra64–ra64. doi: 10.1126/scisignal.2000998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science. 2014;343:84–88. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic Screens in Human Cells Using the CRISPR-Cas9 System. Science. 2014;343:80–85. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cong L, Ann Ran F, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013;339:819–824. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-Guided Human Genome Engineering via Cas9. Science. 2013;339:823–827. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, Nureki O, Zhang F. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang T, Yu H, Hughes NW, Liu B, Kendiril A, Klein K, Chen WW, Lander ES, Sabatini DM. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell. 2017;168:890–903. doi: 10.1016/j.cell.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rodriguez-Viciana P, Oses-prieto J, Burlingame A, Fried M, McCormick F. A Phosphatase Holoenzyme Comprised of Shoc2/Sur8 and the Catalytic Subunit of PP1 Functions as an M-Ras Effector to Modulate Raf Activity. Mol Cell. 2006;22:217–230. doi: 10.1016/j.molcel.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 76.Young LC, Hartig N, Munoz-Alegre M, Oses-Prieto JA, Durdu S, Bender S, Vijayakumar V, Rudan MV, Gewinner C, Henderson S, Jathoul AP, Ghatrora R, Lythgoe MF, Burlingame AL, Rodriguez-Viciana P. An MRAS, SHOC2, and SCRIB Complex Coordinates ERK Pathway Activation with Polarity and Tumorigenic. Growth, Mol Cell. 2013;52:679–692. doi: 10.1016/j.molcel.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 77.Harvey CD, Yasuda R, Zhong H, Svoboda K. The Spread of Ras Activity Triggered by Activation of a Single Dendritic Spine. Science. 2008;321:136–141. doi: 10.1126/science.1159675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kholodenko BN. Spatially distributed cell signalling. FEBS Lett. 2009;583:4006–4012. doi: 10.1016/j.febslet.2009.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tsyganov MA, Kolch W, Kholodenko BN. The topology design principles that determine the spatiotemporal dynamics of G-protein cascades. Mol Biosyst. 2012;8:730–743. doi: 10.1039/c2mb05375f. [DOI] [PubMed] [Google Scholar]
- 80.Muñoz-García J, Kholodenko BN. Signalling over a distance: gradient patterns and phosphorylation waves within single cells. Biochem Soc Trans. 2010;38 doi: 10.1042/BST0381235. [DOI] [PubMed] [Google Scholar]
- 81.Verissimo CS, Overmeer RM, Ponsioen B, Drost J, Mertens S, Verlaan-Klink I, van Gerwen B, Van der Ven M, Van der Wetering M, Egan DA, Bernards R, Clevers H, Bos JL, Snippert HJ. Targeting mutant RAS in patient-derived colorectal cancer organoids by combinatorial drug screening. Elife. 2016:e18489. doi: 10.7554/eLife.18489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Harvey CD, Ehrhardt AG, Cellurale C, Zhong H, Yasuda R, Davis RJ, Svoboda K. A genetically encoded fluorescent sensor of ERK activity. Proc Natl Acad Sci. 2008;105:19264–19269. doi: 10.1073/pnas.0804598105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Albeck JG, Mills GB, Brugge JS. Frequency-Modulated Pulses of ERK Activity Transmit Quantitative Proliferation Signals. Mol Cell. 2013;49:249–261. doi: 10.1016/j.molcel.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ryu H, Chung M, Dobrzynski M, Fey D, Blum Y, Lee SS, Peter M, Kholodenko BN, Jeon NL, Pertz O. Frequency modulation of ERK activation dynamics rewires cell fate. Mol Syst Biol. 2015;11 doi: 10.15252/msb.20156458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vandame P, Spriet C, Riquet F, Trinel D, Cailliau-maggio K. Optimization of ERK Activity Biosensors for both Ratiometric and Lifetime FRET Measurements. Sensors. 2014;14:1140–1154. doi: 10.3390/s140101140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Regot S, Hughey JJ, Bajar BT, Carrasco S, Covert MW. High-Sensitivity Measurements of Multiple Kinase Activities in Live Single Cells. Cell. 2014;157:1724–1734. doi: 10.1016/j.cell.2014.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Toettcher JE, Weiner OD, Lim WA. Using Optogenetics to Interrogate the Dynamic Control of Signal Transmission by the Ras/Erk Module. Cell. 2013;155:1422–1434. doi: 10.1016/j.cell.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Levskaya A, Weiner OD, Lim WA, Voigt CA. Spatiotemporal Control of Cell Signalling Using A Light- Switchable Protein Interaction. Nature. 2009;461:997–1001. doi: 10.1038/nature08446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 90.Deininger MWN, Druker BJ. Specific Targeted Therapy of Chronic Myelogenous Leukemia with Imatinib. Pharmacol Rev. 2003;55:401–423. doi: 10.1124/pr.55.3.4. [DOI] [PubMed] [Google Scholar]
- 91.Flaherty KT, Puzanov I, Kim KB, Ribas A, MacArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. Inhibition of Mutated, Activated BRAF in Metastatic Melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 93.Swaika A, Crozier JA, Joseph RW. Vemurafenib: an evidence-based review of its clinical utility in the treatment of metastatic melanoma. Drug Des Devel Ther. 2014;4:775–787. doi: 10.2147/DDDT.S31143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cohen MH, Williams GA, Sridhara R, Chen G, Pazdur R. FDA Drug Approval Summary: Gefitinib (ZD1839) (Iressa) Tablets. Oncologist. 2003;8:303–306. doi: 10.1634/theoncologist.8-4-303. [DOI] [PubMed] [Google Scholar]
- 95.Khozin S, Blumenthal GM, Jiang X, He K, Boyd K, Murgo A, Justice R, Keegan P, Pazdur R. U.S. Food and Drug Administration Approval Summary: Erlotinib for the First-Line Treatment of Metastatic Non-Small Cell Lung Cancer With Epidermal Growth Factor Receptor Exon 19 Deletions or Exon 21 (L858R) Substitution Mutations. Oncologist. 2014;21:774–779. doi: 10.1634/theoncologist.2014-0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Keating GM. Afatinib: a review of its use in the treatment of advanced non-small cell lung cancer. Drugs. 2014;74:207–221. doi: 10.1007/s40265-013-0170-8. [DOI] [PubMed] [Google Scholar]
- 97.Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci. 2015;36:422–439. doi: 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 98.Stuhlmiller TJ, Miller SM, Zawistowski JS, Nakamura K, Beltran AS, Duncan JS, Angus SP, Collins KAL, Granger DA, Reuther RA, Graves LM, Gomez SM, Kuan P-F, Parker JS, Chen X, Sciaky N, Carey LA, Earp HS, Jin J, Johnson GL. Inhibition of Lapatinib-Induced Kinome Reprogramming in ERBB2-Positive Breast Cancer by Targeting BET Family Bromodomains. Cell Rep. 2015;11:390–404. doi: 10.1016/j.celrep.2015.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu C-Y, Hu M-H, Hsu C-J, Huang C-T, Wang D-S, Tsai W-C, Chen Y-T, Lee C-H, Chu P-Y, Hsu C-C, Chen M-H, Shiau C-W, Tseng L-M, Chen K-F. Lapatinib inhibits CIP2A/PP2A/ p-Akt signaling and induces apoptosis in triple negative breast cancer cells. Oncogene. 2016;7:9135–9149. doi: 10.18632/oncotarget.7035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, Chiarion-Sileni V, Drucis K, Krajsova I, Hauschild A, Lorigan P, Wolter P, Long GV, Flaherty K, Nathan P, Ribas A, Martin AM, Sun P, Crist W, Legos J, Rubin SD, Little SM, Schadendorf D. Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N Engl J Med. 2015;372 doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 101.AstraZeneca. Selumetinib granted Orphan Drug Designation in the US for adjuvant treatment of differentiated thyroid cancer. AstraZeneca. 2016 [Google Scholar]
- 102.Smith I, Procter M, Gelber RD, Guillaume S, Feyereislova A, Dowsett M, Goldhirsch A, Untch M, Mariani G, Baselga J, Kaufmann M, Cameron D, Bell R, Bergh J, Coleman R, Wardley A, Harbeck N, Lopez RI, Mallmann P, Gelmon K, Wilcken N, Wist E, Rovira PS, Piccart-Gebhart MJ. 2-year follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer: a randomised controlled trial. Lancet. 2007;369:29–36. doi: 10.1016/S0140-6736(07)60028-2. [DOI] [PubMed] [Google Scholar]
- 103.Bang Y, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T, Aprile G, Kulikov E, Hill J, Lehle M, Rüschoff J, Kang YK. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–697. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 104.Wu P, Nielsen TE, Clausen MH. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discov Today. 2016;21:5–10. doi: 10.1016/j.drudis.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 105.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission Possible? Nat Rev Drug Discov. 2014;13:828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Patricelli MP, Janes MR, Li L, Hansen R, Peters U, Kessler LV, Chen Y, Kucharski JM, Feng J, Ely T, Chen JH, Firdaus SJ, Babbar A. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016 doi: 10.1158/2159-8290.CD-15-1105. [DOI] [PubMed] [Google Scholar]
- 108.Papke B, Murarka S, Vogel HA, Mart n-Gago P, Kovacevic M, Truxius DC, Fansa EK, Ismail S, Zimmermann G, Heinelt K, Schultz-Fademrecht C, Al Saabi A, Baumann M, Nussbaumer P, Wittinghofer A, Waldmann H, Bastiaens PIH. Identification of pyrazolopyridazinones as PDEδ inhibitors. Nat Commun. 2016 doi: 10.1038/ncomms11360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, Hahn SA, Triola G, Wittinghofer A, Bastiaens PIH, Waldmann H. Small molecule inhibition of the KRAS PDEδ interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638–642. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 110.Dekker FJ, Rocks O, Vartak N, Menninger S, Hedberg C, Balamurugan R, Wetzel S, Renner S, Gerauer M, Schölermann B, Rusch M, Kramer JW, Rauh D, Coates GW, Brunsveld L, Bastiaens PIH, Waldmann H. Small-molecule inhibition of APT1 affects Ras localization and signaling. Nat Chem Biol. 2010;6:449–456. doi: 10.1038/nchembio.362. [DOI] [PubMed] [Google Scholar]
- 111.Najumudeen AK, Jaiswal A, Lectez B, Guzmán C, Siljamäki E, Posada IMD, Lacey E, Aittokallio T, Abankwa D. Cancer stem cell drugs target K-ras signaling in a stemness context. Oncogene. 2016;35:5248–5262. doi: 10.1038/onc.2016.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 113.Garraway LA, Jänne PA. Circumventing Cancer Drug Resistance in the Era of Personalized Medicine. Cancer Discov. 2012;2:214–226. doi: 10.1158/2159-8290.CD-12-0012. [DOI] [PubMed] [Google Scholar]
- 114.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H, et al. Acquired Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib Is Associated with a Second Mutation in the EGFR Kinase Domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Aplin AE, Kaplan FM, Shao Y. Mechanisms of Resistance to RAF Inhibitors in Melanoma. J Invest Dermatol. 2011;131:1817–1820. doi: 10.1038/jid.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Montagut C, Sharma SV, Shioda T, Mcdermott U, Ulman M, Ulkus LE, Dias-santagata D, Stubbs H, Lee DY, Singh A, Drew L, Haber DA, Settleman J. Elevated CRAF as a Potential Mechanism of Acquired Resistance to BRAF Inhibition in Melanoma. Cancer Res. 2008;68:4853–4862. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Johannessen CM, Boehm JS, Kim SY, Thomas SR, Cogdill P, Barretina J, Caponigro G, Hieronymus H, Ryan R, Yang X, Alkan O, Kim S, Harris JL, Christopher J, Flaherty KT, Dummer R, Weber B, Sellers WR, Schlegel R, Wargo J, Hahn WC, Garraway LA. COT/MAP3K8 drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kholodenko BN. Drug Resistance Resulting from Kinase Dimerization Is Rationalized by Thermodynamic Factors Describing Allosteric Inhibitor Effects. Cell Rep. 2015;12:1939–1949. doi: 10.1016/j.celrep.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 119.Iadevaia S, Lu Y, Morales FC, Mills GB, Ram PT. Identification of Optimal Drug Combinations Targeting Cellular Networks: Integrating Phospho-Proteomics and Computational Network Analysis. Cancer Res. 2010;70:6704–6715. doi: 10.1158/0008-5472.CAN-10-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Waters SB, Chen D, Kao AW, Okada S, Holt KH, Pessin JE. Insulin and Epidermal Growth Factor Receptors Regulate Distinct Pools of Grb2-SOS in the Control of Ras Activation. J Biol Chem. 1996;271:18224–18230. doi: 10.1074/jbc.271.30.18224. [DOI] [PubMed] [Google Scholar]
- 121.Borisov N, Aksamitiene E, Kiyatkin A, Legewie S, Berkhout J, Maiwald T, Kaimachnikov NP, Timmer J, Hoek JB, Kholodenko BN. Systems-level interactions between insulin EGF networks amplify mitogenic signaling. Mol Syst Biol. 2009;5:256. doi: 10.1038/msb.2009.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hennig A, Markwart R, Wolff K, Schubert K, Cui Y, Prior IA, Esparza-Franco MA, Ladds G, Rubio I. Feedback activation of neurofibromin terminates growth factor-induced Ras activation. Cell Commun Signal. 2016;14 doi: 10.1186/s12964-016-0128-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Catalanotti F, Reyes G, Jesenberger V, Galabova-Kovacs G, de Matos Simoes R, Carugo O, Baccarini M. A Mek1–Mek2 heterodimer determines the strength and duration of the Erk signal. Nat Struct Mol Biol. 2009;16:294–303. doi: 10.1038/nsmb.1564. [DOI] [PubMed] [Google Scholar]
- 124.Brunet A, Pages G, Pouyssegur J. Growth factor-stimulated MAP kinase induces rapid retrophosphorylation and inhibition of MAP kinase kinase (MEK1) FEBS Lett. 1994;346:299–303. doi: 10.1016/0014-5793(94)00475-7. [DOI] [PubMed] [Google Scholar]
- 125.Corbalan-Garcia S, Yang SS, Degenhardt KR, Bar-Sagi D. Identification of the Mitogen-Activated Protein Kinase Phosphorylation Sites on Human Sos1 That Regulate Interaction with Grb2. Mol Cell Biol. 1996;16:5674–5682. doi: 10.1128/mcb.16.10.5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chen D, Waters SB, Holt KH, Pessin JE. SOS Phosphorylation and Disassociation of the Grb2-SOS Complex by the ERK and JNK Signaling Pathways. J Biol Chem. 1996;271:6328–6332. doi: 10.1074/jbc.271.11.6328. [DOI] [PubMed] [Google Scholar]
- 127.Waters SB, Holt KH, Ross SE, Syu L, Guan K, Saltiel AR, Koretzky GA, Pessin JE. Desensitization of Ras Activation by a Feedback Disassociation of the SOS-Grb2 Complex. J Biol Chem. 1995;270:20883–20887. doi: 10.1074/jbc.270.36.20883. [DOI] [PubMed] [Google Scholar]
- 128.Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, Conrads TP, Veenstra TD, Lu KP, Morrison DK. Regulation of Raf-1 by Direct Feedback Phosphorylation. Mol Cell. 2005;17:215–224. doi: 10.1016/j.molcel.2004.11.055. [DOI] [PubMed] [Google Scholar]
- 129.Ritt DA, Monson DM, Specht SI, Morrison DK. Impact of Feedback Phosphorylation and Raf Heterodimerization on Normal and Mutant B-Raf Signaling. Mol Cell Biol. 2010;30:806–819. doi: 10.1128/MCB.00569-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gross I, Bassit B, Benezra M, Licht JD. Mammalian Sprouty Proteins Inhibit Cell Growth and Differentiation by Preventing Ras Activation. J Biol Chem. 2001;276:46460–46468. doi: 10.1074/jbc.M108234200. [DOI] [PubMed] [Google Scholar]
- 131.Lim J, Sook E, Wong M, Ong SH, Yusoff P, Low BC, Guy GR. Sprouty Proteins Are Targeted to Membrane Ruffles upon Growth Factor Receptor Tyrosine Kinase Activation. J Biol Chem. 2000;275:32837–32845. doi: 10.1074/jbc.M002156200. [DOI] [PubMed] [Google Scholar]
- 132.Masoumi-Moghaddam S, Amini A, Morris DL. The developing story of Sprouty and cancer. Cancer Metastasis Rev. 2014;33:695–720. doi: 10.1007/s10555-014-9497-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mason JM, Morrison DJ, Basson MA, Licht JD. Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol. 2006;16:45–54. doi: 10.1016/j.tcb.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 134.Rubin C, Zwang Y, Vaisman N, Ron D, Yarden Y. Phosphorylation of Carboxyl-terminal Tyrosines Modulates the Specificity of Sprouty-2 Inhibition of Different Signaling Pathways. J Biol Chem. 2005;280:9735–9744. doi: 10.1074/jbc.M408308200. [DOI] [PubMed] [Google Scholar]
- 135.Tsavachidou D, Coleman ML, Athanasiadis G, Li S, Licht JD, Olson MF, Weber BL. SPRY2 Is an Inhibitor of the Ras/Extracellular Signal-Regulated Kinase Pathway in Melanocytes and Melanoma Cells with Wild-Type BRAF but Not with the V599E Mutant. Cancer Res. 2004;64:5556–5559. doi: 10.1158/0008-5472.CAN-04-1669. [DOI] [PubMed] [Google Scholar]
- 136.Tefft D, Lee M, Smith S, Crowe DL, Bellusci S, Warburton D. mSprouty2 inhibits FGF10-activated MAP kinase by differentially binding to upstream target proteins. Am J Physiol Lung Cell Mol Physiol. 2002;283:L700–L706. doi: 10.1152/ajplung.00372.2001. [DOI] [PubMed] [Google Scholar]
- 137.Boykevisch S, Zhao C, Sondermann H, Philippidou P, Halegoua S, Kuriyan J, Bar-sagi D. Regulation of Ras Signaling Dynamics by Sos-Mediated Positive Feedback. Curr Biol. 2006;16:2173–2179. doi: 10.1016/j.cub.2006.09.033. [DOI] [PubMed] [Google Scholar]
- 138.Freedman TS, Sondermann H, Friedland GD, Kortemme T, Bar-sagi D, Marqusee S, Kuriyan J. A Ras-induced conformational switch in the Ras activator Son of sevenless. Proc Natl Acad Sci. 2006;103:16692–16697. doi: 10.1073/pnas.0608127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sondermann H, Soisson SM, Boykevisch S, Yang S, Bar-Sagi D, Kuriyan J. Structural Analysis of Autoinhibition in the Ras Activator Son of Sevenless. Cell. 2004;119:393–405. doi: 10.1016/j.cell.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 140.Kholodenko BN. Negative feedback and ultrasensitivity can bring about oscillations in the mitogen-activated protein kinase cascades. Eur J Biochem. 2000;267:1583–1588. doi: 10.1046/j.1432-1327.2000.01197.x. [DOI] [PubMed] [Google Scholar]
- 141.Das J, Ho M, Zikherman J, Govern C, Yang M, Weiss A, Chakraborty AK, Roose JP. Digital Signaling and Hysteresis Characterize Ras Activation in Lymphoid Cells. Cell. 2009;136:337–351. doi: 10.1016/j.cell.2008.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lun X, Zanotelli VRT, Wade JD, Schapiro D, Tognetti M, Dobberstein N, Bodenmiller B. Influence of node abundance on signaling network state and dynamics analyzed by mass cytometry. Nat Biotechnol. 2017;35:164–172. doi: 10.1038/nbt.3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Birtwistle MR, Rauch J, Kiyatkin A, Aksamitiene E, Dobrzy M, Hoek JB, Kolch W, Ogunnaike BA, Kholodenko BN. Emergence of bimodal cell population responses from the interplay between analog single-cell signaling and protein expression noise. BMC Syst Biol. 2012;6:109. doi: 10.1186/1752-0509-6-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Dobrzynski M, Nguyen LK, Birtwistle MR, von Kriegsheim A, Fernandez AB, Cheong A, Kolch W, Kholodenko BN. Nonlinear signalling networks and cell- to-cell variability transform external signals into broadly distributed or bimodal responses. J R Soc Interface. 2014;11:20140383. doi: 10.1098/rsif.2014.0383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dobrzynski M, Fey D, Nguyen LK, Kholodenko BN. Bimodal Protein Distributions in Heterogeneous Oscillating Systems. Lect Notes Comput Sci. 2012:17–28. [Google Scholar]
- 146.Shin S, Rath O, Choo S, Fee F, McFerran B, Kolch W, Cho K. Positive- and negative-feedback regulations coordinate the dynamic behavior of the Ras-Raf-MEK-ERK signal transduction pathway. J Cell Sci. 2008;122:425–435. doi: 10.1242/jcs.036319. [DOI] [PubMed] [Google Scholar]
- 147.Kochańczyk M, Kocieniewski P, Kozłowska E, Jaruszewicz-Błońska J, Sparta B, Pargett M, Albeck JG, Hlavacek WS, Lipniacki T. Relaxation oscillations and hierarchy of feedbacks in MAPK signaling. Sci Rep. 2017;7:38244. doi: 10.1038/srep38244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bluthgen N, Bruggeman FJ, Legewie S, Herzel H, Westerhoff HV, Kholodenko BN. Effects of sequestration on signal transduction cascades. FEBS J. 2006;273:895–906. doi: 10.1111/j.1742-4658.2006.05105.x. [DOI] [PubMed] [Google Scholar]
- 149.Markevich NI, Hoek JB, Kholodenko BN. Signaling switches and bistability arising from multisite phosphorylation in protein kinase cascades. J Cell Biol. 2004;164:353–359. doi: 10.1083/jcb.200308060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Muller J, Ory S, Copeland T, Piwnica-Worms H, Morrison DK. C-TAK1 Regulates Ras Signaling by Phosphorylating the MAPK Scaffold, KSR1. Mol Cell. 2001;8:983–993. doi: 10.1016/s1097-2765(01)00383-5. [DOI] [PubMed] [Google Scholar]
- 151.Cullis J, Meiri D, Sandi MJ, Radulovich N, Kent OA, Medrano M, Mokady D, Normand J, Larose J, Marcotte R, Marshall CB, Ikura M, Ketela T, Moffat J, Neel BG, Gingras A-C, Tsao M-S, Rottapel R. The RhoGEF GEF-H1 Is Required for Oncogenic RAS Signaling via KSR-1. Cancer Cell. 2014;25:181–195. doi: 10.1016/j.ccr.2014.01.025. [DOI] [PubMed] [Google Scholar]
- 152.Lemmon MA, Schlessinger J. Cell Signaling by Receptor Tyrosine Kinases. Cell. 2010:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Markevich NI, Tsyganov MA, Hoek JB, Kholodenko BN. Long-range signaling by phosphoprotein waves arising from bistability in protein kinase cascades. Mol Syst Biol. 2006;2:61. doi: 10.1038/msb4100108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kiyatkin A, Aksamitiene E, Markevich NI, Borisov NM, Hoek JB, Kholodenko BN. Scaffolding Protein Grb2-associated Binder 1 Sustains Epidermal Growth Factor-induced Mitogenic and Survival Signaling by Multiple Positive Feedback Loops. J Biol Chem. 2006;281:19925–19938. doi: 10.1074/jbc.M600482200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Prior IA, Hancock JF. Ras trafficking, localization and compartmentalized signalling. Semin Cell Dev Biol. 2012;23:145–153. doi: 10.1016/j.semcdb.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Fivaz M, Meyer T. Reversible intracellular translocation of KRas but not HRas in hippocampal neurons regulated by Ca 2. J Cell Biol. 2005;170:429–441. doi: 10.1083/jcb.200409157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kranenburg O, Verlaan I, Moolenaar WH. Regulating c-Ras function: cholesterol depletion affects caveolin association, GTP loading, and signaling. Curr Biol. 2001;11:1880–1884. doi: 10.1016/s0960-9822(01)00582-6. [DOI] [PubMed] [Google Scholar]
- 158.Matallanas D, Sanz-moreno V, Arozarena I, Calvo F, Agudo-iba L, Santos E, Berciano T, Crespo P. Distinct Utilization of Effectors and Biological Outcomes Resulting from Site-Specific Ras Activation: Ras Functions in Lipid Rafts and Golgi Complex Are Dispensable for Proliferation and Transformation. Mol Cell Biol. 2006;26:100–116. doi: 10.1128/MCB.26.1.100-116.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Schmick M, Vartak N, Papke B, Kovacevic M, Truxius DC, Rossmannek L, Bastiaens PIH. KRas Localizes to the Plasma Membrane by Spatial Cycles of Solubilization, Trapping and Vesicular Transport. Cell. 2014;157:459–471. doi: 10.1016/j.cell.2014.02.051. [DOI] [PubMed] [Google Scholar]
- 160.Nakhaeizadeh H, Amin E, Nakhaei-rad S, Dvorsky R. The RAS-Effector Interface: Isoform-Specific Differences in the Effector Binding Regions. PLoS One. 2016;11:e0167145. doi: 10.1371/journal.pone.0167145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Fey D, Halasz M, Dreidax D, Kennedy SP, Hastings JF, Rauch N, Munoz AG, Pilkington R, Fischer M, Westermann F, Kolch W, Kholodenko BN, Croucher DR. Signaling pathway models as biomarkers: Patient-specific simulations of JNK activity predict the survival of neuroblastoma patients. Sci Signal. 2015;8:ra130. doi: 10.1126/scisignal.aab0990. [DOI] [PubMed] [Google Scholar]
- 162.Chmielecki J, Foo J, Oxnard GR, Hutchinson K, Ohashi K, Somwar R, Wang L, Amato KR, Arcila M, Sos ML, Socci ND, Viale A, De Stanchina E, Ginsberg MS, Thomas RK, Kris MG, Inoue A, Ladanyi M, Miller VA, Michor F, Pao W. Optimization of Dosing for EGFR-Mutant Non-Small Cell Lung Cancer with Evolutionary Cancer Modeling. Sci Transl Med. 2011;3 doi: 10.1126/scitranslmed.3002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gallaher JA, Enriquez-Navas PM, Luddy KA, Gatenby RA, Anderson ARA. Spatial heterogeneity and evolutionary dynamics modulate time to recurrence in continuous and adaptive cancer therapies. bioRxiv. 2017 doi: 10.1158/0008-5472.CAN-17-2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Enriquez-Navas PM, Kam Y, Das T, Hassan S, Silva A, Foroutan P, Ruiz E, Martinez G, Minton S, Gillies RJ, Gatenby RA. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci Transl Med. 2016;8 doi: 10.1126/scitranslmed.aad7842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Shain KH, Silva A, Meads MB, Distler A, Jacobson T, Gatenby R, Baz R, Silva M, Rebatchouk D, Cubitt C. A Multidisciplinary Model Predicts Clinical Response in Relapsed Multiple Myeloma. Blood. 2015;126:501. [Google Scholar]
- 166.Altrock PM, Brendel C, Renella R, Orkin SH, Williams DA, Michor F. Mathematical modeling of erythrocyte chimerism informs genetic intervention strategies for sickle cell disease. Am J Hematol. 2016;91:931–937. doi: 10.1002/ajh.24449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Serre R, Benzekry S, Padovani L, Meille C, Andre N, Ciccolini J, Barlesi F, Muracciole X, Muracciole X, Barbolosi D. Mathematical Modeling of Cancer Immunotherapy and Its Synergy with Radiotherapy. Cancer Res. 2016;76:4931–4940. doi: 10.1158/0008-5472.CAN-15-3567. [DOI] [PubMed] [Google Scholar]
- 168.Kim E, Rebecca VW, Smalley KSM, Anderson ARA. Phase i trials in melanoma: A framework to translate preclinical findings to the clinic. Eur J Cancer. 2016;67:213–222. doi: 10.1016/j.ejca.2016.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Fitzgerald JB, Schoeberl B, Nielsen UB, Sorger PK. Systems biology and combination therapy in the quest for clinical efficacy. Nat Chem Biol. 2006;2:458–467. doi: 10.1038/nchembio817. [DOI] [PubMed] [Google Scholar]
- 170.Holbeck SL, Camalier R, Crowell JA, Govindharajulu JP, Hollingshead M, Anderson LW, Polley E, Rubinstein L, Srivastava A, Wilsker D, Collins JM, Doroshow JH. The National Cancer Institute ALMANAC: A Comprehensive Screening Resource for the Detection of Anticancer Drug Pairs with Enhanced Therapeutic Activity. Cancer Res. 2017;77:3564–3576. doi: 10.1158/0008-5472.CAN-17-0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Chakrabarti S, Michor F. Pharmacokinetics and drug interactions determine optimum combination strategies in computational models of cancer evolution. Cancer Res. 2017;77:3908–3921. doi: 10.1158/0008-5472.CAN-16-2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kolch W, Halasz M, Granovskaya M, Kholodenko BN. The dynamic control of signal transduction networks in cancer cells. Nat Rev Cancer. 2015;15:515. doi: 10.1038/nrc3983. [DOI] [PubMed] [Google Scholar]
- 173.Lenzen C, Cool RH, Prinz H, Kuhlmann J, Wittinghofer A. Kinetic Analysis by Fluorescence of the Interaction between Ras and the Catalytic Domain of the Guanine Nucleotide Exchange Factor Cdc25 Mm. Biochemistry. 1998;2960:7420–7430. doi: 10.1021/bi972621j. [DOI] [PubMed] [Google Scholar]
- 174.Eccleston JF, Moore KJM, Brownbridge GG, Webb MR, Lowet PN. Fluorescence approaches to the study of the p21ras GTPase mechanism. Biochem Soc Trans. 1991;19:432–437. doi: 10.1042/bst0190432. [DOI] [PubMed] [Google Scholar]
- 175.Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol Cancer Res. 2015;13:1325–1336. doi: 10.1158/1541-7786.MCR-15-0203. [DOI] [PubMed] [Google Scholar]
- 176.Wey M, Lee J, Jeong SS, Kim J, Heo J. Kinetic Mechanisms of Mutation-Dependent Harvey Ras Activation and Their Relevance for the Development of Costello Syndrome. Biochemistry. 2013;52:8465–8479. doi: 10.1021/bi400679q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Donovan S, Shannon KM, Bollag G. GTPase activating proteins: critical regulators of intracellular signaling. Biochim Biophys Acta. 2002;1602:23–45. doi: 10.1016/s0304-419x(01)00041-5. [DOI] [PubMed] [Google Scholar]
- 178.Sydor JR, Engelhard M, Wittinghofer A, Goody RS, Herrmann C. Transient Kinetic Studies on the Interaction of Ras and the Ras-Binding Domain of c-Raf-1 Reveal Rapid Equilibration of the Complex. Biochemistry. 1998;37:14292–14299. doi: 10.1021/bi980764f. [DOI] [PubMed] [Google Scholar]
- 179.Ahmadian MR, Hoffmann U, Goody RS, Wittinghofer A. Individual Rate Constants for the Interaction of Ras Proteins with GTPase-Activating Proteins Determined by Fluorescence Spectroscopy. Biochemistry. 1997;36:4535–4541. doi: 10.1021/bi962556y. [DOI] [PubMed] [Google Scholar]
- 180.Bollag G, Adler F, Mccabe PC, Conner E, Thompson P, Mccormick F, Shannon K. Biochemical Characterization of a Novel KRAS Insertion Mutation from a Human Leukemia. J Biol Chem. 1996;271:32491–32495. doi: 10.1074/jbc.271.51.32491. [DOI] [PubMed] [Google Scholar]
- 181.John J, Frech M, Wittinghofer A. Biochemical Properties of Ha-ras Encoded p21 Mutants and Mechanism of the Autophosphorylation Reaction. J Biol Chem. 1988;263:11792–11799. [PubMed] [Google Scholar]
- 182.Smith MJ, Ottoni E, Ishiyama N, Goudreault M, Haman A, Meyer C, Tucholska M, Gasmi-Seabrook G, Menezes S, Laister RC, Minden MD, Marschalek R, Gingras A-C, Hoang T, Ikura M. Evolution of AF6-RAS association and its implications in mixed-lineage leukemia. Nat Commun. 2017 doi: 10.1038/s41467-017-01326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Smith MJ, Ikura M. Intergrated RAS signaling defined by parallel NMR detection of effectors and regulators. Nat Chem Biol. 2014;10 doi: 10.1038/nchembio.1435. [DOI] [PubMed] [Google Scholar]