

Abstract
Splenomegaly is a well-known phenomenon typically associated with inflammation. However, the underlying cause of this phenotype has not been well characterized. Furthermore, the splenomegaly phenotype seen in lymphotoxin (LT) signaling-deficient mice is characterized by increased numbers of splenocytes and splenic neutrophils. Splenomegaly, as well as the related phenotype of increased lymphocyte counts in non-lymphoid tissues, is thought to result from the absence of secondary lymphoid tissues in LT-deficient mice. We now present evidence that mice deficient in LTα1β2 or LTβR develop splenomegaly and increased numbers of lymphocytes in non-lymphoid tissues in a microbiota-dependent manner. Antibiotic administration to LTα1β2- or LTβR-deficient mice reduces splenomegaly. Furthermore, re-derived germ-free Ltbr −/− mice do not exhibit splenomegaly or increased inflammation in non-lymphoid tissues compared to specific pathogen-free Ltbr −/− mice. By using various LTβ- and LTβR-conditional knockout mice, we demonstrate that retinoic acid-related orphan receptor γT-positive type 3 innate lymphoid cells provide the required active LT signaling to prevent the development of splenomegaly. Thus, this study demonstrates the importance of LT-mediated immune responses for the prevention of splenomegaly and systemic inflammation induced by microbiota.
Keywords: germ-free, lymphotoxin, microbiota, splenomegaly, type 3 innate lymphoid cells
Introduction
The human microbiota is home to tens of trillions of microbes that are primarily categorized as Bacteria, Archaea and Eukarya.1 Human co-evolution with the microbiota has led to the development of a symbiotic relationship where the microbiota contributes to host physiological processes, such as food metabolism, and the host provides a special niche for the microbiota to inhabit.2 When comparing germ-free mice to those colonized with normal microbiota, it was quickly revealed that the presence of the microbiota is critical to the development of the normal immune system; GF mice develop fewer lymphoid structures in the intestinal tissue and produce less IgA than normal mice.3
Recently, a group of innate cells called innate lymphoid cells (ILCs) has been found to be important in regulating inflammation at barrier sites such as the lung and colon. ILCs originate from common lymphoid progenitors but lack somatic recombination capabilities and develop in the fetal liver and adult bone marrow.4,5,6 ILCs are innate lymphocytes that have the capability to respond quickly to a variety of stimuli to influence adaptive immune cells.5 ILCs are mainly divided into three groups based on the distinct transcription factors that are critical for their development to maturity. Type 3 innate lymphoid cells (ILC3s) express retinoic acid-related orphan receptor γT (RORγt) and mainly produce IL-22, IL-17 and IFNγ.7 ILC3s are found only in the skin, intestinal lamina propria and mucosal immune sites.8
The LT pathway is traditionally thought to be important for the development and function of lymph nodes and gut-associated lymphoid tissue (GALT). One of the primary roles of active lymphotoxin (LT) signaling is to organize immune cells within follicular structures to promote specific immune responses.9,10,11,12,13,14 LT is also known to promote the production of antimicrobial peptides, IgA and IL-22, that are involved in mucosal defense.15 Soluble LTα and membrane LTα1β2 are core members of the tumor necrosis factor (TNF) superfamily.16 LTα forms a soluble homotrimer that can bind TNF receptor 1 and 2.16 The LT signaling pathway is critical for the development and maintenance of secondary lymphoid organs.17,18,19,20,21 Thus, mice deficient in LTα, LTβ or LTβR lack GALT and display compromised mucosal defenses.17,18,19,21,22,23 LTα1β2 is expressed on the surface of T, B, LTi (lymphoid tissue inducer cells) and ILC3 cells and signals via LTβR expressed on stromal cells, myeloid cells, hepatocytes, epithelial cells and endothelial cells.15,16,24
It has been determined that common variable immunodeficiency (CVID) and other forms of autoimmunity are associated with gene variants within the Tnf and Lta locus.25,26 Like CVID patients, it has previously been noted that Ltbr −/− mice demonstrate lymphocytosis, inflammation in the lungs and liver, and enlarged spleens.19,27,28,29 These phenotypes are presumed to be due to the absence of peripheral lymph nodes.30,31,32,33,34 However, our data support the idea that splenomegaly and increased numbers of T cells in non-lymphoid tissues are not caused by lack of secondary lymphoid tissues but can be attributed to the role of LT in controlling microbiota-induced inflammation.
Materials and methods
Mice
C57BL/6, Rag1 −/− and Ragγc −/− mice were originally obtained from Harlan, Jackson and Taconic Labs, respectively, and were bred and maintained in-house for multiple generations. Lta −/− , Ltb −/− , Ltbr −/−, Rorc −/− , Cd4 cre+ Ltb f/f (LTβΔT), Cd19 cre+ Ltb f/f (LTβΔB), Cd4 cre+ Cd19 cre+ Ltb f/f (LTβΔT, B), Rorc cre+ Ltb f/f (LTβΔILC, T), Cd11c Cre+ Ltbr f/f (LTβRΔDC), and LysM Cre+ Ltbr f/f (LTβRΔMφ) mice were bred as littermates in a vivarium at the University of Chicago and were initially characterized elsewhere.4,14,17,19,20,21,35,36,37 Ltb −/− and LTβΔILC,T mice were crossed onto the Rag1 −/− background for this study. Littermates were generated by breeding of a null parent (usually the dam) to a heterozygous mate. In the case of conditionally deficient mice, Cre-negative littermate Ltb f/f or Ltbr f/f mice were used as WT controls. Mice were genotyped before weaning between 21 and 28 days after birth. All mice were bred and kept under specific pathogen-free (SPF) conditions. All mice used were age and sex matched. All SPF mice were maintained on normal chow (Harlan Teklad 2918). All GF mice were maintained on standard autoclaved and irradiated chow (Harlan Teklad 2016S). Fecal samples were collected weekly and underwent culture and 16s PCR to monitor the microbiological status of the experimental isolators. All experimental procedures were approved and carried out in accordance to University of Chicago’s IACUC (Protocol #71866).
Depletion of gut commensal microbiota
Animals were genotyped prior to weaning and separated at 3–4 weeks of age. Animals were either treated at weaning or at 8 weeks of age with broad-spectrum antibiotics in their drinking water for a period of 4 weeks, as initially described elsewhere.38 Briefly, a modified cocktail of antibiotics purchased from Sigma (ampicillin (1 g/l), gentamicin (1 g/l), vancomycin (500 mg/l), neomycin sulfate (1 g/l) and metronidazole (1 g/l)) was dissolved in autoclaved water and given to mice ad libitum. Autoclaved water alone was given to control mice. The antibiotic cocktail was replaced on a weekly basis, and the mice were monitored for health status issues on a biweekly basis.
Stool DNA extraction
Stool samples were freshly collected and frozen at less than −20 °C. Stool DNA extraction was performed with Mini-beadbeater (BioSpec Products, Bartlesville, OK, USA) homogenization with beads, followed by extraction with QIAamp DNA Stool Mini Kit’s (Qiagen, Germantown, MD, USA) protocol. The DNA was eluted and saved at −20 °C until further analysis.
Germ-free derivation of LTβR littermates
Derivation of Ltbr −/− mice was contracted through Taconic Biosciences via the standard germ-free (GF) derivation protocol using GF isolator reared, GF surrogate mothers. Briefly, Taconic Biosciences received male Ltbr −/− mice from us to be bred to C57BL/6 female mice. At day 19–20 of gestation, the ‘uterine package’ was surgically removed from the female donor. Under sterile conditions, the ‘uterine package’ was opened and the Ltbr +/− pups were transferred to a GF isolator housing a surrogate mother. The microbiological status of the isolator was monitored following transfer. Once the Ltbr +/− mice reached adulthood, they were shipped to the University of Chicago’s Gnotobiotic Core Facility using Taconic GF Shipper. Specialized technicians at the Gnotobiotic Core transferred the animals from the Taconic GF Shipper to the designated flexible isolator. GF heterogynous LTβR mice were bred together to generate GF Ltbr +/+ and Ltbr −/− mice. GF Ltbr +/+ and Ltbr −/− were maintained by homozygous breeding in the gnotobiotic facility. Fecal samples were collected weekly to monitor the microbiological status of the experimental isolators by culture and PCR of bacterial 16s ribosomal DNA. All experimental procedures were approved and carried out in accordance with University of Chicago’s IACUC (Protocol #71866).
Flow cytometry and antibodies
Viable cells from spleens and lamina propria lymphocytes (LPL) were counted under a microscope (Zeiss, Jena, Germany) using a hemocytometer. All samples were stained with 7-AAD or ZombieAqua in 1 × PBS following Biolegend’s instructions (San Diego, CA, USA). Cell samples were then washed with FACS wash buffer (1 × PBS+2% fetal bovine serum) before incubating the samples in anti-CD16/CD32 Fc block (clone 2.4g2), in the dark, for at least 15 min at room temperature. As needed, samples were stained with a master mix of antibodies (CD90.2 (clone 30-H12), CD45 (clone 30-F11), GR-1 (clone RB6-8C5), CD11b (clone M1/70), CD3ɛ (clone 145-2C11), CD8α (clone 53-6.3), CD4 (clone GK1.5), CD11c (clone HL3), NK1.1 (clone PK136), F4/80 (clone BM8), Ly-6C (clone HK1.2), Ly-6G (clone 1A8), CD19 (clone 1D3), purchased from BioLegend, for 30 min in the dark at 4 °C. Samples were then washed again with FACS wash buffer before fixation with eBioscience (Thermo Fisher Scientific, Waltham, MA, USA) IC fixation buffer overnight, in the dark at 4 °C. The data were collected on an LSR-Fortessa (BD, Franklin Lakes, NJ, USA) and analyzed using FlowJo software (Ashland, OR, USA).
Sorting and transfer of ILC3s
ILC3s from LPL of Rag1 −/− mice were sorted based on 7AAD, CD45 and CD90 expression using a FACSAria (BD) cell sorter as previously described.39,40 Briefly, the small intestine and colonic tissue, including the cecum, were removed and cleaned using sterile 1 × PBS and mechanical scraping. The mucosal tissue was washed using 1 × HBSS+0.5% dithiothreitol before enzymatic digestion using DNase I (Sigma, St Louis, MO, USA) and Liberase (Roche, Indianapolis, IN, USA). The intestines were further disrupted using gentleMACs C tubes (Miltenyi Biotec, San Diego, CA, USA). Lymphocytes were further purified by gradient centrifugation using a 40/80% Percoll (GE, Pittsburgh, PA, USA) gradient. LPL were collected at the interphase, washed and counted. The cells were stained at 1 × 107 cells per ml before sorting. LPLs were gated by CD45 and CD90 expression where intestinal CD90hiCD45lo LPLs are RORγt+ ILC3s.39,40 Purification checks were performed after each sort. The cells were kept on ice until ⩾2.5 × 105 cells were transferred via retro-orbital, i.v. injection into 3–4-week-old mice. The mice were aged for another 4–6 weeks before analysis.
Blocking LT and IL-22 signaling in vivo
The LTβR:hIg procedure used in this study has been previously described.41,42 Briefly, 100 μg per mouse of LTβR:hIg was administered by intraperitoneal (i.p.) injections weekly from weaning for the duration of the experiment. Anti-IL-22 antibody (generously provided by Wenjun Ouyang, Genentech, South San Francisco, CA, USA) was administered i.p. (150 μg per mouse 3 × per week) from weaning for the duration of the experiment. Mouse weights were monitored before each injection to ensure normal health status.
Real-time PCR
RNA was extracted from colon samples frozen in RNAlater (Ambion, Thermo Fisher Scientific) at −80 °C by using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. The RNA product was treated with amplification-grade DNase I (Qiagen). Product integrity was verified by running samples on 2% agarose gels. We used 2 μg of RNA to make cDNA using Maxima Reverse Transcriptase and associated buffers, dNTPs and oligo(dT) primer (Thermo Scientific) according to the manufacturer’s protocol. Samples were amplified on an iQ5 instrument (Bio-Rad Laboratories, Hercules, CA, USA) using SsoFast EvaGreen Supermix (Bio-Rad Laboratories); primer concentrations were 0.5 μM in the final reaction. Correct melting temperatures for all products were verified after amplification. For all products, amplification in all samples resulted in correct melting temperatures. Replicates were made of all samples, which were analyzed using the 2−ΔΔCT method normalized to the corresponding Gapdh. qSTAR qPCR primer pairs against Mus musculus gene H2 were purchased from OriGene (Rockville, MD, USA). mIL-22 (F: 5′--3′, R: 5′--3′). Gapdh (F: 5′--3′, R: 5′--3′).
Statistical methods
The data were analyzed using Student’s t-test or ANOVA with multiple comparisons correction, when appropriate, using GraphPad Prism 6.0 (La Jolla, CA, USA). The data from experiments are presented and denoted as the mean values±s.e.m. or mean with 95% confidence interval wherever appropriate. P<0.05 was considered statistically significant.
Results
LT beta-receptor prevents gut microbiota-induced splenomegaly and systemic inflammation
To confirm the splenomegaly phenotype in LT-deficient hosts, we measured the spleen size and counted the number of splenocytes. We observed significant differences in spleen size (Figure 1a) and splenocyte number (Figure 1b) in SPF Ltbr −/− mice compared to Ltbr +/+ mice or littermate Ltbr +/− mice. To determine when the splenomegaly phenotype first appears, we performed a time-course experiment. Splenomegaly was measured in SPF Ltbr +/− and Ltbr −/− littermates before weaning until 12 weeks after birth. We found that Ltbr −/− and Ltbr +/− littermate mice had similar spleen sizes before and immediately after weaning. Interestingly, the splenomegaly phenotype became statistically significant only when Ltbr −/− mice reached adulthood (Supplementary Figure 1A). These data suggest that inherent lack of lymph nodes did not result in splenomegaly but that the splenomegaly phenotype develops in Ltbr −/− mice after the adult commensal microbiota has established in these mice.43,44
Figure 1.
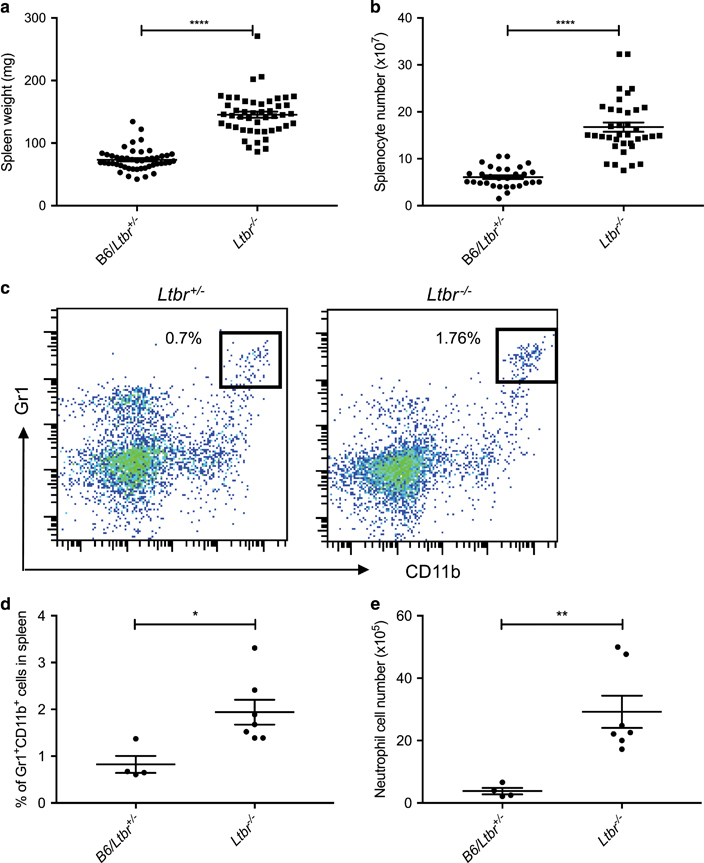
The neutrophilic splenomegaly phenotype in lymphotoxin-deficient mice. (a) Spleens from SPF Ltbr +/+, littermate Ltbr +/− and Ltbr −/− mice were harvested and weighed. n=47, pooled from six repeats. (b) Trypan blue negative splenocytes counted with a hemocytometer from spleens in a. (c) Example flow plots showing the Gr1+CD11b+ population that has been gated on CD45+, CD3− and B220− populations from the spleen of Ltbr +/− and Ltbr −/− mouse, respectively. (d) SPF spleens were examined for percentage of neutrophils (CD45+, CD3−, B220−, Gr1+ and CD11b+) out of total splenocytes and (e) absolute neutrophil cell number. (a, b, d and e) Unpaired t-tests were performed. *P<0.05; **P<0.01; ****P<0.0001.
To determine whether the splenomegaly phenotype comprised one particular immune cell type or the expansion of all immune cells, we analyzed various subsets of splenic immune cells by flow cytometry. When we examined the splenocyte cell population, we noticed a relative increase in all major cell populations. The only significant shift in the differential was noted to be an increase in the percentage of Gr1+CD11b+ cells (Figure 1c), most likely neutrophils, in the SPF Ltbr −/− splenocytes compared to Ltbr +/− littermates. Specifically, both the percentage (Figure 1d) and absolute number (Figure 1e) of neutrophils in the spleens of SPF Ltbr +/− mice were significantly lower compared to the amount of neutrophils found in the spleens of SPF Ltbr −/− mice. Since neutrophils typically respond to bacterial infections, and Ltbr −/− mice are known to be highly susceptible to colonic bacterial infections, we wondered whether exposure to commensal bacteria promoted the splenomegaly phenotype in SPF mice. Thus, we sought to determine whether the splenomegaly phenotype and corresponding expansion of neutrophils is a sign of bacterial-induced inflammation.
To determine whether the bacteria within the microbiota led to the expansion of neutrophils in the spleen of Ltbr −/− mice, we utilized antibiotics to deplete the commensals in our SPF colony.38 To test this hypothesis, broad-spectrum antibiotics were administered ad libitum in drinking water to SPF Ltbr −/− and Ltbr +/− mice, starting at weaning for 4 weeks. ABX treatment had no effect on spleen sizes (Figure 2a) or splenocyte numbers (Figure 2b) of Ltbr +/+ or littermate Ltbr +/− mice. Surprisingly, compared to control, spleen size (Figure 2a) and splenocyte numbers (Figure 2b) were significantly reduced in ABX-treated Ltbr −/− mice. Consistent with the idea that increased neutrophils were dependent on bacteria, we also found a reduction in both percentage (Figure 2c) and absolute cell numbers (Figure 2d) of neutrophils in the spleen of ABX-treated Ltbr −/− mice. Interestingly, if broad-spectrum antibiotics were given to SPF Ltbr −/− ad libitum for 4 weeks starting at 8 weeks of age, the spleen weight (Supplementary Figure 1B) and splenocyte numbers (Supplementary Figure 1C) were similar to untreated control Ltbr −/− mice. Together, these data suggest that the initial exposure of Ltbr −/− mice to bacteria allows for the development of neutrophilic splenomegaly.
Figure 2.
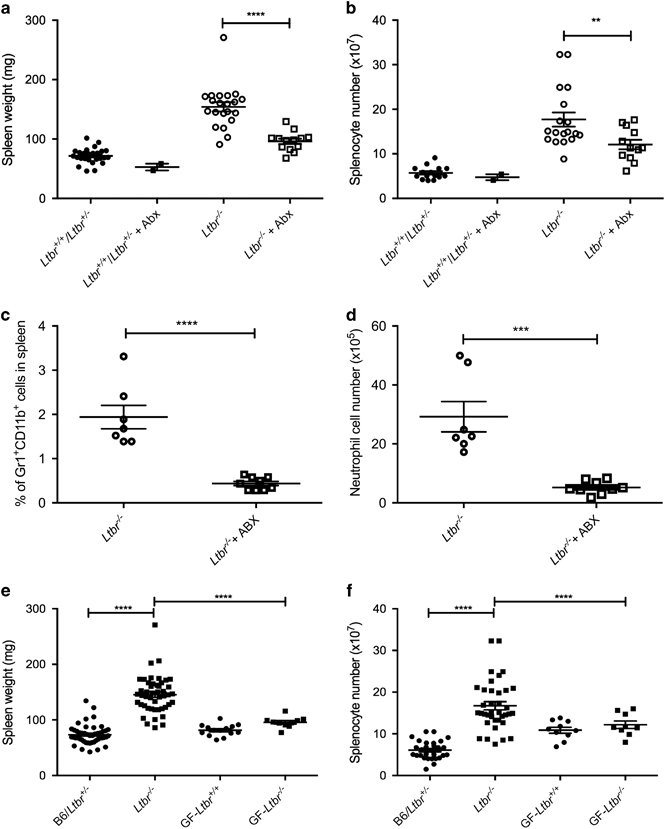
The commensal microbiota influences the splenomegaly phenotype seen in lymphotoxin-deficient mice. SPF Ltbr +/+ and littermate Ltbr +/− and Ltbr −/− mice were placed on antibiotics (ABX) water or water alone at 4 weeks of age. Mice were killed after an additional 4 weeks. The spleens were harvested and (a) weighed, (b) counted, and (c) percentage and (d) absolute cell number of neutrophils were determined. (e) Spleens from SPF Ltbr +/+, littermate Ltbr +/− and Ltbr −/− mice were harvested and weighed (n=47, pooled from six repeats). Spleens from germfree (GF) Ltbr +/+ and Ltbr −/− spleen weights were harvested and weighed. n=10–14, pooled from six repeats. (f) Trypan blue negative splenocytes counted with a hemocytometer from spleens in a. Error bars represent the mean and s.e.m. Student’s t-test was performed on c and d. One-way ANOVA with Tukey correction was performed on a, b, e and f. **P<0.01; ***P<0.001; ****P<0.0001.
Previous work has demonstrated the possibility that disseminating microbiota can promote splenomegaly.45 To determine whether the presence of disseminating microbiota could be promoting splenomegaly in LT-deficient mice, we utilized various culture techniques in both aerobic and anaerobic conditions to try to culture bacteria from spleen and liver tissues (Supplementary Methods). No bacteria were consistently detected from tissues using the culture conditions attempted. To confirm whether the increased basal inflammation in LT-deficient mice was microbiota-dependent, we re-derived the Ltbr −/− mice GF. Intriguingly, the splenomegaly phenotype was no longer observed in the GF mouse colony. Specifically, GF Ltbr −/− mice had spleen sizes (Figure 2e) and splenocyte numbers (Figure 2f) comparable to GF Ltbr +/+ mice. Together, these data suggest that the splenomegaly phenotype originally observed in SPF Ltbr −/− mice might be microbiota-dependent. In other words, LT signaling could prevent peripheral inflammation in the presence of the microbiota.
LT on group 3 innate lymphoid cells prevents splenomegaly
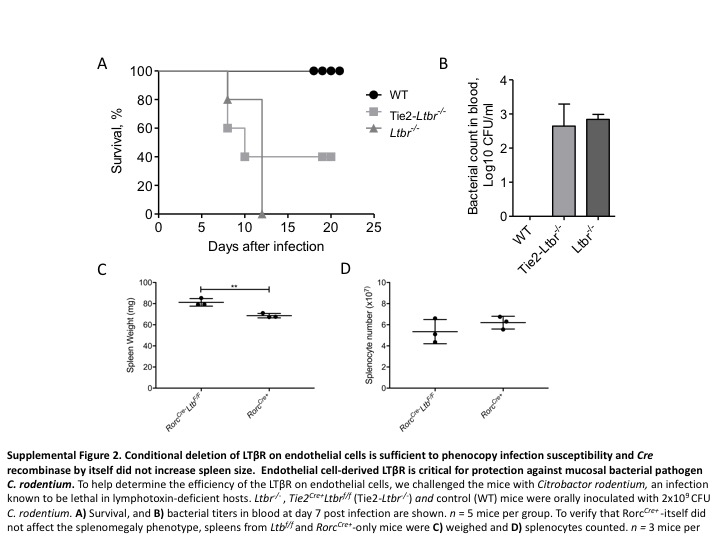
To understand which component of the host immune system responds to bacteria in an LT-dependent manner, we sought to determine which immune cells were involved. To determine which immune cells expressing LTα1β2 ligand and/or LTβR were important in regulating spleen size, we generated various LTβ and LTβR conditional knockout mice. First, we deleted LTβR on myeloid precursors in LysM Cre+ Ltbr f/f mice but found no difference in spleen size, suggesting that LTβR expression on myeloid cell lineages is not essential for development of splenomegaly. We next targeted the endothelial cell lineage using Tie2cre (Supplementary Figures 2A and B). We did not observe major difference in the spleen phenotype of Tie2 Cre+ Ltbr f/f mice (endothelial cells; Figure 3a). To determine which cells expressing LT control microbiota-mediated inflammation, we first deleted LTβ on B cells (Cd19 Cre+ Ltb f/f mice) and all T cells (Cd4 Cre+ Ltb f/f mice) but observed no difference in spleen weight (Figure 3b). In addition, deletion of LTβ in both T cells and B cells (Cd19 Cre+ Cd4 Cre+ Ltb f/f mice) did not result in splenomegaly (Figure 3b).
Figure 3.

The absence of lymphotoxin on RORγt expressing cells results in splenomegaly. (a) Conditional LTβR-deficient mice were generated by crossing Ltbr f/f mice with LysM Cre+ mice and Tie2 Cre+ mice (n=3–4, repeated two times). (b, c) Ltb f/f mice (n=3–13, repeated five times) were bred to Cd4 Cre+ and Cd19 Cre+mice to inactivate LT in T cells and B cells, respectively. Mice were killed, and the spleens were removed and weighed. Error bars represent the mean and s.e.m. One-way ANOVA was performed. **P<0.01; ***P<0.001.
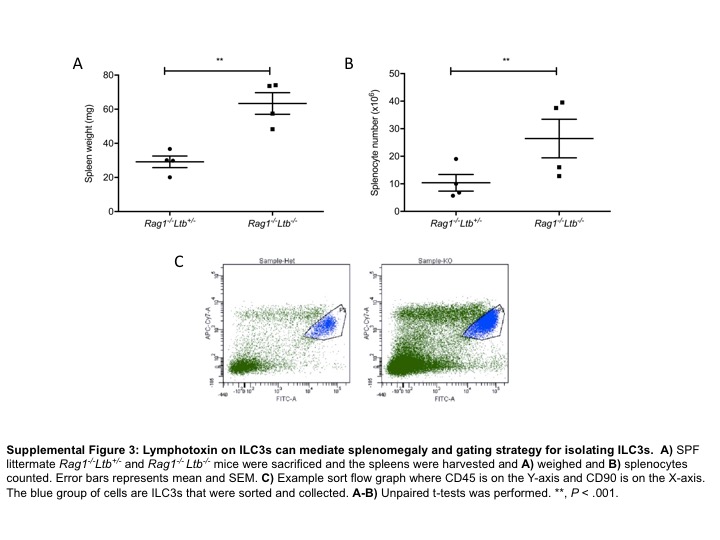
Since RORγt+LTβ+ ILC3s are shown to control gut infection, we next deleted LT on RORγt cells.13,35 Impressively, we observed that Rorc Cre+ Ltb f/f mice had significantly larger spleens compared to littermate controls in this single conditional mouse (Figure 3c). The data suggest that ILC3s expressing LTβ regulate spleen size, since T cells deficient in LT did not have larger spleens than littermate controls (Figure 3b). To ensure that Rorc Cre+ expression did not have a biological impact in this model, we validated that the spleen sizes (Supplementary Figure 2C) and the splenocyte numbers (Supplementary Figure 2D) did not differ between Rorc Cre+ mice and Rorc Cre− Ltb f/f mice.46,47 Since T cells and ILC3s both express LT, we next crossed Rorc Cre+ Ltb f/f mice onto the Rag1 −/− background to determine whether the lack of LTα1β2 on ILC3 was sufficient for responding to the microbiota in the absence of adaptive immunity. Interestingly, Rag1 −/− Rorc Cre+ Ltb f/f mice had significantly larger spleens (Figure 4a) and increased splenocyte numbers (Figure 4b) compared to SPF Rag1 −/− mice. Furthermore, Rag1 −/− Rorc Cre+ Ltb f/f mice had a significantly higher frequency of neutrophils (Figure 4c) and absolute neutrophil cell numbers (Figure 4d) compared to control Rag1 −/− mice (Figure 4e). To verify that ILC3s expressing LT were needed following exposure to the same microbiota, we generated littermate Rag1 −/− Ltb +/− and Rag1 −/− Ltb −/− mice and observed that mice that lack LT on only ILC3s had significantly larger spleens (Supplementary Figure 3A) and higher splenocyte numbers (Supplementary Figure 3B) compared to littermate controls. Together, these data demonstrate that LT-expressing ILC3s are needed to prevent the development of splenomegaly in the presence of the commensal bacteria.
Figure 4.
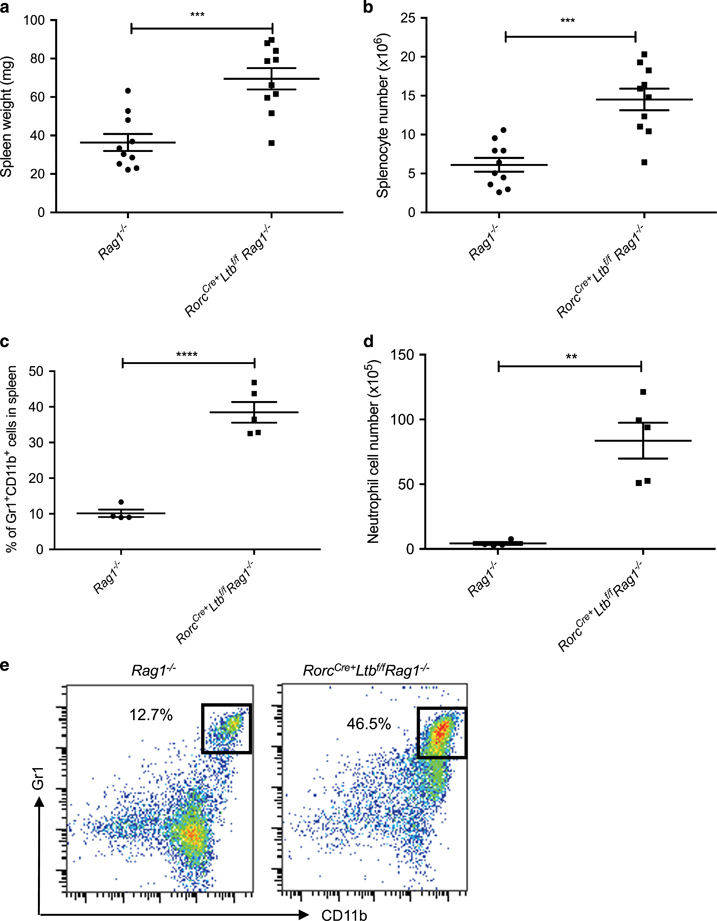
Lymphotoxin expression on ILC3s prevents the development of splenomegaly. Rorc Cre+ Ltb f/f mice were crossed onto B6 Rag1 −/− background. The mice were killed and the spleens were harvested and (a) weighed, (b) counted, and (c) percentage and (d) absolute cell number of neutrophils were determined and compared against spleens from B6 Rag1 −/− mice. (e) Example flow plots showing the Gr1+CD11b+ population that has been gated on CD45+, CD3− and B220− populations from the spleen of Rag1 −/− and Rorc Cre+ Ltb f/f Rag1 −/− mouse, respectively. n=8, repeated two times. Error bars represent the mean and s.e.m. Unpaired t-tests were performed. **P<0.01; ***P<0.001; ****P<0.0001.
Active LT signaling from group 3 innate lymphoid cells is necessary to prevent splenomegaly
Since we determined that LT on ILC3s was needed to prevent the development of splenomegaly and systemic inflammation, we wondered whether ILC3s expressing LTα1β2 by themselves were sufficient for preventing splenomegaly. To determine whether a lack of LT signaling from ILC3 was sufficient to promote splenomegaly, we sorted ILC3s based on CD90 and CD45 expression and transferred them into Ragγc −/− hosts (Supplementary Figure Supplementary Figure 3C).40 Ragγc −/− mice lack T cells, B cells, natural killer cells and most importantly, ILCs, due to lack of functional receptors for many cytokines and recombinase activating gene-2.37,48,49 To test whether ILC3s expressing LTα1β2 were necessary to prevent splenomegaly, we isolated LPLs from littermate Rag1 −/− Ltb +/− and Rag1 −/− Ltb −/− mice.
ILC3s were sorted from the corresponding LPLs based on CD45+ and CD90+ expression and transferred via retro-orbital i.v. injection into littermate Ragγc −/− mice before weaning. The Ragγc −/− mice who received ILC3s sorted from Rag1 −/− Ltb +/− LPLs had significantly smaller spleens than the Ragγc −/− mice who received ILC3s sorted from Rag1 −/− Ltb −/− LPLs (Figure 5a). In addition, the Ragγc −/− mice who received ILC3s sorted from Rag1 −/− Ltb +/− LPLs had significantly fewer splenocytes compared to the Ragγc −/− mice who received ILC3s sorted from Rag1 −/− Ltb −/− LPLs (Figure 5b). Although we did not see a significant increase in the frequency of neutrophils (Figure 5c), when we examined the splenocyte populations, we observed a corresponding significant increase in absolute numbers of neutrophils (Figure 5d) in the Ragγc −/− mice who received ILC3s sorted from Rag1 −/− Ltb −/− LPLs. Given these data, we determined that LTα1β2 expressed on the membrane of ILC3s was necessary and sufficient to prevent the development of neutrophilic splenomegaly in the presence of the microbiota.
Figure 5.
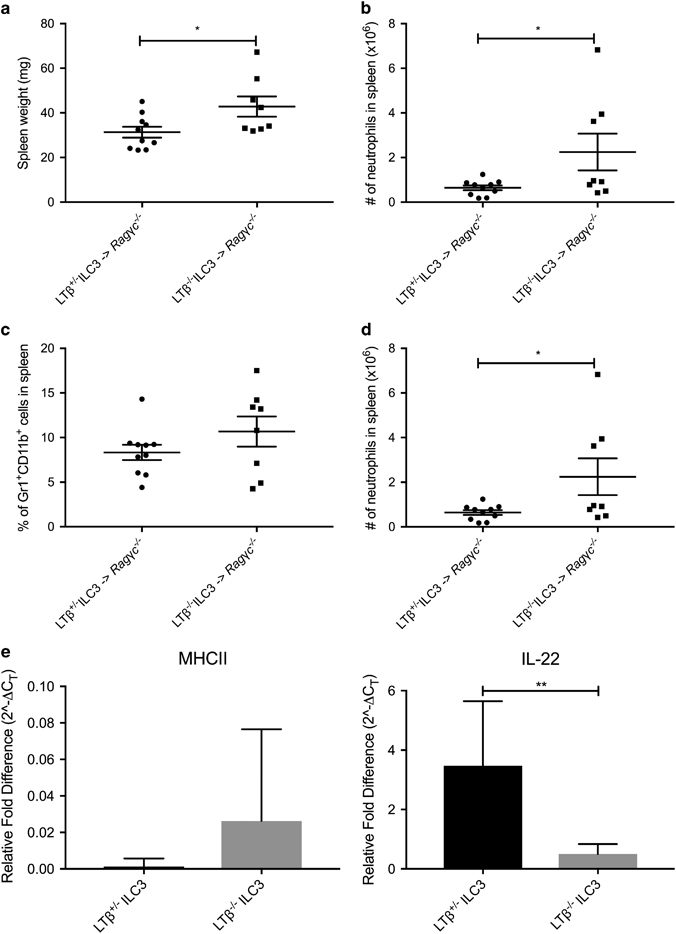
Inactivation of lymphotoxin on ILC3s results in diminished IL-22 expression and promotes the development of neutrophilic splenomegaly. SPF littermate Ragγc −/− were weaned and given ILC3s from Rag1.Ltb +/− or Rag1.Ltb −/− transfer via retro-orbital injections. The mice were killed after 5 weeks and the spleens were harvested and (a) weighed, (b) counted, and (c) percentage and (d) absolute cell number of neutrophils were determined. n=8–10, repeated three times. (e) MHCII and IL22 expression from ILC3s sorted from Rag1.Ltb +/− or Rag1.Ltb −/− mice were measured via qPCR. n=3–5, with technical replicates. (a–d) Error bars represent the mean and s.e.m. (e) Error bars represent mean values with 95% CI. Unpaired t-tests were performed. *P<0.05; **P<0.01.
It is known that Rorγt −/− mice, which also lack ILC3s, have a splenomegaly defect, demonstrating that ILC3s are necessary to maintain and promote a proper mucosal gut barrier.50,51 Recently, expression of MHC-II on ILC3s has been shown to mediate the host’s immune response at the gut mucosal barrier to prevent the development of splenomegaly.8,52 In addition, IL-22 has been shown to play an important role in preventing splenomegaly in an ILC3 and microbiota-dependent manner.7,45,50 Furthermore, it is known that ILC3s producing IL-22 are critical for anti-bacterial defense in a STAT3-dependent manner.39 Since the LT pathway regulates IL-22 signaling in the context of gut inflammation, we wondered whether these signaling pathways were affected by LT expression on ILC3s.13,35
To test whether LTα1β2 expression on ILC3s can influence MHC-II and IL-22 expression, we isolated LPLs from naive littermate Rag1 −/− Ltb +/− and Rag1 −/− Ltb −/− mice. ILC3s were sorted from the corresponding LPLs based on CD45+ and CD90+ expression and collected for RNA isolation. The relative expression of MHC-II on LTα1β2-sufficient ILC3s did not significantly differ from LTα1β2-deficient ILC3s (Figure 5e). Interestingly, LTα1β2-deficient ILC3s had significantly lower IL-22 expression compared to LTα1β2-sufficient ILC3s (Figure 5e). This suggests that LT expression on ILC3s is necessary for normal IL-22 production. This implies that the ability of ILC3s to produce IL-22 at homeostatic conditions is dependent on sufficient LT expression and/or signaling. These data demonstrate that ILC3s expressing LTα1β2 and actively providing LT signaling are needed to prevent the development of splenomegaly in the presence of bacteria.
To determine whether the IL-22 pathway was directly involved in regulating microbiota-induced inflammation, we blocked IL-22 signaling in Rag1 −/− mice. Littermate Rag1 −/− mice were weaned and given αIL-22 or IgG control i.p. 3 × weekly for 4 weeks. Although we did observe a significant difference in spleen size between αIL-22-treated Rag1 −/− mice and control Rag1 −/− mice (Figure 6a), when we examined the splenocyte numbers, we did not observe a significant difference between the mice that received αIL-22 or IgG control (Figure 6b). These data show that only inhibiting IL-22 signaling is insufficient to promote the development of splenomegaly in Rag1 −/− mice. Since expression of LTβ on ILC3s can dampen splenomegaly, we next wondered whether active LT signaling was required. In other words, is the splenomegaly phenotype due to the lack of lymph nodes or active LT?
Figure 6.
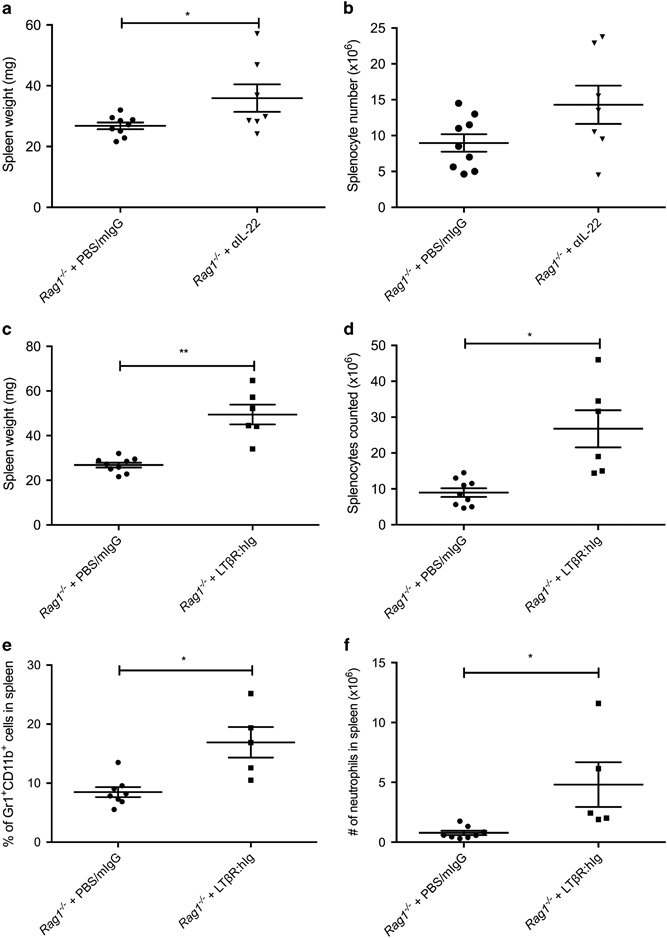
The absence of active signaling from LT results in the development of neutrophilic splenomegaly. SPF littermate Rag1 −/− were weaned and given αIL-22 or mIgG as control for 4 weeks. The mice were killed and the spleens were harvested and (a) weighed and (b) splenocytes counted. SPF littermate Rag1 −/− were weaned and given LTβR:hIg or mIgG as control for 4 weeks. The mice were killed and the spleens were harvested and (c) weighed, (d) counted, and (e) percentage and (f) absolute cell number of neutrophils determined and compared against spleens from control mice. n=5–8, repeated three times. Error bars represent the mean and s.e.m. Unpaired t-tests were performed. *P<0.05; **P<0.01.
To determine whether ILC3s needed to constitutively provide LT to prevent the development of systemic inflammation, we examined whether blocking LT signaling would have an effect on spleen size. We blocked LT signaling in Rag1 −/− mice with LTβR:hIg fusion protein, since in Rag1 −/− mice ILC3s are the main immune cells expressing LTα1β2. Littermate Rag1 −/− mice were treated with LTβR:hIg fusion protein or IgG control for 4 weeks, starting at weaning. In LTβR:hIg treated Rag1 −/− mice, we observed an increase in spleen size (Figure 6c) and splenocyte numbers (Figure 6d) compared to control Rag1 −/− mice. We observed a significant difference not only in the frequency of neutrophils (Figure 6e), but when we examined the splenocyte cell populations, we also found an increase in absolute number of neutrophils (Figure 6f). These data demonstrate that active LT signaling from ILC3s is needed to prevent the development of bacteria-induced neutrophilic splenomegaly in SPF Rag1 −/− mice. These data suggest that ILC3s expressing LTα1β2 and actively providing LT signaling are needed to prevent the development of splenomegaly in the presence of bacteria. In conclusion, this work determined that LT-expressing ILC3s are needed to maintain proper mucosal barrier immunity to prevent the development of microbiota-induced splenomegaly.
Discussion
Splenomegaly and increased lymphocytes in non-lymphoid tissues in LT-deficient mice have been previously attributed to the lack of the secondary lymphoid tissues. This work revisited this issue and determined that splenomegaly is dependent on both LT signaling and the microbiota. Within the splenocyte cell population, there was an increase in the percentage of neutrophils observed only in Ltbr −/− mice and not in their corresponding SPF Ltbr +/− littermates. Oral antibiotics given during the initial exposure to the commensal microbiota were sufficient to prevent the neutrophilic splenomegaly.
By using different conditional mouse models that deleted Ltb and Ltbr on various immune cells, we determined that cells expressing RORγt transcription factor and LTβ were important for mediating the host’s immune response to the microbiota. Specifically, LTβ expression and active LT signaling on ILC3s was necessary and sufficient to prevent the development of neutrophilic splenomegaly. Furthermore, active LT signaling from ILC3s is needed to prevent the development of bacteria-induced neutrophilic splenomegaly in SPF Rag1 −/− mice. These results suggest that membrane LTα1β2 expressed on ILC3s is necessary and sufficient to prevent the development of neutrophilic splenomegaly in the presence of the commensal microbiota. Therefore, the splenomegaly phenotype previously observed in LT-deficient and LT-signaling deficient models develops following exposure to the gut bacteria.
Currently, we have not observed significant spleen size differences in various conditional knockouts of LTβR on either myeloid or endothelial cell lineages. It is unclear which cell type expresses LTβR to receive the LT signal from ILC3s. Since LTβR is expressed on stromal, endothelial and epithelial cells, it is possible that these cell types are also needed to coordinately regulate colonic inflammation and promote tissue homeostasis.19,21,31,32,53,54,55,56 Previous work has proposed that both dendritic cells expressing LTβR interacting with ILC3s and ILC3s interacting with LTβR expressed on epithelial cells can initiate the IL-23/IL-22 pathway needed against pathogens.13,35,36,57,58 Therefore, we propose the following model: LTβ expression on ILC3s binds and activates LTβR on DC and epithelial cells, and this interaction is important for modulating microbiota-induced inflammation at baseline. In other words, active LT signaling from ILC3s is also needed to promote host homeostasis in the presence of commensal bacteria.
The presence of bacterial ligands such as LPS and flagellin has been shown to upregulate intestinal epithelial proliferation, paneth cell proliferation and expression of antimicrobial peptides (AMPs) such as RegIIIγ through the IL-22 pathway.59,60,61,62 Therefore, it is possible that the compartmentalization of the commensals within the gut lumen is compromised in mucosal immune-deficient mice. Mice deficient in IgA antibody production have been shown to have increased systemic antibodies directed against the microbiota, thus implying that bacterial components can be exposed to the systemic immune system.12,59 Previous work has implicated the important role of mucosal lymphoid tissues in containing and regulating the colonic commensal microbiota to prevent inflammation, specifically Th17 immunity.45,63,64,65,66 Despite the fact that we were unable to consistently detect bacteria in systemic tissue from Ltbr −/− mice using the culture methods described herein, it still remains to be determined whether systemic bacteria can be detected and found through the utilization of other culture methods.
Previous research has shown that IL-22 plays an important role in preventing splenomegaly in an ILC3- and microbiota-dependent manner.7,45,50 The IL-22RA pathway has been implicated in the establishment of the microbiota and mucosal homeostasis that prevents dissemination of commensal bacteria during colonic inflammation.67 It has also been demonstrated that the LTβR pathway upregulates IL-22 and antimicrobial peptide expression in a bacterial colitis model, such as the Citrobacter rodentium infection model.13,35,68 Interestingly, SPF IL-22-deficient mice do not display abnormal pathologies compared to WT counterparts.69,70 Furthermore, following the transfer of LTα1β2-deficient ILC3s or LTα1β2-sufficient ILC3s into Ragγc −/− hosts, we did not see a significant difference in expression of the typical AMPs (RegIIIβ and RegIIIγ) associated with downstream IL-22-signaling pathway in the colonic tissues (data not shown). Thus, the current study raises the possibility that LT-signaling could have a dual role in mucosal immune homeostasis: one acting upstream of IL-22 signaling, and another independent of IL-22 signaling. Further experiments will be needed to determine the specific downstream implications of LT signaling that is needed to maintain basal mucosal immunity following initial microbiota exposure.
Recent work has implicated the role of MHC-II expression and signaling on ILC3s to mediate and prevent microbiota-induced splenomegaly.8,52 Based on our work, it appears that LT expression on ILC3s did not significantly alter MHC-II expression on ILC3s. On one hand, this observation could mean that LT signaling has a minimal impact on MHC-II signaling along the mucosal barrier. On the other hand, this raises the possibility that MHC-II signaling and response to the commensal microbiota could be upstream of LT-signaling and induction. This observation is especially interesting given the importance of lymphoid structures and MHC-II signaling in various immune diseases, from autoimmunity to cancer immunology.71 Previous work has shown that LT/LIGHT can indirectly affect MHC-II signaling to promote the prevalence of various autoimmune diseases.72,73,74 However, it remains to be seen whether defects in MHC-II signaling could also indirectly affect LT signaling along the mucosal barrier, and if so, by which mechanism and in which disease states.
Lastly, ILC3s have been demonstrated to play an instrumental role in modulating systemic and colonic inflammation during microbial exposure.8,75 Since cryptopatches, comprising mainly ILC3s, differentiate into isolated lymphoid follicles only in the presence of the commensal microbiota, it is believed that microbial ligands can activate ILC3s.8,76,77 Proper activation of ILC3s is needed to prevent systemic inflammation and the development of splenomegaly.7,50,51,78,79,80 The current study suggests that the maturation/differentiation of ILC3s is dependent on LT signaling and the commensal microbiota. Thus, LT signaling not only has a role in the formation of isolated lymphoid follicles and colonic follicles but is also needed to regulate the microbial signals received from the commensal bacteria. This inability to modulate the commensal bacteria results in the development of systemic inflammation and splenomegaly.
Electronic supplementary material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Dr Cathy Nagler from University of Chicago for insightful discussions. We thank Andrea Crawford and Dr Betty Theriault from Gnotobiotic Research Animal Facility at University of Chicago for technical assistance with our germ-free mouse colony.
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Supplementary Information for this article can be found on the Cellular & Molecular Immunology website 10.1038/cmi.2017.25
References
- 1.Consortium THMP Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol. 2010;10:159–169. doi: 10.1038/nri2710. [DOI] [PubMed] [Google Scholar]
- 3.Smith K, McCoy KD, Macpherson AJ. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin Immunol. 2007;19:59–69. doi: 10.1016/j.smim.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Eberl G, Marmon S, Sunshine M-J, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol. 2004;5:64–73. doi: 10.1038/ni1022. [DOI] [PubMed] [Google Scholar]
- 5.Sonnenberg GF, Artis D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Med. 2015;21:698–708. doi: 10.1038/nm.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sonnenberg GF. Regulation of intestinal health and disease by innate lymphoid cells. Int Immunol. 2014;26:501–507. doi: 10.1093/intimm/dxu052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sawa S, Lochner M, Satoh-Takayama N, Dulauroy S, Bérard M, Kleinschek M, et al. RORγt+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol. 2011;12:320–326. doi: 10.1038/ni.2002. [DOI] [PubMed] [Google Scholar]
- 8.Hepworth MR, Fung TC, Masur SH, Kelsen JR, McConnell FM, Dubrot J, et al. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4+ T cells. Science. 2015;348:1031–1035. doi: 10.1126/science.aaa4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berger DP, Naniche D, Crowley MT, Koni PA, Flavell RA, Oldstone MBA. Lymphotoxin-β-deficient mice show defective antiviral immunity. Virology. 1999;260:136–147. doi: 10.1006/viro.1999.9811. [DOI] [PubMed] [Google Scholar]
- 10.GeurtsvanKessel CH, Willart MAM, Bergen IM, van Rijt LS, Muskens F, Elewaut D, et al. Dendritic cells are crucial for maintenance of tertiary lymphoid structures in the lung of influenza virus-infected mice. J Exp Med. 2009;206:2339–2349. doi: 10.1084/jem.20090410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar V, Scandella E, Danuser R, Onder L, Nitschké M, Fukui Y, et al. Global lymphoid tissue remodeling during a viral infection is orchestrated by a B cell-lymphotoxin-dependent pathway. Blood. 2010;115:4725–4733. doi: 10.1182/blood-2009-10-250118. [DOI] [PubMed] [Google Scholar]
- 12.Macpherson AJ, Gatto D, Sainsbury E, Harriman GR, Hengartner H, Zinkernagel RM. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science. 2000;288:2222–2226. doi: 10.1126/science.288.5474.2222. [DOI] [PubMed] [Google Scholar]
- 13.Ota N, Wong K, Valdez PA, Zheng Y, Crellin NK, Diehl L, et al. IL-22 bridges the lymphotoxin pathway with the maintenance of colonic lymphoid structures during infection with Citrobacter rodentium. Nat Immunol. 2011;12:941–948. doi: 10.1038/ni.2089. [DOI] [PubMed] [Google Scholar]
- 14.Tumanov AV, Kuprash DV, Lagarkova MA, Grivennikov SI, Abe K, Shakhov AN, et al. Distinct role of surface lymphotoxin expressed by B cells in the organization of secondary lymphoid tissues. Immunity. 2002;17:239–250. doi: 10.1016/S1074-7613(02)00397-7. [DOI] [PubMed] [Google Scholar]
- 15.Upadhyay V, Fu Y-X. Lymphotoxin signalling in immune homeostasis and the control of microorganisms. Nat Rev Immunol. 2013;13:270–279. doi: 10.1038/nri3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ware CF, VanArsdale TL, Crowe PD, Browning JL. The ligands and receptors of the lymphotoxin system. Curr Top Microbiol Immunol. 1995;198:175–218. doi: 10.1007/978-3-642-79414-8_11. [DOI] [PubMed] [Google Scholar]
- 17.Alimzhanov MB, Kuprash DV, Kosco-Vilbois MH, Luz A, Turetskaya RL, Tarakhovsky A, et al. Abnormal development of secondary lymphoid tissues in lymphotoxin β-deficient mice. Proc Natl Acad Sci USA. 1997;94:9302–9307. doi: 10.1073/pnas.94.17.9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Togni P, Goellner J, Ruddle NH, Streeter PR, Fick A, Mariathasan S, et al. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science. 1994;264:703–707. doi: 10.1126/science.8171322. [DOI] [PubMed] [Google Scholar]
- 19.Fütterer A, Mink K, Luz A, Kosco-Vilbois MH, Pfeffer K. The lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity. 1998;9:59–70. doi: 10.1016/S1074-7613(00)80588-9. [DOI] [PubMed] [Google Scholar]
- 20.Mariathasan S, Matsumoto M, Baranyay F, Nahm MH, Kanagawa O, Chaplin DD. Absence of lymph nodes in lymphotoxin-alpha(LT alpha)-deficient mice is due to abnormal organ development, not defective lymphocyte migration. J Inflamm. 1995;45:72–78. [PubMed] [Google Scholar]
- 21.Tumanov AV, Grivennikov SI, Shakhov AN, Rybtsov SA, Koroleva EP, Takeda J, et al. Dissecting the role of lymphotoxin in lymphoid organs by conditional targeting. Immunol Rev. 2003;195:106–116. doi: 10.1034/j.1600-065X.2003.00071.x. [DOI] [PubMed] [Google Scholar]
- 22.Onder L, Danuser R, Scandella E, Firner S, Chai Q, Hehlgans T, et al. Endothelial cell-specific lymphotoxin-β receptor signaling is critical for lymph node and high endothelial venule formation. J Exp Med. 2013;210:465–473. doi: 10.1084/jem.20121462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu Y-X, Chaplin DD. Development and maturation of secondary lymphoid tissues. Annu Rev Immunol. 1999;17:399–433. doi: 10.1146/annurev.immunol.17.1.399. [DOI] [PubMed] [Google Scholar]
- 24.van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol. 2010;10:664–674. doi: 10.1038/nri2832. [DOI] [PubMed] [Google Scholar]
- 25.Mullighan CG, Fanning GC, Chapel HM, Welsh KI. TNF and lymphotoxin-alpha polymorphisms associated with common variable immunodeficiency: role in the pathogenesis of granulomatous disease. J Immunol. 1997;159:6236–6241. [PubMed] [Google Scholar]
- 26.Richard AC, Peters JE, Lee JC, Vahedi G, Schäffer AA, Siegel RM et al. Targeted genomic analysis reveals widespread autoimmune disease association with regulatory variants in the TNF superfamily cytokine signalling network. Genome Med 2016 [Internet]. 2016 Jul 19 (cited 2016 Aug 8);8. Available at http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4952362/. [DOI] [PMC free article] [PubMed]
- 27.Mackay F, Majeau GR, Lawton P, Hochman PS, Browning JL. Lymphotoxin but not tumor necrosis factor functions to maintain splenic architecture and humoral responsiveness in adult mice. Eur J Immunol. 1997;27:2033–2042. doi: 10.1002/eji.1830270830. [DOI] [PubMed] [Google Scholar]
- 28.Kang H-S, Blink SE, Chin RK, Lee Y, Kim O, Weinstock J, et al. Lymphotoxin Is required for maintaining physiological levels of serum IgE that minimizes Th1-mediated airway inflammation. J Exp Med. 2003;198:1643–1652. doi: 10.1084/jem.20021784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spahn TW, Müller MK, Domschke W, Kucharzik T. Role of lymphotoxins in the development of Peyer’s patches and mesenteric lymph nodes. Ann NY Acad Sci. 2006;1072:187–193. doi: 10.1196/annals.1326.029. [DOI] [PubMed] [Google Scholar]
- 30.Zhu M, Yang Y, Wang Y, Wang Z, Fu Y-X. LIGHT regulates inflamed draining lymph node hypertrophy. J Immunol. 2011;186:7156–7163. doi: 10.4049/jimmunol.1002097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Browning JL, Allaire N, Ngam-Ek A, Notidis E, Hunt J, Perrin S, et al. Lymphotoxin-beta receptor signaling is required for the homeostatic control of HEV differentiation and function. Immunity. 2005;23:539–550. doi: 10.1016/j.immuni.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 32.Chyou S, Ekland EH, Carpenter AC, Tzeng T-CJ, Tian S, Michaud M, et al. Fibroblast-type reticular stromal cells regulate the lymph node vasculature. J Immunol. 2008;181:3887–3896. doi: 10.4049/jimmunol.181.6.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liao S, Ruddle NH. Synchrony of High Endothelial venules and lymphatic vessels revealed by immunization. J Immunol. 2006;177:3369–3379. doi: 10.4049/jimmunol.177.5.3369. [DOI] [PubMed] [Google Scholar]
- 34.Scheu S, Alferink J, Pötzel T, Barchet W, Kalinke U, Pfeffer K. Targeted disruption of LIGHT causes defects in costimulatory T cell activation and reveals cooperation with lymphotoxin beta in mesenteric lymph node genesis. J Exp Med. 2002;195:1613–1624. doi: 10.1084/jem.20020215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tumanov AV, Koroleva EP, Guo X, Wang Y, Kruglov A, Nedospasov S, et al. Lymphotoxin controls the IL-22 protection pathway in gut innate lymphoid cells during mucosal pathogen challenge. Cell Host Microbe. 2011;10:44–53. doi: 10.1016/j.chom.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Koroleva EP, Kruglov AA, Kuprash DV, Nedospasov SA, Fu Y-X, et al. Lymphotoxin beta receptor signaling in intestinal epithelial cells orchestrates innate immune responses against mucosal bacterial infection. Immunity. 2010;32:403–413. doi: 10.1016/j.immuni.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DiSanto JP, Müller W, Guy-Grand D, Fischer A, Rajewsky K. Lymphoid development in mice with a targeted deletion of the interleukin 2 receptor gamma chain. Proc Natl Acad Sci USA. 1995;92:377–381. doi: 10.1073/pnas.92.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by Toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 39.Guo X, Qiu J, Tu T, Yang X, Deng L, Anders RA, et al. Induction of innate lymphoid cell-derived interleukin-22 by the transcription factor STAT3 mediates protection against intestinal infection. Immunity. 2014;40:25–39. doi: 10.1016/j.immuni.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo X, Muite K, Wroblewska J, Fu Y-XPurification and adoptive transfer of group 3 gut innate lymphoid cellsIn:Ivanov AI(ed.). Gastrointestinal Physiology and Diseases. Springer: New York, NY, USA. 2016 pp 189–196. [DOI] [PubMed]
- 41.Wu Q, Wang Y, Wang J, Hedgeman EO, Browning JL, Fu Y-X. The requirement of membrane lymphotoxin for the presence of dendritic cells in lymphoid tissues. J Exp Med. 1999;190:629–638. doi: 10.1084/jem.190.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Browning JL, Sizing ID, Lawton P, Bourdon PR, Rennert PD, Majeau GR, et al. Characterization of lymphotoxin-alpha beta complexes on the surface of mouse lymphocytes. J Immunol. 1997;159:3288–3298. [PubMed] [Google Scholar]
- 43.Johansson MEV, Jakobsson HE, Holmén-Larsson J, Schütte A, Ermund A, Rodríguez-Piñeiro AM, et al. Normalization of host intestinal mucus layers requires long-term microbial colonization. Cell Host Microbe. 2015;18:582–592. doi: 10.1016/j.chom.2015.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Costello EK, Stagaman K, Dethlefsen L, Bohannan BJM, Relman DA. The application of ecological theory toward an understanding of the human microbiome. Science. 2012;336:1255–1262. doi: 10.1126/science.1224203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science. 2012;336:1321–1325. doi: 10.1126/science.1222551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee J-Y, Ristow M, Lin X, White MF, Magnuson MA, Hennighausen L. RIP-Cre revisited, evidence for impairments of pancreatic β-cell function. J Biol Chem. 2006;281:2649–2653. doi: 10.1074/jbc.M512373200. [DOI] [PubMed] [Google Scholar]
- 47.Smith L. Good planning and serendipity: exploiting the Cre/Lox system in the testis. Reproduction. 2011;141:151–161. doi: 10.1530/REP-10-0404. [DOI] [PubMed] [Google Scholar]
- 48.Garcia S, DiSanto J, Stockinger B. Following the development of a CD4 T cell response in vivo. Immunity. 1999;11:163–171. doi: 10.1016/S1074-7613(00)80091-6. [DOI] [PubMed] [Google Scholar]
- 49.Greenberg PD, Riddell SR. Deficient Cellular Immunity—finding and fixing the defects. Science. 1999;285:546–551. doi: 10.1126/science.285.5427.546. [DOI] [PubMed] [Google Scholar]
- 50.Garidou L, Pomié C, Klopp P, Waget A, Charpentier J, Aloulou M, et al. The gut microbiota regulates intestinal CD4 T cells expressing RORγt and controls metabolic disease. Cell Metab. 2015;22:100–112. doi: 10.1016/j.cmet.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 51.Zhang N, Guo J, He Y-W. Lymphocyte accumulation in the spleen of retinoic acid receptor-related orphan receptor gamma-deficient mice. J Immunol. 2003;171:1667–1675. doi: 10.4049/jimmunol.171.4.1667. [DOI] [PubMed] [Google Scholar]
- 52.Hepworth MR, Monticelli LA, Fung TC, Ziegler CGK, Grunberg S, Sinha R, et al. Innate lymphoid cells regulate CD4+ T cell responses to intestinal commensal bacteria. Nature. 2013;498:113–117. doi: 10.1038/nature12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ehlers S, Holscher C, Scheu S, Tertilt C, Hehlgans T, Suwinski J, et al. The Lymphotoxin Receptor Is Critically Involved in Controlling Infections with the Intracellular Pathogens Mycobacterium tuberculosis and Listeria monocytogenes. J Immunol. 2003;170:5210–5218. doi: 10.4049/jimmunol.170.10.5210. [DOI] [PubMed] [Google Scholar]
- 54.Hu D, Mohanta SK, Yin C, Peng L, Ma Z, Srikakulapu P, et al. Artery tertiary lymphoid organs control aorta immunity and protect against atherosclerosis via vascular smooth muscle cell lymphotoxin β receptors. Immunity. 2015;42:1100–1115. doi: 10.1016/j.immuni.2015.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schneider K, Loewendorf A, De Trez C, Fulton J, Rhode A, Shumway H, et al. Lymphotoxin-mediated crosstalk between B cells and splenic stroma promotes the initial type I interferon response to cytomegalovirus. Cell Host Microbe. 2008;3:67–76. doi: 10.1016/j.chom.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun T, Rojas OL, Li C, Philpott DJ, Gommerman JL. Hematopoietic LTβR deficiency results in skewed T cell cytokine profiles during a mucosal viral infection. J Leukoc Biol. 2016;100:103–110. doi: 10.1189/jlb.4MAB0715-294R. [DOI] [PubMed] [Google Scholar]
- 57.Spahn TW, Maaser C, Eckmann L, Heidemann J, Lügering A, Newberry R, et al. The lymphotoxin-β receptor is critical for control of murine Citrobacter rodentium-induced colitis. Gastroenterology. 2004;127:1463–1473. doi: 10.1053/j.gastro.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 58.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 59.Peterson DA, Planer JD, Guruge JL, Xue L, Downey-Virgin W, Goodman AL, et al. Characterizing the interactions between a naturally primed immunoglobulin A and its conserved Bacteroides thetaiotaomicron species-specific epitope in gnotobiotic mice. J Biol Chem. 2015;290:12630–12649. doi: 10.1074/jbc.M114.633800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deplancke B, Gaskins HR. Microbial modulation of innate defense: goblet cells and the intestinal mucus layer. Am J Clin Nutr. 2001;73:1131S–1141S. doi: 10.1093/ajcn/73.6.1131S. [DOI] [PubMed] [Google Scholar]
- 61.Loonen LMP, Stolte EH, Jaklofsky MTJ, Meijerink M, Dekker J, van Baarlen P, et al. REG3γ-deficient mice have altered mucus distribution and increased mucosal inflammatory responses to the microbiota and enteric pathogens in the ileum. Mucosal Immunol. 2014;7:939–947. doi: 10.1038/mi.2013.109. [DOI] [PubMed] [Google Scholar]
- 62.Specian RD, Oliver MG. Functional biology of intestinal goblet cells. Am J Physiol. 1991;260:C183–C193. doi: 10.1152/ajpcell.1991.260.2.C183. [DOI] [PubMed] [Google Scholar]
- 63.Fung TC, Bessman NJ, Hepworth MR, Kumar N, Shibata N, Kobuley D, et al. Lymphoid-tissue-resident commensal bacteria promote members of the IL-10 cytokine family to establish mutualism. Immunity. 2016;44:634–646. doi: 10.1016/j.immuni.2016.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Withers DR, Hepworth MR, Wang X, Mackley EC, Halford EE, Dutton EE, et al. Transient inhibition of ROR-γt therapeutically limits intestinal inflammation by reducing TH17 cells and preserving group 3 innate lymphoid cells. Nat Med. 2016;22:319–323. doi: 10.1038/nm.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sonnenberg GF, Monticelli LA, Elloso MM, Fouser LA, Artis D. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity. 2011;34:122–134. doi: 10.1016/j.immuni.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sonnenberg GF, Fouser LA, Artis D. Functional biology of the IL-22-IL-22R pathway in regulating immunity and inflammation at barrier surfaces. Adv Immunol. 2010;107:1–29. doi: 10.1016/B978-0-12-381300-8.00001-0. [DOI] [PubMed] [Google Scholar]
- 67.Pham TAN, Clare S, Goulding D, Arasteh JM, Stares MD, Browne HP, et al. Epithelial IL-22RA1-mediated fucosylation promotes intestinal colonization resistance to an opportunistic pathogen. Cell Host Microbe. 2014;16:504–516. doi: 10.1016/j.chom.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Satoh-Takayama N, Lesjean-Pottier S, Sawa S, Vosshenrich CAJ, Eberl G, Di Santo JP. Lymphotoxin-β receptor-independent development of intestinal IL-22-producing NKp46+ innate lymphoid cells. Eur J Immunol. 2011;41:780–786. doi: 10.1002/eji.201040851. [DOI] [PubMed] [Google Scholar]
- 69.Zenewicz LA, Yin X, Wang G, Elinav E, Hao L, Zhao L, et al. IL-22 Deficiency alters colonic microbiota to be transmissible and colitogenic. J Immunol. 2013;190:5306–5312. doi: 10.4049/jimmunol.1300016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity. 2008;29:947–957. doi: 10.1016/j.immuni.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tang H, Zhu M, Qiao J, Fu Y-X. Lymphotoxin signalling in tertiary lymphoid structures and immunotherapy. Cell Mol Immunol. 2017 doi: 10.1038/cmi.2017.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McHugh NJ, Owen P, Cox B, Dunphy J, Welsh K. MHC class II, tumour necrosis factor alpha, and lymphotoxin alpha gene haplotype associations with serological subsets of systemic lupus erythematosus. Ann Rheum Dis. 2006;65:488–494. doi: 10.1136/ard.2005.039842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stemme S, Fager G, Hansson GK. MHC class II antigen expression in human vascular smooth muscle cells is induced by interferon-gamma and modulated by tumour necrosis factor and lymphotoxin. Immunology. 1990;69:243–249. [PMC free article] [PubMed] [Google Scholar]
- 74.Wang J, Fu Y-X. LIGHT (a cellular ligand for herpes virus entry mediator and lymphotoxin receptor)-mediated thymocyte deletion is dependent on the interaction between TCR and MHC/self-peptide. J Immunol. 2003;170:3986–3993. doi: 10.4049/jimmunol.170.8.3986. [DOI] [PubMed] [Google Scholar]
- 75.Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol201112: 383–390. [DOI] [PubMed]
- 76.Lorenz RG, Chaplin DD, McDonald KG, McDonough JS, Newberry RD. Isolated lymphoid follicle formation is inducible and dependent upon lymphotoxin-sufficient b lymphocytes, lymphotoxin β receptor, and TNF receptor I function. J Immunol. 2003;170:5475–5482. doi: 10.4049/jimmunol.170.11.5475. [DOI] [PubMed] [Google Scholar]
- 77.Giacomin PR, Moy RH, Noti M, Osborne LC, Siracusa MC, Alenghat T, et al. Epithelial-intrinsic IKK expression regulates group 3 innate lymphoid cell responses and antibacterial immunity. J Exp Med. 2015;212:1513–1528. doi: 10.1084/jem.20141831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mortha A, Chudnovskiy A, Hashimoto D, Bogunovic M, Spencer SP, Belkaid Y, et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science. 2014;343:1249288. doi: 10.1126/science.1249288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Qiu J, Guo X, Chen Z-ME, He L, Sonnenberg GF, Artis D, et al. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity. 2013;39:386–399. doi: 10.1016/j.immuni.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sanos SL, Vonarbourg C, Mortha A, Diefenbach A. Control of epithelial cell function by interleukin-22-producing RORγt+ innate lymphoid cells. Immunology. 2011;132:453–465. doi: 10.1111/j.1365-2567.2011.03410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.