

Abstract
The withdrawal of pharmaceutical companies from developing drugs against Alzheimer's highlights the inherent difficulties in tackling this devastating disease. In the meantime, biotech companies and basic research explore new targets and drug combinations.

Subject Categories: Molecular Biology of Disease, Neuroscience, S&S: Health & Disease
A diagnosis of Alzheimer's disease (AD) is a devastating experience, both for patients and their families. The most common type of dementia, AD, slowly builds up in the brain, creeping in the parts that control memory, thought, and language, and ultimately destroys the patient's ability to interact with the human and physical environment. About 50 million people in the world suffer from dementia, and the number is set to triplicate by 2050 (Fig 1). As the risk of AD rapidly increases beyond the age of 65, and as the human population is aging, much of the forecasted increase of people with dementia will take place in low‐ and middle‐income countries, according to the World Health Organization (Fig 1).
Figure 1. Dementia, a public health priority.

WHO infographic about the symptoms of dementia, the cause, the number of people affected and its cost.Source: World Health Organization. Reproduced with permission.
Given the increasing prevalence and the severity of AD, enormous financial and human resources—both public and private—have been devoted to finding a cure; in addition, a successful drug would reap immense revenues. So far, however, scientists still do not fully understand the causes of the disease, and treatment is mainly focused at slowing or delaying symptoms to help patients maintain their mental functions.
Challenges and failures
The hallmark of AD is the accumulation of extracellular plaques from amyloid‐β (Aβ) protein—which is derived from a membrane protein called amyloid precursor protein (APP)—and of intracellular neurofibrillary tangles of tau protein in brain cells (Fig 2). These aggregates gradually lead to neuron loss, synaptic dysfunction, and progressive cognitive degeneration (Fig 3). This so‐called amyloid cascade hypothesis has been the reference point for most research and drug development. Most attempts to slow the progression of the disease or to revert its effects target the amyloid‐β and tau proteins to prevent the formation of fibrils and plaques—but with no tangible results so far.
Figure 2. Molecular architecture of amyloid plaques and neurofibrillary tangles in Alzheimer's disease.
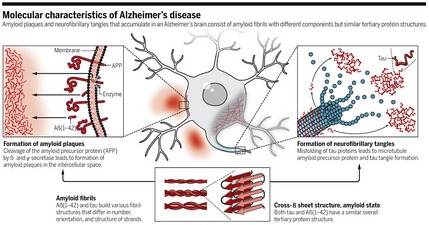
Reproduced from 1, with permission.
Figure 3. The sequence of major pathogenic events leading to AD proposed by the amyloid cascade hypothesis.
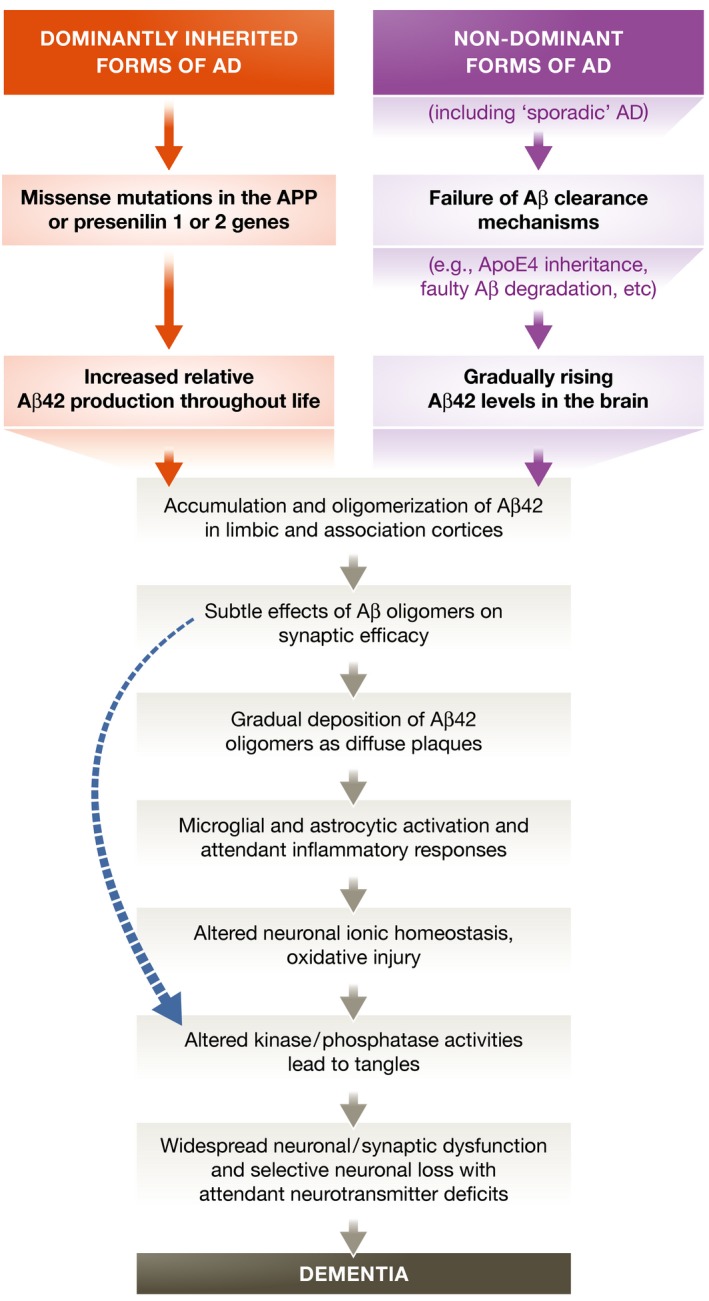
The curved blue arrow indicates that Aβ oligomers may directly injure the synapses and neurites of brain neurons, in addition to activating microglia and astrocytes. Reproduced from 2, with permission.
[W]hen it comes to treating AD, the per se difficult drug‐discovery enterprise becomes a titanic challenge
Indeed, when it comes to treating AD, the per se difficult drug discovery enterprise becomes a titanic challenge. Any candidate must be able to cross the blood–brain barrier, without exerting any toxicity to normal nerve cells while breaking down or preventing Aβ plaques and tau tangles. These plaques accumulate over decades, which make it difficult to identify patients with early‐stage AD who might benefit from therapies that prevent further damage and loss of neurons. AD diagnosis is not straightforward either, as symptoms overlap with other conditions that affect cognition. As a result, trials are difficult to organize and extremely expensive. The last Alzheimer's approved drug was memantine in 2003, and it is only used for symptomatic treatment; since then some 400 clinical trials have failed.
In January this year, US pharmaceutical giant Pfizer announced that it abandons its efforts to develop drugs to treat Alzheimer's and Parkinson's disease. “This was an exercise to re‐allocate spend across our portfolio, to focus on those areas where our pipeline, and our scientific expertise, is strongest”, the company explained in the statement (https://www.reuters.com/article/us-pfizer-alzheimers/pfizer-ends-research-for-newalzheimers-parkinsons-drugs-idUSKBN1EW0TN). This decision came after years of massive investment—including the sponsorship of a specialist venture capital fund called Dementia Discovery Fund (http://theddfund.com/)—and disappointing results.
Pfizer is not abandoning neurological diseases altogether though. It still pursues its research programs on rare neurological diseases, such as transthyretin amyloidosis, a condition characterized by the progressive buildup of amyloid deposits in several organs and tissues, including the peripheral and central nervous systems. Development of chronic pain treatment tanezumab, a monoclonal antibody targeting nerve growth factor, that is currently in phase 3, will also continue.
In January this year, US pharmaceutical giant Pfizer announced that it abandons its efforts to develop drugs to treat Alzheimer's and Parkinson's disease.
Within days after Pfizer's announcement, Axovant Sciences published negative results for its clinical trial of intepirdine to treat dementia with Lewy bodies, a progressive neurodegenerative disorder characterized by the abnormal deposits of a protein called alpha‐synuclein (http://investors.axovant.com/news-releases/news-release-details/axovant-announces-negative-results-intepirdine-phase-2b-headway), and JAMA published findings showing that idalopirdine, a selective 5‐hydroxytryptamine‐6 receptor antagonist, was not able to improve cognitive change in patients with mild to moderate Alzheimer in three clinical trials 3. At the same time, Eli Lilly published the last round of data that brought it to abandon development of solanezumab, a monoclonal antibody designed to target β‐amyloid protein, that failed to improve cognition in patients with mild Alzheimer's disease 4.
New players stepping in
These setbacks came at a time when other key players pledged renewed efforts to find a cure against dementia. In November last year, Bill Gates invested US$100 million in the Dementia Discovery Fund, motivated by the fact that “men in my family have suffered from Alzheimer's”, (https://www.gatesnotes.com/Health/Digging-Deep-Into-Alzheimers). “The longer you live the more likely you are to develop a chronic condition. Your risk of getting arthritis, Parkinson's or another non‐infectious disease that diminishes your quality of life increases with each year. But of all the disorders that plague us late in life one stands out as a particularly big threat to society: Alzheimer's disease”, Gates noted.
The World Health Organization also now recognizes dementia as a public health priority. In May 2017, the World Health Assembly endorsed the Global Action Plan on the public health response to dementia 2017–2025. The Plan is intended as a comprehensive blueprint for action, to guide policymakers and WHO partners and members in areas such as increasing awareness of dementia and support for dementia caretakers, and strengthening policies and service planning. Later the same year, WHO launched the Global Dementia Observatory, “a data and knowledge exchange platform that offers easy access to key dementia data from Member States across the following three domains: policies, service delivery, and information and research”, (http://www.who.int/mental_health/neurology/dementia/Global_Observatory/en/).
Biotech picking up
“In view of the current failures of big pharma to conduct well thought‐through, modern and state‐of‐the‐art drug trials in the field of AD, I think the future lies in the hand of small biotech companies that work closely together with academic frontrunners and key opinion leaders in designing and carrying out proof of concept trials with new compounds that may or not may target Aβ or tau pathways”, said Philip Scheltens at the Alzheimer Center, VU University Amsterdam in the Netherlands. “Governments should invest much more than before in drug development themselves together with academia, instead of leaving it all to pharma”.
Indeed, while large drug companies are somehow retreating from dementia research, various startup companies have stepped up to the challenge by pursuing avenues that diverge or are collateral to the mainstream Aβ/tau hypothesis. And, surprisingly, big venture investment is flowing into these companies.
Japanese pharmaceutical company Takeda recently signed a US$1 billion deal with San Francisco‐based biotech Denali that has a preclinical pipeline of drugs for treating AD. At the heart of the Denali's strategy lies rigorous genetic definition of the patient population and the identification of specific biomarkers to monitor drug effects. The platform technology Denali has developed hinges on antibody transport vehicles to help drug candidates penetrate the blood–brain barrier and deliver sufficient concentration of the drug to the brain. Two of the running Alzheimer's programs target BACE1—an aspartic protease that initiates the formation of Aβ from APP—and TREM2, an immune receptor found in brain microglia that has been linked to the risk of developing Alzheimer's.
… various startup companies have stepped up to the challenge by pursuing avenues that diverge or are collateral to the mainstream Aβ/tau hypothesis
Moreover, a recent study resulting from a collaboration between Denali and Washington University School of Medicine in St Louis, MS, USA, showed that an antibody targeting the apoE protein found in brain plaques cuts the level of plaques by half, raising hopes that this could limit the debilitating symptoms of the disease 5. The researchers expressed the human APOE gene—variants of which are considered the most significant risk factor for AD—in APOE‐knocked mice. Intriguingly, the apoE‐directed antibody, HAE‐4, did not bind to the product of the gene found in the blood, where it is involved in the transport of lipids and cholesterol.
A different strategy is pursued by Rodin Therapeutics, a biotech firm based in Cambridge, MA, USA. Their main idea is “synaptic resilience through targeted epigenetics”, that is, trying to restore synaptic function in degenerative brain diseases through modulation of histone deacetylases (HDAC) with tailored, proprietary molecules. In particular, Rodin is targeting HDAC2, that when bound to transcription factor Sp3, is known to negatively regulate synaptic function in neurons and blocks memory‐related gene expression. A recent study has shown that inhibition of HDAC2‐Sp3 binding ameliorates memory deficit in a mouse model of AD 6.
Rodin is not the only player to believe that epigenetics could be a key mechanism for treating AD. In April this year, biotech Oryzon Genomics, based in Barcelona, Spain, received approval to start a phase IIa clinical trial. Their candidate compound ORY‐2001 is designed to selectively inhibit Lysine‐specific histone demethylase 1 (LSD1), a histone‐modifying enzyme that is an important regulator of neural stem cell proliferation and neuronal development, and monoamine oxidase B (MAO‐B), the levels of which are elevated in AD brain and thought to regulate Aβ production in neurons via γ‐secretase (https://www.oryzon.com/sites/default/files/PRESS_RELEASE_11-2018.pdf).
Probiodrug, based in Halle, Germany, is another biotech that is trying to tackle AD from a different angle. The target here is not Aβ itself, but rather pyroglutamate‐Aβ, which has enhanced aggregation potential and propensity to form toxic oligomers. Probiodrug's lead candidate PQ912, now in phase 2b trials, is an inhibitor of glutaminyl cyclase that catalyzes pyroglutamate formation. Preclinical studies have also shown that another molecule, a pyroglutamate‐Aβ‐specific antibody, reduces plaques and improves cognition in a mouse model.
Exploring alternative targets
An increasing number of researchers are also walking off the beaten track to reach the promised land of AD therapeutics [see Box 1]. For instance, the finding that mitochondrial dysfunction is an early event in AD pathogenesis offers new clues to understand what makes neuronal cells go haywire. The mitochondrial membrane protein VDAC1 (voltage‐dependent anion channel 1), in particular, has been repeatedly linked to AD, given its role in calcium homeostasis, oxidative stress, and mitochondria‐mediated apoptosis 7. A related, intriguing observation is the inverse association between cancer and AD, namely that AD patients have lower rates of cancer and, conversely, cancer survivors experience a decreased risk of AD.
A related, intriguing observation is the inverse association between cancer and AD, namely that AD patients have lower rates of cancer and, conversely, cancer survivors experience a decreased risk of AD
“With respect to the inverse association between cancer and Alzheimer's disease, metabolism can be the link. Highly metabolic activity in cancer and the opposite for AD‐associated cells, would result in different products of metabolism, with a different pattern of expression/repression of genes in a cell, leading to the different diseases”, commented Varda Shoshan‐Barmatz from Ben Gurion University, Beersheva, Israel. “I believe that as in cancer, where metabolism‐related studies make a comeback, Alzheimer's researchers will soon focus on unrevealing the network of metabolism pathway to find out what goes awry in Alzheimer's diseases”.
While there is a relative scarcity of therapeutics in clinical development, basic and preclinical research continues to grow driven by the increasing prevalence of neurodegenerative diseases. In total, more than 100 agents (including symptomatic agents) are at the drug‐testing stage, with a wide range of mechanisms of action even if the main focus is still on anti‐amyloid or anti‐tau agents, especially for what concerns the leads in phase III clinical trials (Fig 4).
Figure 4. Alzheimer's disease drug development pipeline.
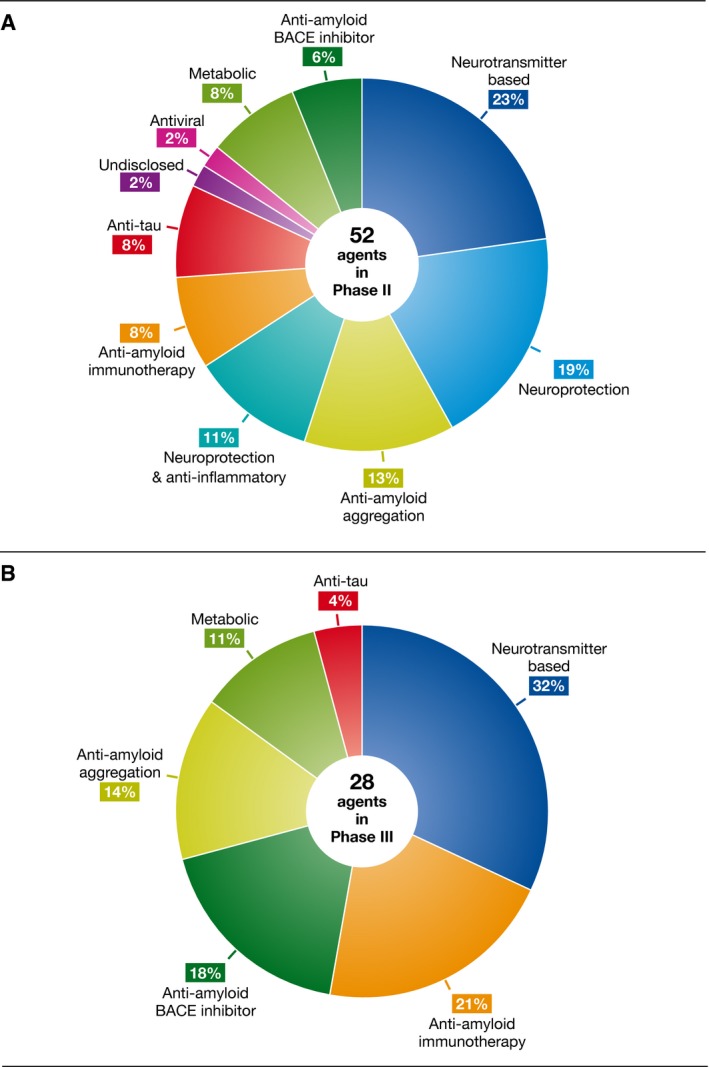
Mechanisms of action of agents in phase II (A) and phase III (B). BACE, β‐site amyloid precursor protein cleaving enzyme. Original data were retrieved from http://www.clinicaltrials.gov. Reproduced from 8, with permission.
Nonetheless, as most trials so far have failed, the low success rate of drugs remains a major concern. “There are many factors contributing to the currently low rate of success of drug development for AD. The understanding of the biology of AD is incomplete, the emphasis on testing single therapies where combinations may be required, the few candidates entering phase I, the lack of predictive validity of animal models, lack of efficacy of candidate therapies, and the emergence of unacceptable side effects all limit successful treatment development”, commented Jeffrey Cummings and colleagues, in a recent review 8. In particular, the slow recruitment of patients for clinical trials is a critical point. Cummings and colleagues noted that, in many cases, the recruitment period is longer than the treatment period, and failure of meeting the recruitment goal is not rare 8.
Box 1. Tapping into regenerative medicine to heal AD brain.
Nearly all efforts to identify therapeutics for AD have just generated frustration and disappointment. While the currently prevailing emphasis is on preventing the neurodegeneration purportedly caused by the deposition of Aβ plaques and the formation of tau tangles, a growing number of researchers believe modulation of neurogenesis might hold the key to mend AD‐damaged brains.
“To tackle the current lack of effective drugs and the continuous clinical failures, we are called to shift from the prevailing drug discovery paradigm targeting pathogenesis to the one rather targeting the brain endogenous regenerative process”, said medicinal chemist Maria Laura Bolognesi at the University of Bologna, Italy, an expert in AD drug discovery. The concept focuses on stimulating the regeneration of neural stem cells and the activation of neuroprotective pathways through small molecules and nanomedicines. In regard to small molecules, two main approaches have been pursued: target‐based strategies to inhibit signaling pathways and proteins such as HDAC, kinases, and neutrophins, and cell‐based phenotypic screens of small‐molecule libraries 10.
Interesting case stories indeed support the view that neuroregeneration could untangle the Gordian knot of AD. Biotech Neuronascent has an orally administered molecule in preclinical stage, NNI‐362, that “significantly increased the number of neurons in a key memory region of the brain, which was associated with the actual reversal of memory impairment in elderly mice”, (http://www.neuronascent.com/r&d-alzheimers.htm). Another example is P7C3: Discovered about a decade ago by Andrew Pieper and colleagues at the University of Iowa, USA, the molecule is neuroprotective, preventing both newborn and mature neurons from cell death in animal models of neurodegenerative diseases. A recent study showed that a P7C3‐based compound helped maintain normal neurologic function in AD rats, even in the presence of the pathological traits, such as amyloid plaques, tau tangles, and neuroinflammation (https://medicine.uiowa.edu/content/saving-neurons-may-offer-new-approach-treating-alzheimer%E2%80%99s-disease-0).
“Although in their infancy, small molecules triggering neurogenesis and neuroprotection have huge therapeutic potential in counteracting neurodegeneration and showing disease‐modifying effects”, said Bolognesi. “Surely, we must proceed with caution until they prove effective and safe in patients, critically challenge ourselves and look at the question from all possible angles. However, as academic scientists, we should be willing to take the risk to invest in less conventional pharmaceutical tools, which, if successful, will have tremendous health benefits”.
Combination therapies
Given the evident difficulties in finalizing the canonical drug discovery process in AD, several key players across the field now believe combination therapy might offer new hopes. This approach involves the use of two or more treatments, each targeting one of the factors involved in AD initiation and progression: the accumulation of misfolded proteins, oxidative stress, or mitochondrial dysfunction 9. To spur researchers along this path, the Alzheimer's Association and Alzheimer's Drug Discovery Foundation recently partnered to create a new grant scheme, called the Alzheimer's Combination Therapy Opportunities (ACTO). With this initiative, the partners “[c]hallenge the research community to propose combinations of repurposed and/or repositioned drugs that have the potential to be disease‐modifying for Alzheimer's disease and are hypothesized to work synergistically to provide a large effect in slowing or reversing Alzheimer's disease progression”, (https://www.alzdiscovery.org/assets/content/static/ACTO_rfa_2016.pdf). Focusing on compounds that have already been tested for other conditions and found safe will eventually hasten delivery of the combination in the AD clinical practice, they believe.
The first combination therapy project funded under the new mechanism is the clinical trial of AMX0035, an oral formulation of sodium phenylbutyrate and tauroursodeoxycholic acid, developed by Amylyx Pharmaceuticals (http://amylyx.com/innovative-alzheimers-disease-combination-therapy-trial-supported-by-new-joint-funding-initiative/). Preclinical studies have shown that AMX0035 is able to block nerve cell death and neuroinflammation in models of Alzheimer's. Further, the combination will be shortly tested in a clinical trial as a treatment for amyotrophic lateral sclerosis.
“The main challenge to developing successful Alzheimer's combination therapies is that there currently are no disease modifying drugs for Alzheimer's disease that are approved by the US Food and Drug Administration. Therefore, we will need to combine experimental drugs that have not yet been shown effective as a mono‐therapy”, explained James Hendrix, Alzheimer's Association Director of Global Science Initiatives. “Second, very few companies have more than one drug candidate that they would be able to combine. As such, we may need to combine drugs from different, competing companies. This means there are business challenges as well as scientific challenges”.
Beyond amyloid and tau
Notwithstanding the recent attempts to look beyond amyloid and tau, these elements really define AD as a unique neurodegenerative disorder, and the quest for treatments cannot leave aside a better understanding of the interplay between the two. “While the recent failures are disheartening, we learn from each of these trials. With advances in neuroimaging, now allowing monitoring of amyloid, tau and neurodegeneration, in addition to bioinformatics and biotechnology, we are poised to discover new treatments for this complex disease”, commented Krista Lanctôt, at Sunnybrook Health Sciences Centre in Toronto, Canada, whose research focus is on the neurobiology of dementia.
Lanctôt pointed to the framework recently drafted by the US National Institute on Aging and Alzheimer's Association, that, she believes, will pave the way forward for a responsive research agenda based on biomarkers (https://www.alzheimersanddementia.com/article/S1552-5260(18)30072-4/fulltext). The document outlines the “ATN” biomarker classification that defines validated biomarkers for A‐amyloid beta, T‐tau, and N‐neurodegeneration. The underlying vision of the biomarker‐based research framework is that defining AD as a biological construct will permit a better characterization and understanding of the sequence of events that lead to cognitive impairment, enabling a more targeted research on pharmaceutical interventions. “The classification defines a third category, nonspecific neurodegeneration. These additional processes, though not specific to the diagnostic features of AD, likely play a substantial role and should not be underestimated”, Lanctôt added.
Given the evident difficulties in finalizing the canonical drug discovery process in AD, several key players across the field now believe combination therapy might offer new hopes.
Even with the most optimistic view, the road to an effective treatment for AD will be long and bumpy. However, given the number of sufferers at stake, giving up is not an option. When looking at a black future, the late Stephen Hawking commented: “black holes ain't as black as they are painted. They are not the eternal prisons they were once thought. Things can get out of a black hole both on the outside and possibly to another universe. So if you feel you are in a black hole, don't give up—there's a way out”.
EMBO Reports (2018) 19: e46714
References
- 1. Pospich S, Raunser S (2017) The molecular basis of Alzheimer's plaques. Science 358: 45–46 [DOI] [PubMed] [Google Scholar]
- 2. Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med 8: 595–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Atri A, Frölich L, Ballard C, Tariot PN, Molinuevo JL, Boneva N, Windfeld K, Raket LL, Cummings JL (2018) Effect of idalopirdine as adjunct to cholinesterase inhibitors on change in cognition in patients with Alzheimer disease: three randomized clinical trials. JAMA 319: 130–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Honig L, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, Hager K, Andreasen N, Scarpini E, Liu‐Seifert H et al (2018) Trial of Solanezumab for mild dementia due to Alzheimer's disease. N Engl J Med 378: 321–330 [DOI] [PubMed] [Google Scholar]
- 5. Liao F, Li A, Xiong M, Bien‐Ly N, Jiang H, Zhang Y, Finn MB, Hoyle R, Keyser J, Lefton KB et al (2018) Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J Clin Invest 128: 2144–2155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamakawa H, Cheng J, Penney J, Gao F, Rueda R, Wang J, Yamakawa S, Kritskiy O, Gjoneska E, Tsai LH (2017) The transcription factor Sp3 cooperates with HDAC2 to regulate synaptic function and plasticity in neurons. Cell Rep 20: 1319–1334 [DOI] [PubMed] [Google Scholar]
- 7. Shoshan‐Barmatz V, Nahon‐Crystal E, Shteinfer‐Kuzmine A, Gupta R (2018) VDAC1, mitochondrial dysfunction, and Alzheimer's disease. Pharmacol Res 131: 87–101 [DOI] [PubMed] [Google Scholar]
- 8. Cummings J, Lee G, Mortsdorf T, Ritter A, Zhong K (2017) Alzheimer's disease drug development pipeline: 2017. Alzheimers Dement 3: 367–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hendrix JA, Bateman RJ, Brashear HR, Duggan C, Carrillo MC, Bain LJ (2016) Challenges, solutions, and recommendations for Alzheimer's disease combination therapy. Alzheimers Dement 12: 623–630 [DOI] [PubMed] [Google Scholar]
- 10. Uliassi E, Gandini A, Perone RC, Bolognesi ML (2017) Neuroregeneration versus neurodegeneration: toward a paradigm shift in Alzheimer's disease drug discovery. Future Med Chem 9: 995–1013 [DOI] [PubMed] [Google Scholar]