

Abstract
The cytokine interferon‐γ (IFNγ) can induce expression of MHC class II (MHCII) on many different cell types, leading to antigen presentation to CD4+ T cells and immune activation. This has also been linked to anti‐tumour immunity and graft‐versus‐host disease. The extent of MHCII upregulation by IFNγ is cell type‐dependent and under extensive control of epigenetic regulators and signalling pathways. Here, we identify novel genetic and chemical factors that control this form of MHCII expression. Loss of the oxidative stress sensor Keap1, autophagy adaptor p62/SQSTM1, ubiquitin E3‐ligase Cullin‐3 and chromatin remodeller BPTF impair IFNγ‐mediated MHCII expression. A similar phenotype is observed for arsenite, an oxidative stressor. Effects of the latter can be reversed by the inhibition of HDAC1/2, linking oxidative stress conditions to epigenetic control of MHCII expression. Furthermore, dimethyl fumarate, an antioxidant used for the treatment of several autoimmune diseases, impairs the IFNγ response by manipulating transcriptional control of MHCII. We describe novel pathways and drugs related to oxidative conditions in cells impacting on IFNγ‐mediated MHCII expression, which provide a molecular basis for the understanding of MHCII‐associated diseases.
Keywords: dimethyl fumarate, interferon‐γ, Keap1, MHC class II, oxidative stress
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Immunology; Signal Transduction
Introduction
Antigen presentation by major histocompatibility complex class II (MHCII) molecules is critical for the initiation of an adaptive CD4+ helper T‐cell response and for efficient CTL responses to infections and cancer 1, 2. Many MHCII alleles are correlated to specific autoimmune disorders, for example, HLA‐DRB1*1501 to multiple sclerosis 3, 4, and it is anticipated that MHCII antigen presentation sensitizes to many autoimmune diseases. Expression of MHCII is limited to antigen‐presenting immune cells such as dendritic cells and B cells. However, under inflammatory conditions normal tissue cells can also express and present peptides on MHCII following release of various cytokines, predominantly interferon gamma (IFNγ) 5. This is important in a series of pathologies, including the onset of graft‐versus‐host disease (GVHD) 6, 7, transplant rejection 8, 9, autoimmune diseases 10, as well as T‐cell priming by tumour cells 11, 12.
Transcription of MHCII, as well as the associated invariant chain (Ii), which aides MHCII trafficking and occupies the peptide binding groove before antigen loading, is governed by transcriptional master regulator CIITA 13, 14. CIITA does not bind the MHCII promoter directly, but rather assembles a complex of transcription factors at the MHCII promoter, which includes RFX5, CREB and NF‐Y 15. Furthermore, CIITA alters the chromatin environment by recruiting remodelling factors such as BRG‐1, histone acetyltransferases (HATs) and deacetylates (HDACs) 16, 17, 18, as well as by its intrinsic HAT activity 19. CIITA itself is transcribed from different promoters in different cell types, with its IFNγ‐induced isoform being initiated by transcription factor IRF‐1 20.
Besides upregulating MHC class II antigen presentation, IFNγ induces a broad pro‐inflammatory gene signature in both immune and non‐immune cells and is important for clearance of viral and bacterial infections 21. For these reasons, cancer cells promote resistance to immunotherapy by altering their IFNγ signalling pathway 22, 23, illustrating the importance of an intact IFNγ response for immune recognition. Sustained IFNγ signalling can lead to uncontrolled activation of the immune system, causing MHCII‐dependent transplant rejection 24 as well as autoimmunity, but its importance in the disease pathology for different autoimmune diseases is ambiguous 25, 26, 27, 28, 29. At the molecular level, engagement of the IFNγ receptor by IFNγ leads to the activation of JAK kinases, which phosphorylate and stimulate nuclear translocation of transcription factor STAT1 30. STAT1 subsequently induces transcription of IRF‐1 that controls the expression of many pro‐inflammatory genes including CIITA 31. While the central pathway leading to transcription of IRF‐1, CIITA and MHCII is conserved in most cells, many IFNγ‐induced genes are expressed in a cell type‐specific manner 16, 32, 33, 34, suggesting additional regulation by epigenetic modifiers and signalling pathways to steer the response. This is illustrated by the observation that HDAC inhibitors increase the expression of IFNγ‐target genes in different tumours, thereby sensitizing tumour cells to immune checkpoint inhibition 35, 36. Understanding the factors regulating the IFNγ response and MHCII expression could thus provide novel means of interfering with this important signalling pathway.
Here, we identified several novel regulators of IFNγ‐mediated MHCII expression, including oxidative stress sensor Keap1, autophagy adaptor p62, E3‐ligase Cullin‐3 and chromatin remodeller BPTF. We illustrate the role of oxidative stress on MHCII expression with the immunotoxic agent arsenite and the autoimmune suppressive drug dimethyl fumarate, providing additional mechanisms of action for these compounds. Our experiments show that the expression MHCII is controlled by complex pathways, allowing chemical intervention for controlling MHCII‐based pathologies.
Results
Keap1 regulates IFNγ‐induced expression of MHCII
The highly variable induction of MHCII by IFNγ in different cell types implies that additional proteins are in charge of regulating this process. Previously, using an RNAi screen focused on de‐ubiquitinating enzymes, we identified OTUD1 as a regulator of constitutive MHCII transcription in melanoma cells (A. Sapmaz, I. Berlin, E. Bos, R. H. Wijdeven, H. Janssen, R. Konietzny, A. E. Erson‐Bensan, R. I. Koning, B. M. Kessler, J. Neefjes & H. Ovaa, submitted). A secondary siRNA screen for potential interactors of OTUD1 37 in different cell types yielded E3‐ligase adaptor Keap1 as the most prominent hit. Keap1 is a multifunctional protein that is best known for inhibiting oxidative stress responses by facilitating Cullin‐3‐dependent ubiquitination and degradation of Nrf2, a transcription factor for antioxidant genes 38. In addition, Keap1 regulates NF‐κB signalling 39, 40, autophagy 41, DNA repair 42, drug resistance 43 and cell migration 44, through binding to a variety of substrates, but it has not been linked to MHCII expression. Silencing Keap1 attenuated IFNγ‐induced MHCII expression in HeLa and U118 cells, but not constitutive expression in MelJuSo melanoma cells, setting it apart from OTUD1 (Figs 1A and B, and EV1A). Similarly, no downregulation of constitutive MHCII expression in THP‐1 cells and U937 cells was observed after Keap1 depletion (Fig EV1B). However, IFNγ‐induced MHCII expression on both cells was also not (THP‐1) or only marginally (U937) affected in these cells, suggesting that Keap1 is more important in non‐hematopoietic cells or cells not expressing MHCII constitutively.
Figure 1. Keap1 positively regulates IFNγ‐mediated MHCII expression.
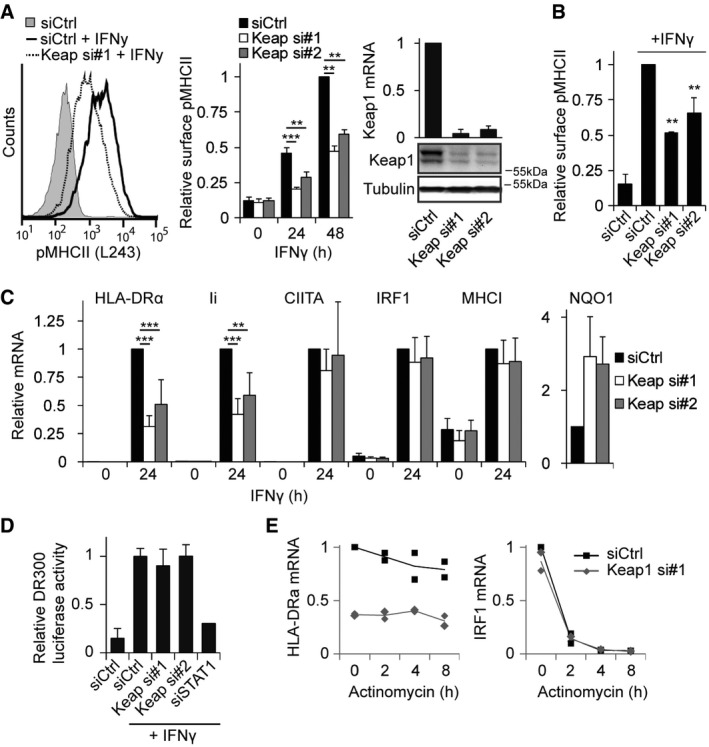
- HeLa cells were transfected with siCtrl or siRNAs targeting Keap1 and stained 72 h later for peptide‐loaded MHCII (L243‐cy5), stimulated or not with 100 ng/ml IFNγ for the indicated time. Representative histogram and quantifications are shown. Right: Keap1 silencing was determined by Western blot analysis (bottom) and qRT–PCR (top, normalized to GAPDH).
- U118 cells were analysed for MHCII levels according to the same protocol as in (A).
- HeLa cells transfected with siCtrl or siKeap1 were either or not exposed to IFNγ for 24 h, and mRNA expression of the indicated genes was analysed using qRT–PCR.
- MHCII promoter activity was analysed using a luciferase under control of the MHCII promoter (DR300) in cells transfected with the indicated siRNAs and treated with IFNγ when indicated. siSTAT1 was used as a positive control, and signals were normalized to a Renilla control plasmid.
- Cells transfected with siCtrl or siKeap1 were treated with IFNγ for 24 h and lysed, or actinomycin D (2 μM) was added and cells were lysed 2, 4 or 8 h later. mRNA expression level of HLA‐DRα and IRF1 was analysed using qRT–PCR, and IRF1 was used as a control for effectivity of actinomycin D. Individual data points are represented by dots, and the line is the average of the two experiments.
Figure EV1. Keap1 regulates IFNγ‐induced MHCII expression.
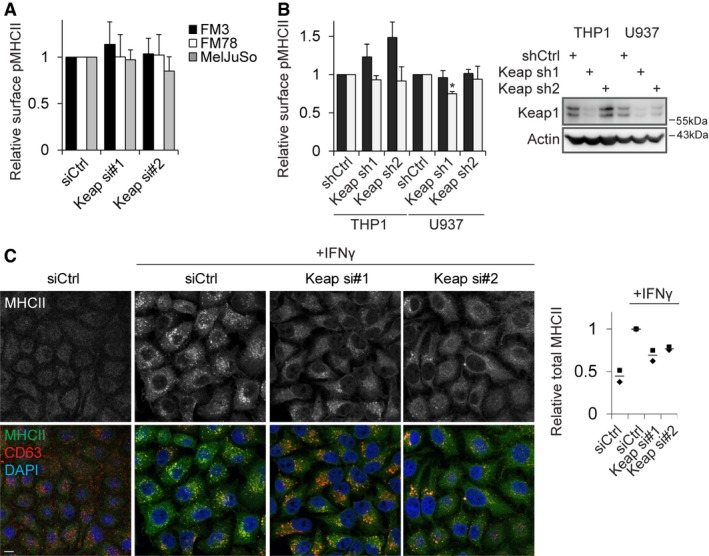
- FM3, FM78 and MelJuSo melanoma cells were transfected with siCtrl or siRNAs targeting Keap1 and stained 72 h later for peptide‐loaded MHCII (L243‐cy5) before analysis by flow cytometry.
- THP‐1 and U937 cells stably transfected with the indicated shRNAs were stimulated or not with IFNγ for 48 h, and surface expression of peptide‐loaded MHCII was assessed using flow cytometry. For stimulated samples, the signal for the unstimulated cells was subtracted to allow analysis of the IFNγ‐induced expression. Right: Keap1 silencing was determined by Western blot analysis.
- HeLa cells transfected with the indicated siRNAs were stimulated or not for 24 h with IFNγ and stained for MHCII, CD63 (late endosomal marker) and DAPI (DNA). For quantification, average signal intensity of at least six fields was quantified over two independent experiments. Individual experimental averages are represented by dots, and the line is the average of the two experiments.
We then deciphered in which step of the MHCII pathway Keap1 controls MHCII surface expression. To analyse defects in biosynthesis and cell surface transport of MHCII, the subcellular localization of MHCII was visualized. Cells silenced for Keap1 contained significantly reduced total amounts of MHCII, but the intracellular distribution was not affected (Fig EV1C). qPCR analyses of different genes in HeLa cells stimulated with IFNγ for 24 h indicated that Keap1 silencing inhibited the transcription of HLA‐DRα, as well as Ii, but not CIITA (Fig 1C). To assess whether Keap1 controls the activity of CIITA or the associated transcription factors, a luciferase construct under control of the MHCII promoter was utilized. In contrast to the control knockdown of STAT1, depletion of Keap1 had no effect on MHCII promoter activity (Fig 1D), indicating that Keap1 does not control any of the factors involved in promoter activation. Furthermore, Keap1 did not affect the stability of HLA‐DRα transcripts, since inhibition of RNA polymerase II by actinomycin D followed by culturing did not yield any differences in degradation rates (Fig 1E). Thus, Keap1 is a novel regulator of MHCII transcription, independently of promoter activation or mRNA stability.
HDAC1/2 inhibition negates effect of Keap1 on MHCII expression
The discrepancy between the effect of Keap1 on endogenous MHCII transcription and the effect of Keap1 on exogenous MHCII promoter activity suggested a role for epigenetic regulation controlled by Keap1 to influence IFNγ‐induced MHCII transcription. MHCII expression is reportedly controlled by various epigenetic markers, including H3K27me3, DNA methylation and histone deacetylation 18, 45, 46. Treatment of cells with inhibitors of EZH2, which prevent H3K27me3 modifications 47, 48, as well as HDAC inhibitors, induced MHCII expression in HeLa cells (Fig 2A), whereas inhibition of DNA methylation by decitabine had no effect (data not shown). Whereas the relative effect of Keap1 depletion remained intact upon treatment with EZH2‐inhibitors, all three HDAC inhibitors corrected the inhibition of IFNγ‐induced MHCII expression following Keap1 silencing. This suggested that Keap1 controls MHCII expression by manipulating histone acetylation/deacetylation activity.
Figure 2. Keap1 regulates MHCII expression through HDACs.
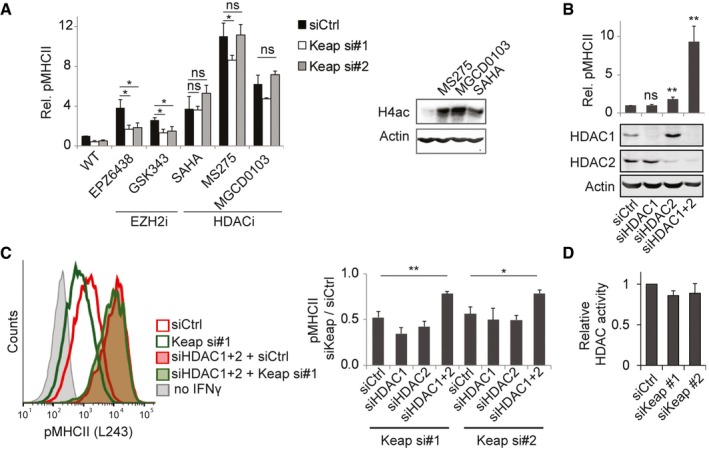
- HeLa cells transfected with the indicated siRNAs were treated for 48 h with IFNγ and indicated EZH2 or HDAC inhibitors; expression of MHCII was analysed by flow cytometry and normalized to MFI of control HeLa cells. Maximum non‐toxic doses of the inhibitors were used: EPZ6438 (2 μM), GSK343 (10 μM), SAHA (5 μM), MS‐275 (0.1 μM) and MGCD0103 (1 μM). Right: Western blot for H4ac, to test effectivity of the HDAC inhibitors.
- Cells transfected with siCtrl (75 nM), siHDAC1 (37.5 nM siHDAC1 + 37.5 nM siCtrl), siHDAC2 (37.5 nM siHDAC2 + 37.5 nM siCtrl) or siHDAC1 + 2 (37.5 nM siHDAC1 + 37.5 nM siHDAC2). Cells were exposed for 48 h to IFNγ, and expression of MHCII was analysed by flow cytometry and MFI normalized to siCtrl. Bottom: silencing efficiency was evaluated using Western blot.
- Cells transfected with the indicated siRNAs (37.5 nM HDAC1, 37.5 nM HDAC2 and 37.5 nM Keap1 or siCtrl) were stimulated for 48 h with IFNγ, and expression of MHCII was determined by flow cytometry. Left: representative histogram. Right: bar graph of the average of three independent experiments. Signal for siKeap1 was normalized to the respective siCtrl.
- HeLa cells transfected with indicated siRNAs were lysed after 3 days, and HDAC activity was determined using Fluor de Lys assay reagents.
SAHA is a pan‐HDAC inhibitor, while MGCD0103 specifically targets HDAC1/2 and MS‐275 inhibits HDAC1 and to a minor extent HDAC2 49, arguing that HDAC1 or HDAC2 is the primary regulator of MHCII expression, which is in line with data that overexpression of HDAC1 or HDAC2 represses MHCII expression 18. However, silencing of HDAC1 did not upregulate MHCII expression, while knockdown of HDAC2 had only a minor effect (Fig 2B and C). HDAC1 and HDAC2 have partly overlapping functions and can compensate for each other 50; therefore, we simultaneously silenced both HDACs. This strongly increased IFNγ‐induced MHCII expression and decreased the sensitivity of cells to Keap1 depletion (Fig 2B and C). This suggests that both HDAC1 and HDAC2 are involved in the inhibition of IFNγ‐induced MHCII expression in a pathway that intermingles with Keap1. However, Keap1 does not directly regulate the activity of HDAC1 and HDAC2, as concluded by determining HDAC activity in cells either or not depleted of Keap1 (Fig 2D).
Keap1 interactors p62/SQSTM1, Cul3 and BPTF regulate MHCII expression
How does Keap1 interfere with MHCII expression in non‐professional antigen‐presenting cells (APCs)? Keap1 mainly serves as a substrate adaptor for Cul3 and binds many proteins via its Kelch‐domain, including Nrf2 and other substrates containing an ETGE motif, to properly position these for Cul3‐mediated ubiquitination 51. Two point mutants of Keap1 were generated, one that renders Keap1 unable to bind ETGE motif‐containing proteins (Y572A) 52 and one that eliminates ubiquitin transfer to substrate proteins (G186R) 53. Stable expression of wild‐type RNAi resistant Keap1 allowed rescue of IFNγ‐induced MHCII expression upon Keap1 silencing, confirming that the effect of the siRNAs relied on Keap1 depletion. Yet, MHCII expression could not be rescued by either point mutant of Keap1 (Fig 3A and B), indicating a role for both substrate binding and ubiquitination in IFNγ‐induced MHCII expression by Keap1. This effect was independent of the canonical substrate Nrf2, as co‐depletion of Nrf2 did not restore IFNγ‐induced MHCII expression to normal levels (Fig 3C).
Figure 3. Keap1 interaction partners regulate MHCII expression.
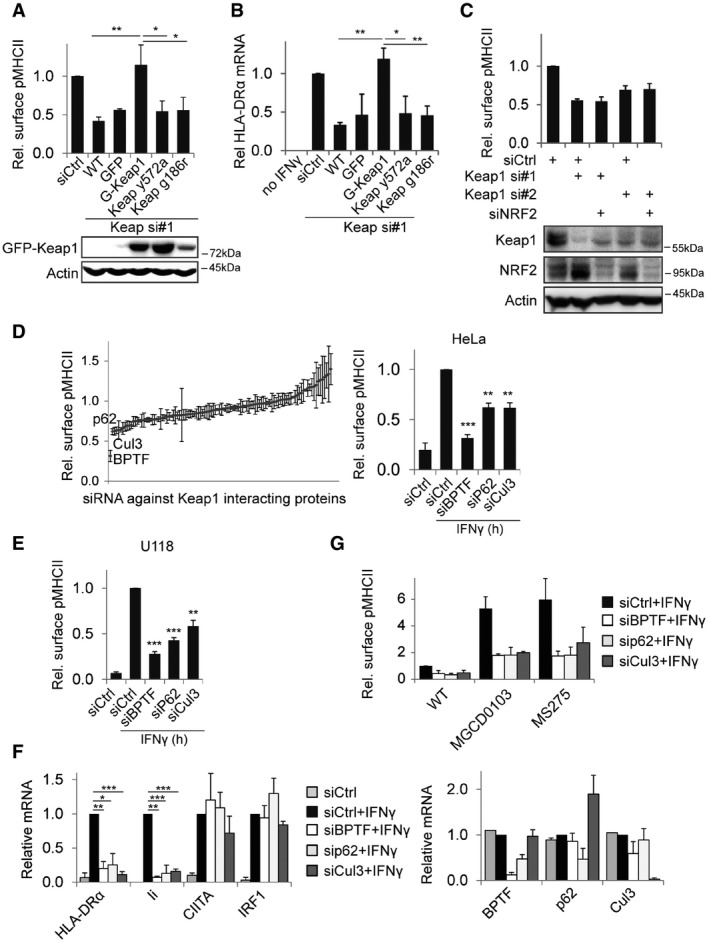
- HeLa cells stably expressing GFP or RNAi resistant GFP‐Keap1 with the indicated mutations were transfected with siRNAs and stimulated with IFNγ for 48 h before analysis by flow cytometry. Shown is MFI relative to siCtrl. Bottom panel: Western blot for expression of the indicated GFP‐Keap1 constructs.
- HeLa cells as in (A) were stimulated for 24 h with IFNγ, and mRNA levels of HLA‐DRα were measured using qRT–PCR and related to siCtrl.
- MHCII expression on HeLa cells transfected with the indicated siRNAs and stimulated with IFNγ for 48 h was measured using flow cytometry and related to siCtrl. Bottom: Western blot analyses for expression of the indicated proteins.
- Screen for effect of silencing Keap1 interacting proteins on MHCII surface levels. HeLa cells transfected with 106 different siRNAs targeting Keap1‐interacting proteins were stimulated with IFNγ for 48 h and analysed by flow cytometry. Right: summary of screening data for the indicated proteins.
- U118 cells were transfected with the indicated siRNAs and the next day stimulated with IFNγ. 48 h later, MHCII expression was analysed by flow cytometry.
- HeLa cells transfected with the indicated siRNAs were stimulated for 24 h with IFNγ and mRNA transcript levels were quantified using qRT–PCR, signal was normalized to GAPDH and siCtrl + IFNγ for each sample. Right graph: knockdown efficiency of the different siRNAs.
- HeLa cells transfected with the indicated siRNAs were stimulated for 48 h with IFNγ and the indicated HDAC inhibitors, followed by MHCII expression by flow cytometry.
To identify proteins that cooperate with Keap1 in the control IFNγ‐induced MHCII transcription, we performed an RNAi screen targeting all 106 described Keap1 interactors (gene search on http://www.ncbi.nlm.nih.gov) and measured the effect on IFNγ‐induced MHCII surface expression by flow cytometry. Using this screen, several proteins were identified as regulating IFNγ‐induced MHCII expression (Fig 3D). Of the top ten hits, only the three most significant hits—BPTF/FALZ, p62/SQSTM1 and Cullin‐3—regulated MHCII at the transcriptional level. BPTF is a chromatin remodeller that binds histone modifications H3K4me3 and H4K16ac and unwinds local chromatin for transcription 54, 55, p62 is an adaptor protein involved in autophagy, perinuclear endosome positioning and cell signalling 56, 57, 58, and Cullin‐3 (Cul3) the ubiquitin ligase that executes the ubiquitination reaction of proteins selected by adaptors such as Keap1 59. Like for Keap1, depletion of these genes also reduced IFNγ‐induced MHCII expression in U118 cells (Fig 3E) and controlled Ii but not CIITA or IRF1 expression (Fig 3F). However, the depletion phenotype of none of these three genes could be restored using HDAC inhibitors (Fig 3G), suggesting that the function of these genes does not fully overlap with that of Keap1.
Arsenite regulates IFNγ‐induced MHCII expression
Keap1 contains several cysteine residues that are modified during oxidative stress, rendering it inactive and facilitating NRF2‐dependent expression of antioxidant genes. Oxidative stress could then also impair IFNγ‐induced MHCII expression by inactivating Keap1. In vivo exposure to sodium arsenite (AS(III)), an oxidative stressor that activates NRF2, has already been reported to decrease the expression of different MHCII alleles and is linked to an impaired immune response 60, 61. To assess a direct role for AS(III) in IFNγ‐induced MHCII expression, HeLa and U118 cells were exposed to different concentrations of AS(III) during stimulation with IFNγ. A dose‐dependent decrease in MHCII expression was observed, indicating a role for AS(III) in the regulation of IFNγ‐induced MHCII expression (Fig 4A). Arsenite indeed targeted Keap1, since Nrf2 target NQO1 was upregulated in a dose‐dependent manner (Fig 4B). Similar to Keap1 depletion, this decrease was transcription‐dependent and confined to MHCII and Ii, but not CIITA (Fig 4B). Furthermore, treatment with HDAC inhibitor MGCD0103 fully restored IFNγ‐induced MHCII expression (Fig 4A). However, AS(III) can also target the H4K16‐specific histone acetyltransferase MYST1 62, suggesting it could exert its effect also via MYST1. In support of this, MYST1 knockdown reduced IFNγ‐induced MHCII expression (Fig 4C). When cells were depleted for either Keap1 or MYST1 and exposed to AS(III), a very minimal additional effect was observed (Fig 4D), substantiating the notion that AS(III) acts through these molecules. Thus, sodium arsenite impaired IFNγ‐mediated MHCII expression, probably via Keap1 and MYST1, and this effect could be negated by HDAC inhibitors.
Figure 4. Arsenite controls IFNγ and histone acetylation‐dependent MHCII expression.
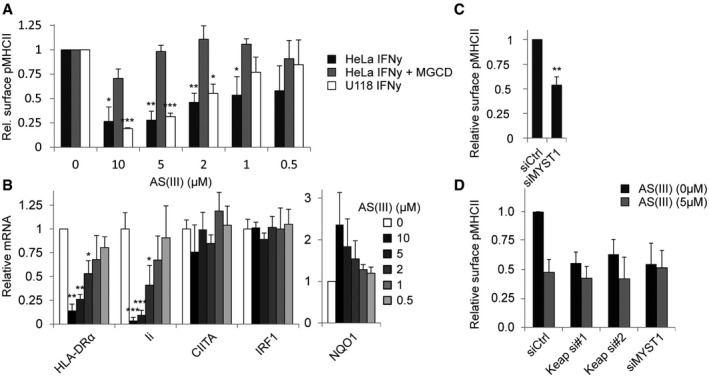
- HeLa and U118 cells were stimulated with IFNγ for 48 h in combination with the indicated concentration of NaAs2O3 in the presence or absence of 1 μM MGCD0103 and analysed for MHCII expression by flow cytometry. MGCD0103‐treated samples were normalized to corresponding measured in the absence of NaAs2O3.
- HeLa cells either or not exposed to NaAs2O3 were stimulated for 24 h with IFNγ, and mRNA levels were measured by qRT–PCR. Data normalized within each sample to condition lacking NaAs2O3.
- MHCII levels on HeLa cells transfected with the indicated siRNAs and stimulated with IFNγ for 48 h were determined by flow cytometry. Data were normalized to siCtrl condition.
- HeLa cells transfected with the indicated siRNAs and stimulated with IFNγ for 48 h in the presence or absence of 5 μM NaAs2O3 were analysed for MHCII expression by flow cytometry. Data were normalized to untreated siCtrl condition.
Antioxidants control IFNγ‐induced MHCII expression
Besides oxidative stress, Keap1 is also a primary target for antioxidants such as tert‐butylhydroquinone (tBHQ) and dimethyl fumarate (DMF) 38, 63. Both of these drugs display immunomodulatory activity, with their mechanism of action not fully understood 64, 65. DMF has been approved by the FDA for the treatment of psoriasis and multiple sclerosis (MS) 66, 67, 68, both autoimmune diseases that have been linked to IFNγ expression and activation of CD4+ T cells 26, 69, 70, implying a possible role for IFNγ‐induced MHCII expression in disease pathology. To test whether DMF, like Keap1 inhibition, reduces IFNγ‐induced MHC class II expression, we exposed various cell lines to IFNγ in the absence or presence of DMF. DMF reduced IFNγ‐induced MHCII expression in all cell lines, whereas constitutive MHCII expression by monocyte‐like THP‐1 cells was unaffected (Fig 5A). Similarly, tBHQ specifically reduced IFNγ‐induced MHCII expression (Fig 5A). In multiple sclerosis (MS), as well as experimental autoimmune encephalomyelitis (EAE, a mouse model for MS), macrophages play an important role in the initiation of the inflammatory response 71. To assess whether DMF also affects MHCII expression by these cells, monocyte‐derived macrophages (MDMs), as well as MDMs that were cultured in the presence of myelin to generate foamy macrophages, which are present in brain lesions of MS patients 72, were treated with different doses of DMF. In both types of macrophages, DMF caused a dose‐dependent decrease in IFNγ‐induced MHCII expression but not constitutive MHCII expression (Fig 5B and C). Constitutive MHCII expression by B cells was also not affected by these drugs (Fig 5D).
Figure 5. Dimethyl fumarate inhibits IFNγ‐induced MHCII and chemokine expression.
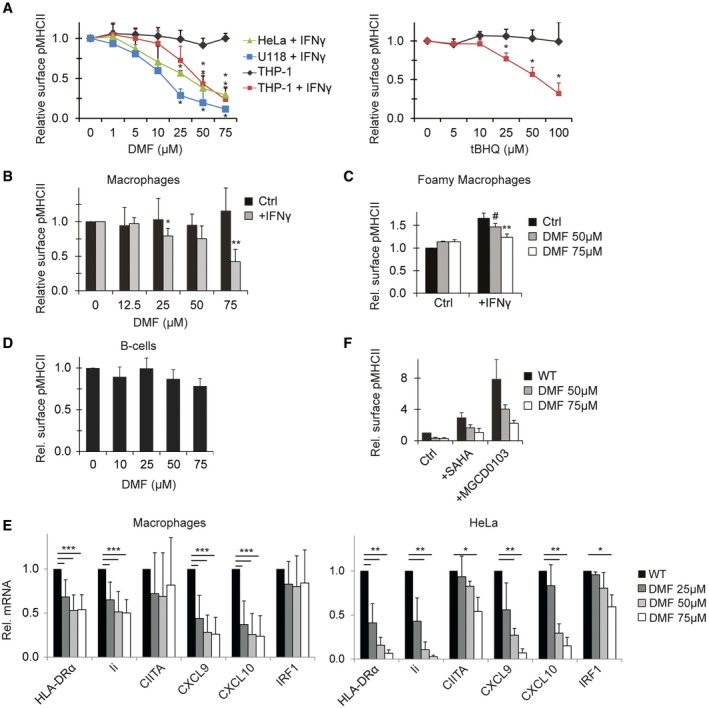
- THP‐1, HeLa and U118 cells either or not exposed to IFNγ for 48 h were cultured in the presence of DMF (left) or tBHQ (right) at indicated concentrations before analyses of MHCII expression by flow cytometry. Since THP1 express constitutive MHCII, MHCII expression is normalized by subtracting MFI of non‐IFNγ exposed THP1. Data normalized to MFI measured in the absence of drugs. Shown is mean + SD, n = 3.
- Monocyte‐derived macrophages were treated with the indicated concentration of DMF and either or not stimulated with IFNγ for 48 h. MHCII expression was determined by flow cytometry. Shown is mean + SD of four independent experiments. For IFNγ‐treated samples, MFI of control unstimulated macrophages was subtracted.
- Foamy macrophages were treated with the indicated concentrations of DMF and either or not activated with IFNγ for 24 h when indicated before MHCII expression was determined by flow cytometry. Shown is mean + SD, n = 3.
- Primary human B cells were cultured for 24 h in different concentrations of DMF as indicated, and surface MHCII expression was analysed by flow cytometry. Shown is mean + SD, n = 4.
- Macrophages (left) or HeLa cells (right) were cultured for 24 h in the presence of IFNγ and DMF when indicated before mRNA expression analysis of the indicated proteins using qRT–PCR. Shown is mean + SD of experiments repeated eightfold (macrophages) or threefold (HeLa).
- HeLa cells were cultured for 48 h with IFNγ in the presence or absence of the indicated inhibitors, and expression of MHCII was determined by flow cytometry. Concentrations: SAHA (5 μM), MGCD0103 (1 μM). Shown is mean MHCII expression + SD of triplicate experiments.
mRNA analysis of macrophages and HeLa cells revealed that the effect was transcriptional, since levels of HLA‐DRα and Ii transcripts were reduced (Fig 5E). No significant decrease was observed for IRF1, whereas the response of CIITA was highly variable between donors and in HeLa cells only observed for the highest concentration of DMF. As reported for keratinocytes and peripheral blood mononuclear cells (PBMCs) 73, DMF also reduced the expression of the IFNγ‐inducible pro‐inflammatory chemokines CXCL9 and CXCL10 in macrophages, suggesting a broad reduction in the IFNγ signature following DMF treatment (Fig 5E). We then tested whether DMF would control MHCII expression in a Keap1‐HDAC‐dependent manner. Inhibition of MHCII expression by DMF could not be relieved by HDAC inhibitors (Fig 5F). Thus, DMF is a broad regulator of IFNγ‐induced MHCII and chemokine expression, via a mechanism independent of HDACs.
Discussion
Cytokine‐stimulated non‐hematopoietic cells have often been discarded as functional contributors to MHCII‐dependent immune activation, due to their lack of co‐stimulatory receptors. Yet, tumour recognition, transplantation rejection, graft‐versus‐host disease and specific autoimmune diseases have been shown to rely at least in part on MHCII expression by non‐professional APCs. The details of this MHCII control are unresolved but would provide interesting leads for drug development or explain the activity of already existing drugs. Here, we identified several genetic factors and compounds that interfere with IFNγ‐induced MHCII expression (Fig 6). One of these, DMF, is an antioxidant used in the treatment of psoriasis and MS and acts as a broad inhibitor of the IFNγ response, offering an additional mechanism of action for this immunomodulatory drug.
Figure 6. The complex transcriptional regulation of IFNγ‐induced regulation of MHCII expression.
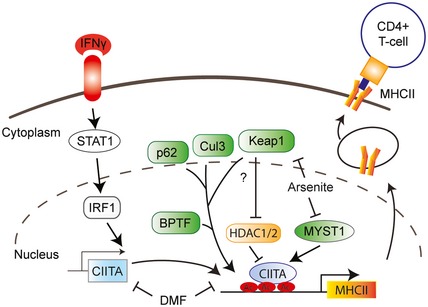
Transcription of MHCII genes is controlled by master regulator CIITA. This study adds various factors and compounds involved in oxidative stress conditions including Keap1, Cul3, p62 and BPTF, as depicted in green. Regulation by Keap1 could involve HDAC1 and HDAC2. Arsenite controls IFNγ‐induced MHCII expression by inhibiting MYST1, as well as Keap1, while the antioxidant and immunosuppressive compound dimethyl fumarate (DMF) inhibits IFNγ‐induced expression of MHCII via a different mechanism.
Dimethyl fumarate (DMF) has recently been FDA approved for the treatment of relapsing‐remitting MS, but its exact mechanism of action is still under debate 74, 75. Several immunomodulatory actions have been ascribed to this drug, including skewing dendritic cell differentiation to a protective type II subset 76, 77, inhibition of dendritic cell and monocyte maturation 65, 78, inhibition of T‐cell activation 79, skewing T cells to a Th2 subtype 80, 81 and inhibition of leucocyte infiltration and migration 82. Our data show that DMF also acts as an inhibitor of IFNγ‐induced chemokine and MHCII expression. DMF also inhibited IFNγ‐induced expression of the chemokines CXCL9 and CXCL10 in macrophages and tumour cells, as also demonstrated for keratinocytes and PBMCs 73. Interestingly, in MS patients, DMF treatment most strongly decreased the number of CXCR3+ helper T cells. CXCR3 is a receptor for CXCL9 and CXCL10 83. The inhibitory effect of DMF extends to IFNγ‐induced expression of MHCII, suggesting that DMF dampens both antigen presentation and immune cell recruitment to inflammatory sites. Since DMF did not affect constitutive expression of MHCII on macrophages and B cells, it probably acts as a context‐specific inhibitor of MHCII expression. The sensitivity of different cell types to DMF varied, with non‐hematopoietic cell types significantly reducing MHCII expression at concentrations around 10 μM, while macrophages required at least 50 μM DMF for a similar response, although the latter is still below the clinical dose of 70 μM 84. This could be the result of the higher expression of antioxidant genes in macrophages, which quench DMF activity 85. Recent data in a mouse model for MS suggest that the immunomodulatory and protective activity of DMF does not involve NRF2 65. This observation is in line with our data that suggest that the effect of DMF on the IFNγ‐axis is independent of the Keap1‐NRF2 antioxidant response pathway. Besides targeting Keap1, DMF is known to inhibit many proteins and signalling pathways 78, 79, 86. It is therefore not surprising that we failed to link any of the common signalling pathways (NF‐κB, MAPK, AKT, ERK1) to the effect observed for DMF on MHCII expression. Thus, DMF controls the IFNγ response and its immunomodulatory action could result from interfering with multiple pathways.
Besides antioxidants, other drugs and pathways are also operational in controlling IFNγ‐induced MHCII expression. This includes the oxidative stressor arsenite. Arsenic contamination of drinking water is a persistent problem in many countries and induces immunotoxicity 87. Our data suggest that attenuated IFNγ‐induced expression of MHCII could contribute to these effects. Arsenite impaired MHCII expression was restored by inhibition of HDAC1/2, arguing that arsenite alters epigenetic regulation of MHCII expression in the context of high cellular HDAC activity. Molecularly, arsenite targets both Keap1 88, 89 and MYST1, an H4K16 histone acetyltransferase 62, 90, 91. IFNγ‐induced MHCII expression was impaired after depletion of either Keap1 or MYST1, suggesting that arsenite control of MHCII expression involves both of these proteins. The effects of oxidative stress induced by arsenite on MHCII expression raise the possibility that other forms of oxidative stress, such as ionizing irradiation, could also control MHCII expression. Indeed, oxidative stress can inhibit T‐cell responses in the tumour microenvironment 92, and ionizing irradiation modulates MHCII expression 93. However, since many cellular pathways are initiated by this form of stress, including mTOR activation and the production of detoxifying enzymes, it remains to be established whether oxidative stress per se attenuates MHCII expression and the concurrent relevance for anti‐tumour immunity in vivo.
We also identified various factors controlling IFNγ‐induced MHCII transcription: Keap1 and its interactors p62/SQSTM1, BPTF and Cullin‐3. Keap1 did not affect MHCII promoter activity or mRNA stability, leaving (epi‐)genetic control of MHCII expression. Indeed, chemical or genetic removal of HDAC1/2 activity from cells neutralized the necessity for Keap1 to properly induce MHCII expression. Surprisingly, this revival was not observed for any of its interaction partners. This could be because these genes act independently of each other in this process. However, Keap1 mutants failing to bind Cul3 or substrates could not rescue the effect of Keap1 depletion, suggesting that it requires both Cul3 and a substrate for full activity. Cul3 partners with many substrate adaptors to control ubiquitination and potentially regulates MHCII expression via several different pathways, explaining why its effect is not dependent on HDAC activity. In line with this, the reduction in MHCII mRNA levels with Cul3 depletion is stronger than that of Keap1 depletion (90% versus 65%). Thus, Keap1 and Cul3 could act together in regulation of MHCII expression, with Cul3 having additional functions independently of Keap1. The HDAC dependence of Keap1 depletion suggests that Keap1 participates in (de‐)acetylation processes. Since the total cellular HDAC activity is not affected by Keap1 depletion, Keap1 could affect targeting of HDACs to a specific protein or genomic region, or regulate activity of acetyltransferases. Of note, our studies were performed on cell lines, but since the four genes identified in this study are widely expressed 94, it is likely that they also regulate IFNγ‐induced MHCII expression in non‐transformed cells and tissues.
Interestingly, our study supports a role for two epigenetic regulators that revolve around H4K16ac: H4K16 acetyltransferase MYST1 and BPTF, a chromatin remodeller that specifically binds H4K16ac (in combination with H3K4me3) to support chromatin accessibility at enhancer and transcription start site regions 54, 95. These two factors could act in tandem where BPTF could function at the MHCII promoter or enhancer region to open its chromatin by binding H4K16ac.
Collectively, our data identify several novel regulators of IFNγ‐induced MHCII expression, as occurs in inflamed tissue. Four genetic factors, Keap1, p62/SQSTM1, Cullin‐3 and BPTF, mediate IFNγ‐mediated MHCII expression. Arsenite, an immunosuppressive compound that induces oxidative stress, also controls IFNγ‐mediated MHCII expression. The effects of Keap1 depletion and arsenite on MHCII expression could be overcome by HDAC inhibitors, implying epigenetic regulation by these factors. Finally, we demonstrate that the drug DMF impairs IFNγ‐induced MHCII and chemokine expression, providing an additional mechanism of action for a drug that is used in the treatment of several MHCII‐linked autoimmune diseases.
Materials and Methods
Cell culture, treatments and constructs
HeLa, U118, FM3 and FM78 cells were cultured in DMDM supplemented with 10% FCS, MelJuSo cells were cultured in IMDM with 10% FCS and THP‐1 and U937 in RPMI with 10% FCS. Cells were stimulated with 100 ng/ml IFNγ (eBioscience) for the indicated times. For generation of stable cell lines, Keap1 was cloned into a GFP‐C1 vector and mutagenized to avoid targeting by Keap1 siRNA#1 using the primers: fw 5′‐ggggctttgacgggacaaatcgtctaaactcagctgagtgttac‐3′, rv 5′‐gggtagtaacactcagctgagtttagacgatttgtcccgtcaaagc‐3′. Subsequent mutagenesis was performed using the following primers: Y572A fw: 5′‐ctacgtccttggaggcgctgatggtcacacgttc‐3′, Y572A rv: 5′‐actgtccaggaacgtgtgaccatcagcgcctccaaggacg‐3′ G186R fw: 5′‐cccagcaatgccatccgcatcgccaacttcg‐3′, G186R rv: 5′‐gctcagcgaagttggcgatgcggatggcattg‐3′. GFP and GFP‐Keap1 mutants were recloned into a retroviral pMX vector and upon retroviral transduction cells selected using puromycin (4 μg/ml, Gibco). Keap1 stable knockdown cells were generated by transduction with lentiviral vectors containing an shRNA sequence targeting Keap1. Keap1 sh1 targeted the 5′‐GCGAATGATCACAGCAATGAA‐3′ sequence of Keap1 and Keap1 sh2 the 5′‐CGGGAGTACATCTACATGCAT‐3′ sequence while cultured in the presence of puromycin (2.5 μg/ml).
Macrophage and B‐cell cultures
Peripheral blood mononuclear cells were separated from buffy coats (Sanquin, Amsterdam) by density gradient centrifugation using Ficoll‐Paque Plus (GE Healthcare). Monocytes and B cells were isolated by positive selection using CD14 and CD19 microbeads, respectively, and the autoMacs Pro Separator (Miltenyi Biotec). Cell purities were validated by FACS using anti‐CD3, CD19, CD45, CD14 and CD56 mAbs. For differentiation into macrophages, monocytes were cultured for at least 5 days in Teflon conical flasks (Nalgene; 2*10E6/ml) in RPMI 1640 without HEPES (Lonza) supplemented with 5% human AB serum (Sanquin) and 1% penicillin/streptomycin (Lonza). To obtain foamy macrophages, myelin was isolated from human white matter tissue (Netherlands Brain Bank, Amsterdam), sonicated and added to these macrophage cultures (50 μg/ml) for 48 h. Cells (8*10E5/ml) were stimulated with DMF at different concentrations in the absence or presence of IFNγ (0.1 mg/ml; eBioscience) for 24 h.
Transfections
For expression studies, HeLa cells were transfected using Effectene (Qiagen) according to the manufacturer's instructions. For siRNA silencing, cells were reverse‐transfected according to the manufacturer's protocol with DharmaFECT transfection reagent #1 and 50 nM total siRNA. Briefly, siRNAs and DharmaFECT were mixed and incubated for 20 min in a culture well, after which cells were added and left to adhere. Three days later, cells were harvested for analysis. Catalog numbers: siCtrl: D00120613‐20, siSTAT1 D‐003543, siHDAC1 D‐003493, siHDAC2 D‐003495, siBPTF M‐004025‐01‐0005, sip62 D‐010230 of the Human siGenome SMARTpool, Dharmacon. siRNA sequences targeting Keap1: #1 GGACAAACCGCCUUAAUUC and #2 GGGCGUGGCUGUCCUCAAU, siRNA sequence targeting p62 for validation: #4 GAAGUGGACCCGUCUACAG, all from Dharmacon.
Reagents and antibodies
Rabbit αHDAC1 NB100‐56340 (Novus Biologicals), rabbit αHDAC2 SC‐7899, mouse αp62 SC‐28359 (both from Santa Cruz), rabbit αH3ac 06599 (Millipore), rabbit αH4ac ab177790, rabbit αH3 ab1791 (both from Abcam), mouse αKeap1 60027‐1‐IG (Proteintech), mouse αActin A5441 (Sigma), rabbit αGFP (as described before 96). SAHA, sodium (meta)arsenite, dimethyl fumarate and GSK343 were acquired from Sigma, and EPZ6438, MS‐275 and MGCD0103 were acquired from Selleckchem.
Flow cytometry
Three days after siRNA transfection, HeLa and U118 cells were trypsinized and stained with Cy5‐labelled L243 antibody 97, before analysis using flow cytometry (BD FACSArray or BD FACS Calibur for GFP co‐detection). For macrophages and B cells, cells were first detached from the culture plates using PBS (Westburg) supplemented with 0.03% EDTA (Sigma‐Aldrich) and blocked using FACS buffer (PBS/0.2%BSA/0.01% sodium azide) containing 10% AB serum (Sanquin) at 4°C. Surface staining was performed using mAbs against HLA‐DR APC‐H7 (L243; BD Biosciences). Anti‐CD68 APC mAb (BioLegend) was used to confirm macrophage differentiation. Stained cells were measured using an LSRII flow cytometer and analysed by FACSDiva software (both BD).
Microscopy
Cells were seeded on coverslips with siRNA transfection reagents. After 2 days, cells were stimulated with IFNγ and the next day fixed in 3.7% formaldehyde for 10 min and permeabilized with 0.1% Triton X‐100 before blocking and staining with the antibodies indicated above in combination with 4′,6‐diamidino‐2‐phenylindole (DAPI, Invitrogen) to stain DNA. Images were acquired using a Leica TCS SP8 confocal microscope (Leica Microsystems, Wetzlar, Germany) at 63× magnification. Quantification was performed using ImageJ, and images were processed using Adobe Photoshop and Illustrator.
Co‐immunoprecipitation and Western blotting
For protein expression analysis, cells were directly lysed in SDS–PAGE loading buffer and proteins were separated by SDS–PAGE and transferred to nitrocellulose or PVDF filters by Western blotting. Antibody incubations and blocking were done in PBS supplemented with 0.1 (v/v)% Tween and 5% (w/v) milk powder. Blots were imaged using the Odyssey Imaging System (LI‐COR) or Chemidoc (Bio‐Rad).
RNA isolation, cDNA synthesis and qPCR
RNA isolation, cDNA synthesis and quantitative RT–PCR were performed according to the manufacturer's (Roche) instructions. For macrophages, total RNA was extracted using the GenElute RNA Purification kit (Sigma‐Aldrich). Signal was normalized to GAPDH and calculated using the Pfaffl formula. Primers used for detection were as follows: GAPDH fw: 5′‐TGTTGCCATCAATGACCCCTT‐3′, GAPDH rv: 5′‐CTCCACGACGTACTCAGCG‐3′, HLA‐DRα fw: 5′‐CATGGGCTATCAAAGAAGAAC‐3′, HLA‐DRα rv: 5′‐CTTGAGCCTCAAAGCTGGC‐3′, Ii fw: 5′‐CACCTGCTCCAGAATGCTG‐3′, Ii rv: 5′‐CAGTTCCAGTGACTCTTTCG‐3′, IRF1 fw: 5′‐GCACCAGTGATCTGTACAAC‐3′, IRF1 rv: 5′‐GCTCCTCCTTACAGCTAAAG‐3′, CIITA fw: 5′‐CCTGCTGTTCGGGACCTAAA‐3, CIITA rv: 5′‐GGATCCGCACCAGTTTGG‐3′, Keap1 fw: 5′‐CTGGAGGATCATACCAAGCAGG‐3′, Keap1 rv: 5′‐GAACATGGCCTTGAAGACAGG‐3′, NQO1 fw: 5′‐GGGCAAGTCCATCCCAACTG‐3′, NQO1 rv: 5′‐GCAAGTCAGGGAAGCCTGGA‐3′, CXCL9 fw: 5′‐ GTGGTGTTCTTTTCCTCTTG‐3′, CXCL9 rv: 5′‐GTAGGTGGATAGTCCCTTGG‐3′, CXCL10 fw: 5′‐ TGATTTGCTGCCTTATCTTTCTGA‐3′, CXCL10 rv: 5′‐ CAGCCTCTGTGTGGTCCATCCTTG‐3′.
Luciferase assays
HeLa cells reverse transfected with the indicated siRNAs were transfected the next day with a luciferase construct under the control of the MHCII locus (pGL3‐DRA) 98, as well as a SV40‐Renilla pGL3 reporter construct 99. Twenty‐four hours later, cells were stimulated with IFNγ for 24 h when indicated and lysed and analysed the day after using the dual‐luciferase reporter assay (Promega). Data were normalized to Renilla luciferase signal.
HDAC activity assay
Three days after transfection, HDAC activity in HeLa cells was analysed using the Fluor de Lys assay (Enzo Life Sciences), according to the manufacturer's instructions. Read‐out was performed on the BMG Labtech Clariostar.
Statistical analysis and experimental set‐up
All experiments shown in the paper were performed independently at least three times. Statistical significance was calculated using a two‐sided paired Student's t‐test for all the normalized data, for the non‐normalized signals an unpaired two‐sided t‐test was done. All error bars represent the standard deviation (SD). Statistical values are as following: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Author contributions
RHW and JN designed the research. RHW performed most of the experiments. MMvL, AFW‐W and RQH performed experiments for Fig 5B–E. PJvdE provided the DR300‐luciferase construct and helpful advice. JJA cloned constructs. RHW and JN interpreted the data and wrote the manuscript, with input from all other authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Acknowledgements
We thank members of the Neefjes group for critical discussions, I.Berlin for reading the manuscript and the LUMC and NKI Flow Cytometry Facilities and NKI Robotics Facility for support, with special thanks to Ben Morris. This work was supported by the Institute of Chemical Immunology, an NWO Gravitation project funded by the Ministry of Education, Culture and Science of the Government of the Netherlands and an ERC Advanced Grant awarded to J.N, as well as the MS foundation for R.H.
EMBO Reports (2018) 19: e45553
References
- 1. Neefjes J, Jongsma ML, Paul P, Bakke O (2011) Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol 11: 823–836 [DOI] [PubMed] [Google Scholar]
- 2. Ahrends T, Babala N, Xiao Y, Yagita H, van Eenennaam H, Borst J (2016) CD27 agonism plus PD‐1 blockade recapitulates CD4+ T‐cell help in therapeutic anticancer vaccination. Can Res 76: 2921–2931 [DOI] [PubMed] [Google Scholar]
- 3. Fernando MM, Stevens CR, Walsh EC, De Jager PL, Goyette P, Plenge RM, Vyse TJ, Rioux JD (2008) Defining the role of the MHC in autoimmunity: a review and pooled analysis. PLoS Genet 4: e1000024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rioux JD, Goyette P, Vyse TJ, Hammarstrom L, Fernando MM, Green T, De Jager PL, Foisy S, Wang J, de Bakker PI et al (2009) Mapping of multiple susceptibility variants within the MHC region for 7 immune‐mediated diseases. Proc Natl Acad Sci USA 106: 18680–18685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kambayashi T, Laufer TM (2014) Atypical MHC class II‐expressing antigen‐presenting cells: can anything replace a dendritic cell? Nat Rev Immunol 14: 719–730 [DOI] [PubMed] [Google Scholar]
- 6. Koyama M, Kuns RD, Olver SD, Raffelt NC, Wilson YA, Don AL, Lineburg KE, Cheong M, Robb RJ, Markey KA et al (2011) Recipient nonhematopoietic antigen‐presenting cells are sufficient to induce lethal acute graft‐versus‐host disease. Nat Med 18: 135–142 [DOI] [PubMed] [Google Scholar]
- 7. MacDonald KP, Shlomchik WD, Reddy P (2013) Biology of graft‐versus‐host responses: recent insights. Biol Blood Marrow Transplant 19: S10–S14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kim J, Park S, Kim HA, Jung D, Kim HJ, Choi HJ, Cho HR, Kwon B (2010) Roles of host nonhematopoietic cells in autoimmunity and donor cell engraftment in graft‐versus‐host disease. Immune Netw 10: 46–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abrahimi P, Qin L, Chang WG, Bothwell AL, Tellides G, Saltzman WM, Pober JS (2016) Blocking MHC class II on human endothelium mitigates acute rejection. JCI Insight 1: e85293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thelemann C, Haller S, Blyszczuk P, Kania G, Rosa M, Eriksson U, Rotman S, Reith W, Acha‐Orbea H (2016) Absence of nonhematopoietic MHC class II expression protects mice from experimental autoimmune myocarditis. Eur J Immunol 46: 656–664 [DOI] [PubMed] [Google Scholar]
- 11. Meazza R, Comes A, Orengo AM, Ferrini S, Accolla RS (2003) Tumor rejection by gene transfer of the MHC class II transactivator in murine mammary adenocarcinoma cells. Eur J Immunol 33: 1183–1192 [DOI] [PubMed] [Google Scholar]
- 12. Johnson DB, Estrada MV, Salgado R, Sanchez V, Doxie DB, Opalenik SR, Vilgelm AE, Feld E, Johnson AS, Greenplate AR et al (2016) Melanoma‐specific MHC‐II expression represents a tumour‐autonomous phenotype and predicts response to anti‐PD‐1/PD‐L1 therapy. Nat Commun 7: 10582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Steimle V, Otten LA, Zufferey M, Mach B (1993) Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome). Cell 75: 135–146 [PubMed] [Google Scholar]
- 14. Masternak K, Muhlethaler‐Mottet A, Villard J, Zufferey M, Steimle V, Reith W (2000) CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev 14: 1156–1166 [PMC free article] [PubMed] [Google Scholar]
- 15. Reith W, LeibundGut‐Landmann S, Waldburger JM (2005) Regulation of MHC class II gene expression by the class II transactivator. Nat Rev Immunol 5: 793–806 [DOI] [PubMed] [Google Scholar]
- 16. Mudhasani R, Fontes JD (2002) The class II transactivator requires brahma‐related gene 1 to activate transcription of major histocompatibility complex class II genes. Mol Cell Biol 22: 5019–5026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beresford GW, Boss JM (2001) CIITA coordinates multiple histone acetylation modifications at the HLA‐DRA promoter. Nat Immunol 2: 652–657 [DOI] [PubMed] [Google Scholar]
- 18. Zika E, Greer SF, Zhu XS, Ting JP (2003) Histone deacetylase 1/mSin3A disrupts gamma interferon‐induced CIITA function and major histocompatibility complex class II enhanceosome formation. Mol Cell Biol 23: 3091–3102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Raval A, Howcroft TK, Weissman JD, Kirshner S, Zhu XS, Yokoyama K, Ting J, Singer DS (2001) Transcriptional coactivator, CIITA, is an acetyltransferase that bypasses a promoter requirement for TAF(II)250. Mol Cell 7: 105–115 [DOI] [PubMed] [Google Scholar]
- 20. van den Elsen PJ (2011) Expression regulation of major histocompatibility complex class I and class II encoding genes. Front Immunol 2: 48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schoenborn JR, Wilson CB (2007) Regulation of interferon‐gamma during innate and adaptive immune responses. Adv Immunol 96: 41–101 [DOI] [PubMed] [Google Scholar]
- 22. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE et al (2016) Loss of IFN‐gamma pathway genes in tumor cells as a mechanism of resistance to Anti‐CTLA‐4 therapy. Cell 167: 397–404.e399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zaretsky JM, Garcia‐Diaz A, Shin DS, Escuin‐Ordinas H, Hugo W, Hu‐Lieskovan S, Torrejon DY, Abril‐Rodriguez G, Sandoval S, Barthly L et al (2016) Mutations associated with acquired resistance to PD‐1 blockade in melanoma. N Engl J Med 375: 819–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ring GH, Saleem S, Dai Z, Hassan AT, Konieczny BT, Baddoura FK, Lakkis FG (1999) Interferon‐gamma is necessary for initiating the acute rejection of major histocompatibility complex class II‐disparate skin allografts. Transplantation 67: 1362–1365 [DOI] [PubMed] [Google Scholar]
- 25. Pollard KM, Cauvi DM, Toomey CB, Morris KV, Kono DH (2013) Interferon‐gamma and systemic autoimmunity. Discov Med 16: 123–131 [PMC free article] [PubMed] [Google Scholar]
- 26. Arellano G, Ottum PA, Reyes LI, Burgos PI, Naves R (2015) Stage‐specific role of interferon‐gamma in experimental autoimmune encephalomyelitis and multiple sclerosis. Front Immunol 6: 492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Oon S, Wilson NJ, Wicks I (2016) Targeted therapeutics in SLE: emerging strategies to modulate the interferon pathway. Clin Transl Immunology 5: e79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Harden JL, Johnson‐Huang LM, Chamian MF, Lee E, Pearce T, Leonardi CL, Haider A, Lowes MA, Krueger JG (2015) Humanized anti‐IFN‐gamma (HuZAF) in the treatment of psoriasis. J Allergy Clin Immunol 135: 553–556 [DOI] [PubMed] [Google Scholar]
- 29. Jackson SW, Jacobs HM, Arkatkar T, Dam EM, Scharping NE, Kolhatkar NS, Hou B, Buckner JH, Rawlings DJ (2016) B cell IFN‐gamma receptor signaling promotes autoimmune germinal centers via cell‐intrinsic induction of BCL‐6. J Exp Med 213: 733–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Platanias LC (2005) Mechanisms of type‐I‐ and type‐II‐interferon‐mediated signalling. Nat Rev Immunol 5: 375–386 [DOI] [PubMed] [Google Scholar]
- 31. Schroder K, Hertzog PJ, Ravasi T, Hume DA (2004) Interferon‐gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75: 163–189 [DOI] [PubMed] [Google Scholar]
- 32. Kamma H, Yazawa T, Ogata T, Horiguchi H, Iijima T (1991) Expression of MHC class II antigens in human lung cancer cells. Virchows Arch B Cell Pathol Incl Mol Pathol 60: 407–412 [DOI] [PubMed] [Google Scholar]
- 33. Wroblewski JM, Bixby DL, Borowski C, Yannelli JR (2001) Characterization of human non‐small cell lung cancer (NSCLC) cell lines for expression of MHC, co‐stimulatory molecules and tumor‐associated antigens. Lung Cancer 33: 181–194 [DOI] [PubMed] [Google Scholar]
- 34. Hiroi M, Ohmori Y (2003) Constitutive nuclear factor kappaB activity is required to elicit interferon‐gamma‐induced expression of chemokine CXC ligand 9 (CXCL9) and CXCL10 in human tumour cell lines. Biochem J 376: 393–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. West AC, Mattarollo SR, Shortt J, Cluse LA, Christiansen AJ, Smyth MJ, Johnstone RW (2013) An intact immune system is required for the anticancer activities of histone deacetylase inhibitors. Can Res 73: 7265–7276 [DOI] [PubMed] [Google Scholar]
- 36. Zheng H, Zhao W, Yan C, Watson CC, Massengill M, Xie M, Massengill C, Noyes DR, Martinez GV, Afzal R et al (2016) HDAC inhibitors enhance T‐cell chemokine expression and augment response to PD‐1 immunotherapy in lung adenocarcinoma. Clin Cancer Res 22: 4119–4132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sowa ME, Bennett EJ, Gygi SP, Harper JW (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138: 389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Taguchi K, Motohashi H, Yamamoto M (2011) Molecular mechanisms of the Keap1‐Nrf2 pathway in stress response and cancer evolution. Genes Cells 16: 123–140 [DOI] [PubMed] [Google Scholar]
- 39. Lee DF, Kuo HP, Liu M, Chou CK, Xia W, Du Y, Shen J, Chen CT, Huo L, Hsu MC et al (2009) KEAP1 E3 ligase‐mediated downregulation of NF‐kappaB signaling by targeting IKKbeta. Mol Cell 36: 131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim JE, You DJ, Lee C, Ahn C, Seong JY, Hwang JI (2010) Suppression of NF‐kappaB signaling by KEAP1 regulation of IKKbeta activity through autophagic degradation and inhibition of phosphorylation. Cell Signal 22: 1645–1654 [DOI] [PubMed] [Google Scholar]
- 41. Fan W, Tang Z, Chen D, Moughon D, Ding X, Chen S, Zhu M, Zhong Q (2010) Keap1 facilitates p62‐mediated ubiquitin aggregate clearance via autophagy. Autophagy 6: 614–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Orthwein A, Noordermeer SM, Wilson MD, Landry S, Enchev RI, Sherker A, Munro M, Pinder J, Salsman J, Dellaire G et al (2015) A mechanism for the suppression of homologous recombination in G1 cells. Nature 528: 422–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wijdeven RH, Pang B, van der Zanden SY, Qiao X, Blomen V, Hoogstraat M, Lips EH, Janssen L, Wessels L, Brummelkamp TR et al (2015) Genome‐wide identification and characterization of novel factors conferring resistance to topoisomerase II poisons in cancer. Can Res 75: 4176–4187 [DOI] [PubMed] [Google Scholar]
- 44. Ito A, Shimazu T, Maeda S, Shah AA, Tsunoda T, Iemura S, Natsume T, Suzuki T, Motohashi H, Yamamoto M et al (2015) The subcellular localization and activity of cortactin is regulated by acetylation and interaction with Keap1. Sci Signal 8: ra120 [DOI] [PubMed] [Google Scholar]
- 45. Abou El Hassan M, Yu T, Song L, Bremner R (2015) Polycomb repressive complex 2 confers BRG1 dependency on the CIITA locus. J Immunol 194: 5007–5013 [DOI] [PubMed] [Google Scholar]
- 46. Morris AC, Spangler WE, Boss JM (2000) Methylation of class II trans‐activator promoter IV: a novel mechanism of MHC class II gene control. J Immunol 164: 4143–4149 [DOI] [PubMed] [Google Scholar]
- 47. Verma SK, Tian X, LaFrance LV, Duquenne C, Suarez DP, Newlander KA, Romeril SP, Burgess JL, Grant SW, Brackley JA et al (2012) Identification of potent, selective, cell‐active inhibitors of the histone lysine methyltransferase EZH2. ACS Med Chem Lett 3: 1091–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA et al (2013) Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci USA 110: 7922–7927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N et al (2008) Determination of the class and isoform selectivity of small‐molecule histone deacetylase inhibitors. Biochem J 409: 581–589 [DOI] [PubMed] [Google Scholar]
- 50. Wilting RH, Yanover E, Heideman MR, Jacobs H, Horner J, van der Torre J, DePinho RA, Dannenberg JH (2010) Overlapping functions of Hdac1 and Hdac2 in cell cycle regulation and haematopoiesis. EMBO J 29: 2586–2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hast BE, Goldfarb D, Mulvaney KM, Hast MA, Siesser PF, Yan F, Hayes DN, Major MB (2013) Proteomic analysis of ubiquitin ligase KEAP1 reveals associated proteins that inhibit NRF2 ubiquitination. Can Res 73: 2199–2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, Johansen T (2010) p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element‐driven gene transcription. J Biol Chem 285: 22576–22591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hast BE, Cloer EW, Goldfarb D, Li H, Siesser PF, Yan F, Walter V, Zheng N, Hayes DN, Major MB (2014) Cancer‐derived mutations in KEAP1 impair NRF2 degradation but not ubiquitination. Can Res 74: 808–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ruthenburg AJ, Li H, Milne TA, Dewell S, McGinty RK, Yuen M, Ueberheide B, Dou Y, Muir TW, Patel DJ et al (2011) Recognition of a mononucleosomal histone modification pattern by BPTF via multivalent interactions. Cell 145: 692–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J, Kauer M, Tackett AJ, Chait BT, Badenhorst P et al (2006) A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 442: 86–90 [DOI] [PubMed] [Google Scholar]
- 56. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282: 24131–24145 [DOI] [PubMed] [Google Scholar]
- 57. Jongsma ML, Berlin I, Wijdeven RH, Janssen L, Janssen GM, Garstka MA, Janssen H, Mensink M, van Veelen PA, Spaapen RM et al (2016) An ER‐associated pathway defines endosomal architecture for controlled cargo transport. Cell 166: 152–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz‐Meco MT, Moscat J (2008) The signaling adaptor p62 is an important NF‐kappaB mediator in tumorigenesis. Cancer Cell 13: 343–354 [DOI] [PubMed] [Google Scholar]
- 59. Furukawa M, Xiong Y (2005) BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3‐Roc1 ligase. Mol Cell Biol 25: 162–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Andrew AS, Jewell DA, Mason RA, Whitfield ML, Moore JH, Karagas MR (2008) Drinking‐water arsenic exposure modulates gene expression in human lymphocytes from a U.S. population. Environ Health Perspect 116: 524–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sikorski EE, Burns LA, McCoy KL, Stern M, Munson AE (1991) Suppression of splenic accessory cell function in mice exposed to gallium arsenide. Toxicol Appl Pharmacol 110: 143–156 [DOI] [PubMed] [Google Scholar]
- 62. Liu D, Wu D, Zhao L, Yang Y, Ding J, Dong L, Hu L, Wang F, Zhao X, Cai Y et al (2015) Arsenic trioxide reduces global histone H4 acetylation at lysine 16 through direct binding to histone acetyltransferase hMOF in human cells. PLoS One 10: e0141014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Takaya K, Suzuki T, Motohashi H, Onodera K, Satomi S, Kensler TW, Yamamoto M (2012) Validation of the multiple sensor mechanism of the Keap1‐Nrf2 system. Free Radic Biol Med 53: 817–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rockwell CE, Zhang M, Fields PE, Klaassen CD (2012) Th2 skewing by activation of Nrf2 in CD4(+) T cells. J Immunol 188: 1630–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schulze‐Topphoff U, Varrin‐Doyer M, Pekarek K, Spencer CM, Shetty A, Sagan SA, Cree BA, Sobel RA, Wipke BT, Steinman L et al (2016) Dimethyl fumarate treatment induces adaptive and innate immune modulation independent of Nrf2. Proc Natl Acad Sci USA 113: 4777–4782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mrowietz U, Christophers E, Altmeyer P (1999) Treatment of severe psoriasis with fumaric acid esters: scientific background and guidelines for therapeutic use. The German Fumaric Acid Ester Consensus Conference. Br J Dermatol 141: 424–429 [DOI] [PubMed] [Google Scholar]
- 67. Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M, Yang M, Raghupathi K, Novas M, Sweetser MT et al (2012) Placebo‐controlled phase 3 study of oral BG‐12 or glatiramer in multiple sclerosis. N Engl J Med 367: 1087–1097 [DOI] [PubMed] [Google Scholar]
- 68. Gold R, Kappos L, Arnold DL, Bar‐Or A, Giovannoni G, Selmaj K, Tornatore C, Sweetser MT, Yang M, Sheikh SI et al (2012) Placebo‐controlled phase 3 study of oral BG‐12 for relapsing multiple sclerosis. N Engl J Med 367: 1098–1107 [DOI] [PubMed] [Google Scholar]
- 69. Kryczek I, Bruce AT, Gudjonsson JE, Johnston A, Aphale A, Vatan L, Szeliga W, Wang Y, Liu Y, Welling TH et al (2008) Induction of IL‐17+ T cell trafficking and development by IFN‐gamma: mechanism and pathological relevance in psoriasis. J Immunol 181: 4733–4741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Karczewski J, Dobrowolska A, Rychlewska‐Hanczewska A, Adamski Z (2016) New insights into the role of T cells in pathogenesis of psoriasis and psoriatic arthritis. Autoimmunity 49: 435–450 [DOI] [PubMed] [Google Scholar]
- 71. Fan X, Zhang H, Cheng Y, Jiang X, Zhu J, Jin T (2016) Double roles of macrophages in human neuroimmune diseases and their animal models. Mediators Inflamm 2016: 8489251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Boven LA, Van Meurs M, Van Zwam M, Wierenga‐Wolf A, Hintzen RQ, Boot RG, Aerts JM, Amor S, Nieuwenhuis EE, Laman JD (2006) Myelin‐laden macrophages are anti‐inflammatory, consistent with foam cells in multiple sclerosis. Brain 129: 517–526 [DOI] [PubMed] [Google Scholar]
- 73. Stoof TJ, Flier J, Sampat S, Nieboer C, Tensen CP, Boorsma DM (2001) The antipsoriatic drug dimethylfumarate strongly suppresses chemokine production in human keratinocytes and peripheral blood mononuclear cells. Br J Dermatol 144: 1114–1120 [DOI] [PubMed] [Google Scholar]
- 74. Al‐Jaderi Z, Maghazachi AA (2016) Utilization of dimethyl fumarate and related molecules for treatment of multiple sclerosis, cancer, and other diseases. Front Immunol 7: 278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dubey D, Kieseier BC, Hartung HP, Hemmer B, Warnke C, Menge T, Miller‐Little WA, Stuve O (2015) Dimethyl fumarate in relapsing‐remitting multiple sclerosis: rationale, mechanisms of action, pharmacokinetics, efficacy and safety. Expert Rev Neurother 15: 339–346 [DOI] [PubMed] [Google Scholar]
- 76. Ghoreschi K, Bruck J, Kellerer C, Deng C, Peng H, Rothfuss O, Hussain RZ, Gocke AR, Respa A, Glocova I et al (2011) Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. J Exp Med 208: 2291–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhu K, Mrowietz U (2001) Inhibition of dendritic cell differentiation by fumaric acid esters. J Invest Dermatol 116: 203–208 [DOI] [PubMed] [Google Scholar]
- 78. Peng H, Guerau‐de‐Arellano M, Mehta VB, Yang Y, Huss DJ, Papenfuss TL, Lovett‐Racke AE, Racke MK (2012) Dimethyl fumarate inhibits dendritic cell maturation via nuclear factor kappaB (NF‐kappaB) and extracellular signal‐regulated kinase 1 and 2 (ERK1/2) and mitogen stress‐activated kinase 1 (MSK1) signaling. J Biol Chem 287: 28017–28026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Blewett MM, Xie J, Zaro BW, Backus KM, Altman A, Teijaro JR, Cravatt BF (2016) Chemical proteomic map of dimethyl fumarate‐sensitive cysteines in primary human T cells. Sci Signal 9: rs10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ockenfels HM, Schultewolter T, Ockenfels G, Funk R, Goos M (1998) The antipsoriatic agent dimethylfumarate immunomodulates T‐cell cytokine secretion and inhibits cytokines of the psoriatic cytokine network. Br J Dermatol 139: 390–395 [DOI] [PubMed] [Google Scholar]
- 81. de Jong R, Bezemer AC, Zomerdijk TP, van de Pouw‐Kraan T, Ottenhoff TH, Nibbering PH (1996) Selective stimulation of T helper 2 cytokine responses by the anti‐psoriasis agent monomethylfumarate. Eur J Immunol 26: 2067–2074 [DOI] [PubMed] [Google Scholar]
- 82. Gill AJ, Kolson DL (2013) Dimethyl fumarate modulation of immune and antioxidant responses: application to HIV therapy. Crit Rev Immunol 33: 307–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wu Q, Wang Q, Mao G, Dowling CA, Lundy SK, Mao‐Draayer Y (2017) Dimethyl fumarate selectively reduces memory T cells and shifts the balance between Th1/Th17 and Th2 in multiple sclerosis patients. J Immunol 198: 3069–3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Loewe R, Pillinger M, de Martin R, Mrowietz U, Groger M, Holnthoner W, Wolff K, Wiegrebe W, Jirovsky D, Petzelbauer P (2001) Dimethylfumarate inhibits tumor‐necrosis‐factor‐induced CD62E expression in an NF‐kappa B‐dependent manner. J Invest Dermatol 117: 1363–1368 [DOI] [PubMed] [Google Scholar]
- 85. Zheng L, Cardaci S, Jerby L, MacKenzie ED, Sciacovelli M, Johnson TI, Gaude E, King A, Leach JD, Edrada‐Ebel R et al (2015) Fumarate induces redox‐dependent senescence by modifying glutathione metabolism. Nat Commun 6: 6001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kastrati I, Siklos MI, Calderon‐Gierszal EL, El‐Shennawy L, Georgieva G, Thayer EN, Thatcher GR, Frasor J (2016) Dimethyl fumarate inhibits the nuclear factor kappab pathway in breast cancer cells by covalent modification of p65 protein. J Biol Chem 291: 3639–3647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jadhav SV, Bringas E, Yadav GD, Rathod VK, Ortiz I, Marathe KV (2015) Arsenic and fluoride contaminated groundwaters: a review of current technologies for contaminants removal. J Environ Manage 162: 306–325 [DOI] [PubMed] [Google Scholar]
- 88. Lau A, Whitman SA, Jaramillo MC, Zhang DD (2013) Arsenic‐mediated activation of the Nrf2‐Keap1 antioxidant pathway. J Biochem Mol Toxicol 27: 99–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. He X, Chen MG, Lin GX, Ma Q (2006) Arsenic induces NAD(P)H‐quinone oxidoreductase I by disrupting the Nrf2 × Keap1 × Cul3 complex and recruiting Nrf2 × Maf to the antioxidant response element enhancer. J Biol Chem 281: 23620–23631 [DOI] [PubMed] [Google Scholar]
- 90. Arita A, Shamy MY, Chervona Y, Clancy HA, Sun H, Hall MN, Qu Q, Gamble MV, Costa M (2012) The effect of exposure to carcinogenic metals on histone tail modifications and gene expression in human subjects. J Trace Elem Med Biol 26: 174–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Jo WJ, Ren X, Chu F, Aleshin M, Wintz H, Burlingame A, Smith MT, Vulpe CD, Zhang L (2009) Acetylated H4K16 by MYST1 protects UROtsa cells from arsenic toxicity and is decreased following chronic arsenic exposure. Toxicol Appl Pharmacol 241: 294–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chen X, Song M, Zhang B, Zhang Y (2016) Reactive oxygen species regulate T cell immune response in the tumor microenvironment. Oxid Med Cell Longev 2016: 1580967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Gallegos CE, Michelin S, Dubner D, Carosella ED (2016) Immunomodulation of classical and non‐classical HLA molecules by ionizing radiation. Cell Immunol 303: 16–23 [DOI] [PubMed] [Google Scholar]
- 94. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A et al (2015) Proteomics. Tissue‐based map of the human proteome. Science 347: 1260419 [DOI] [PubMed] [Google Scholar]
- 95. Frey WD, Chaudhry A, Slepicka PF, Ouellette AM, Kirberger SE, Pomerantz WCK, Hannon GJ, Dos Santos CO (2017) BPTF maintains chromatin accessibility and the self‐renewal capacity of mammary gland stem cells. Stem Cell Reports 9: 23–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Rocha N, Kuijl C, van der Kant R, Janssen L, Houben D, Janssen H, Zwart W, Neefjes J (2009) Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7‐RILP‐p150 Glued and late endosome positioning. J Cell Biol 185: 1209–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Paul P, van den Hoorn T, Jongsma ML, Bakker MJ, Hengeveld R, Janssen L, Cresswell P, Egan DA, van Ham M, Ten Brinke A et al (2011) A Genome‐wide multidimensional RNAi screen reveals pathways controlling MHC class II antigen presentation. Cell 145: 268–283 [DOI] [PubMed] [Google Scholar]
- 98. Gobin SJ, Peijnenburg A, Keijsers V, van den Elsen PJ (1997) Site alpha is crucial for two routes of IFN gamma‐induced MHC class I transactivation: the ISRE‐mediated route and a novel pathway involving CIITA. Immunity 6: 601–611 [DOI] [PubMed] [Google Scholar]
- 99. Voorhoeve PM, le Sage C, Schrier M, Gillis AJ, Stoop H, Nagel R, Liu YP, van Duijse J, Drost J, Griekspoor A et al (2006) A genetic screen implicates miRNA‐372 and miRNA‐373 as oncogenes in testicular germ cell tumors. Cell 124: 1169–1181 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3