

Abstract
Most Mage family members code for antigens on melanoma tumor cells. Maged1 is the black sheep, promiscuously found in normal adult cells, including neurons of the basal ganglia and prefrontal cortex. In this issue of EMBO reports, De Backer et al 1 propose an unexpected role for Maged1. Acute effects of cocaine, such as enhanced locomotion and reinforcement, are gone in mice in which the gene is deleted. In a painstaking combinatorial approach comparing several conditional gene knockout (KO) mouse lines, the authors parse the relevant neural circuits.
Subject Categories: Neuroscience
Addiction is a behavioral disease caused by maladaptive adaptations of neural activity in defined brain regions driven by the chronic consumption of a drug of abuse. While major advances in understanding these adaptations have led to the emergence of a circuit model of addiction 2, little is known about the genetic determinants. Rodent models can reproduce core components of addictive behavior and are therefore extremely useful to probe the genes and molecules involved. For example, the use of conditional gene knockout (KO) in mice highlights the importance of monoamine membrane transporters for cocaine reinforcement 3, and genome‐wide analyses in mice show that cocaine self‐administration triggers adaptive gene regulation in the nucleus accumbens (NAc), a brain region essential for reward‐seeking behaviors 4. However, the functional relevance of these genes for circuit adaptations and eventually drug addiction remains elusive. In this issue of EMBO reports, De Backer et al 1 provide evidence that Maged1 is necessary for neuronal adaptations in the NAc, which underlie enhanced locomotion and reinforcement in response to cocaine (Fig 1).
Figure 1. Maged1 KO mice are immune to cocaine locomotor and reinforcement properties.
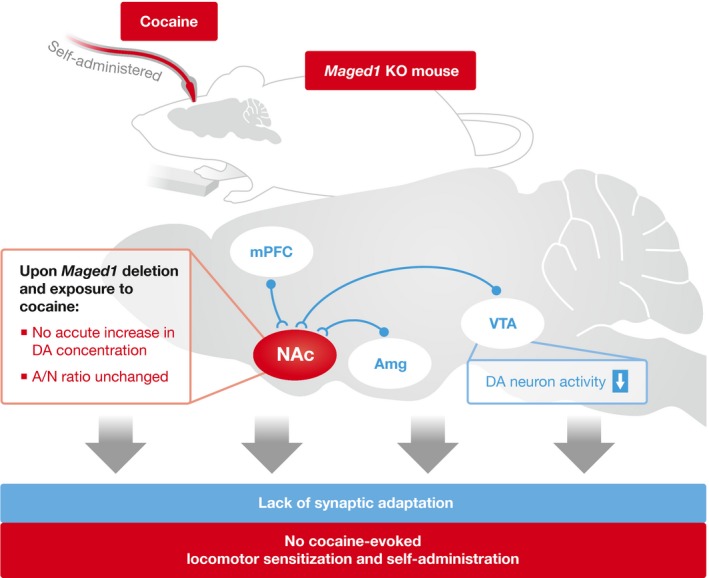
De Backer et al 1 characterize the contribution of Maged1 to drug addiction by using either constitutive knockout mice or site‐ and cell‐type specific deletion of Maged1. Genome‐wide Maged1 KO abolished drug‐induced synaptic plasticity and increased DA concentrations in the NAc after repeated exposure to cocaine. At the behavioral level, Maged1‐deleted mice showed an absence of cocaine‐induced locomotor sensitization and increased cocaine self‐administration. Furthermore, De Backer et al 1 found that Maged1 deletion specifically in the mPFC or Amg, but not in VTA DA neurons or the striatum, recapitulates the absence of cocaine's impact on locomotion.
Maged1, also known as NRAGE in humans, belongs to the type II Mage family, whose members are expressed in various tissues and have been implicated in neuronal development. Two reports suggest that Maged1 may also play a role in addiction. In the NAc of mice exposed to cocaine, Maged1 co‐precipitates with the transcription factor CREB (cAMP response element‐binding protein) implicated in addictive behaviors 4. Furthermore, Maged1 deletion alters memory formation 5, a cognitive function usurped by addictive drugs. To directly challenge the functional necessity of Maged1 for addictive behaviors, De Backer et al 1 tested the locomotor sensitization and reinforcement of cocaine in Maged1 KO mice. Maged1‐deleted mice failed to self‐administer cocaine and exhibited no locomotor sensitization after repeated exposure to cocaine.
Acute effects of cocaine […] are gone in mice in which Maged1 is deleted
Increase in striatal dopamine (DA) concentration is a defining initial effect of addictive drugs required for the induction of addiction 6. This surge in DA can arise from an increase in either the activity of DA neurons from the ventral tegmental area (VTA) or by blocking DA reuptake in striatal synapses. De Backer et al 1 pursued their investigation by monitoring DA concentration in the NAc and neuronal activity of DA neurons in the VTA of Maged1 KO mice before and after cocaine administration. While the activity of VTA DA neurons after cocaine exposure decreased through activation of autoreceptors, this was surprisingly not associated with an acute increase in accumbal DA concentration, suggesting a dissociation between the midbrain and the striatum. This latter result not only supports a fundamental role for Maged1 in neuronal adaptation linked to addictive behaviors but also confirms the pivotal role of dopamine for the onset of addiction.
A third important finding by De Backer et al 1 is an alteration of cocaine‐evoked synaptic plasticity in Maged1 KO mice. Indeed, addictive drugs are known to cause long‐lasting potentiation of excitatory synaptic transmission in NAc neurons expressing DA receptors 7. Current mechanistic models posit that this potentiation is due to a change in the subunit composition of glutamatergic AMPAR (α‐amino‐3‐hydroxy‐5‐methyl‐4‐ isoxazolepropionic acid receptors) or insertion of more receptors in NAc neurons. This interpretation is inferred from ex vivo neuronal recordings and computation of the ratio of AMPAR/NMDAR‐mediated postsynaptic current amplitudes, the so‐called A/N ratio. De Backer et al 1 found the expected increase in A/N ratio at cortico‐accumbal connections in wild‐type but not in Maged1 KO mice injected with cocaine. Thus, the impact of Maged1 deletion in mice on cocaine locomotor sensitization and self‐administration can now be explained by the absence of synaptic adaptation of circuits that converge in the NAc.
To parse the relevant afferents connections, De Backer et al 1 deployed a variety of conditional Maged1 KO mouse lines, using both cell‐type and region‐specific gene inactivation strategies. Surprisingly, Maged1 deletion neither in VTA DA neurons, nor in the striatum, has an impact on the behaviors described above. But when the authors deleted Maged1 in the medial prefrontal cortex (mPFC) or the amygdala, both prominently sending off excitatory afferent connections to the NAc, cocaine locomotor sensitization was reduced, particularly in the amygdala KO. Deletion of Maged1 in a single input to the NAc might not be sufficient. However, when Maged1 was deleted both in the mPFC and BLA (basolateral amygdala), mice still self‐administered cocaine, leaving the circuit driving this behavior to be discovered. A region not explored in the present study is the orbitofrontal cortex (OFC). This may be a prime suspect as inactivation of the OFC reduces the fraction of animals that develop compulsive VTA DA neuron self‐stimulation 8. Thus, exploring the effects of Maged1 deletion specifically in the OFC on the transition from controlled to compulsive drug intake would further enhance our understanding of Maged1 implications for addiction.
In conclusion, De Backer et al 1 convincingly demonstrate the necessity of Maged1 for cocaine‐induced synaptic plasticity at cortico‐accumbal synapses and increased DA concentrations in the NAc, two core neuronal mechanisms leading to addictive behaviors. Site‐specific gene deletions implicate the mPFC and the amygdala in cocaine locomotor sensitization but not reinforcement, in line with much literature. Future studies will aim at elucidating the intracellular mechanisms linking Maged1 function and alterations in neuronal transmission underlying compulsive drug of abuse consumption. The present study may thus fundamentally redefine the function for Maged1, turning the black sheep into a black swan.
Acknowledgements
ML is funded by the Société académique de Genève, and CL holds grants from the ERC and the Swiss NSF.
EMBO Reports (2018) 19: e46743
See also: https://doi.org/10.15252/embr.201745089 (September 2018)
References
- 1. De Backer J, Monlezun S, Detraux B et al (2018) EMBO Rep 19: e45089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lüscher C (2016) Annu Rev Neurosci 39: 257–276 [DOI] [PubMed] [Google Scholar]
- 3. Sora I, Hall FS, Andrews AM et al (2001) Proc Natl Acad Sci USA 98: 5300–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Renthal W, Kumar A, Xiao G et al (2009) Neuron 62: 335–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang JJ, Lai BB, Xu AL et al (2014) Mol Neurobiol 51: 8–18. [DOI] [PubMed] [Google Scholar]
- 6. Lüscher C, Ungless MA (2006) PLoS Med 3: 2005–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Terrier J, Lüscher C, Pascoli V (2016) Neuropsychopharmacology 41: 1779–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pascoli V, Terrier J, Hiver A et al (2015) Neuron 88: 1054–1066. [DOI] [PubMed] [Google Scholar]