

Abstract
Impaired cell polarity is a hallmark of diseased tissue. In the cardiovascular system, laminar blood flow induces endothelial planar cell polarity, represented by elongated cell shape and asymmetric distribution of intracellular organelles along the axis of blood flow. Disrupted endothelial planar polarity is considered to be pro‐inflammatory, suggesting that the establishment of endothelial polarity elicits an anti‐inflammatory response. However, a causative relationship between polarity and inflammatory responses has not been firmly established. Here, we find that a cell polarity protein, PAR‐3, is an essential gatekeeper of GSK3β activity in response to laminar blood flow. We show that flow‐induced spatial distribution of PAR‐3/aPKCλ and aPKCλ/GSK3β complexes controls local GSK3β activity and thereby regulates endothelial planar polarity. The spatial information for GSK3β activation is essential for flow‐dependent polarity to the flow axis, but is not necessary for flow‐induced anti‐inflammatory response. Our results shed light on a novel relationship between endothelial polarity and vascular homeostasis highlighting avenues for novel therapeutic strategies.
Keywords: atherosclerosis, cell polarity, endothelial cell, flow, PAR‐3
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Immunology; Vascular Biology & Angiogenesis
Introduction
Cell polarity is the asymmetric organization of cellular components, such as organelles and cytoskeleton. Intrinsic cellular asymmetry is crucial for cell type‐specific functions. Consequently, the establishment and maintenance of cell polarity are stringently regulated during tissue morphogenesis and in homeostasis. Conversely, loss of cell polarity is often a hallmark of diseases including cancer and cardiovascular dysfunction 1. In vertebrates, endothelial cells (ECs), the cells forming the inner lining of the vasculature, have apical–basal polarity to establish a barrier between blood and the rest of the body 2, 3, 4. Furthermore, ECs have profound morphological adaptation to hemodynamic shear stress and possess planar cell polarity to the direction of blood flow 5, 6. To establish functional vessels, dynamic changes of microtubules and the actin cytoskeleton are observed with maintaining blood flow. Cell shape is elongated along the axis of flow, and the microtubule organization centers (MTOCs) and the Golgi apparatus are aligned in front or behind the nuclei toward the flow direction 7, 8, 9, 10. In contrast, regions of disrupted flow in the aorta often show roundish cell shape and disorganized Golgi orientation, which is highly associated with a pro‐inflammatory condition 5. Importantly, atherosclerotic plaques occur at specific sites in arteries where blood flow is slow and patterns are disturbed 6. However, a causal link between compromised endothelial polarity toward the flow direction and vascular inflammatory responses has not been firmly established.
Cell polarization is achieved by integration of both extra‐ and intracellular signaling cascades controlling cytoskeletal dynamics. The signaling crosstalk among Rho family small GTPases, Cdc42, Rac1, and RhoA, regulates cytoskeletal reorganization mediated by key polarity regulators, the PAR polarity protein complex 11. The PAR complex is composed of PAR‐3, PAR‐6, and atypical PKC (aPKC) and functions in various cell polarization events across species 12. Activated Cdc42 binds to the PAR‐6/aPKC complex, leading to aPKC activation and promoting association with PAR‐3 13. PAR‐3 directly interacts with Rac1 activation factor, Tiam1/2, and further forms a complex with aPKC, PAR‐6, and Cdc42, thereby mediating Cdc42‐induced Rac1 activation for actin cytoskeletal reorganization 14, 15. aPKC forms a protein complex with glycogen synthase kinase‐3 beta (GSK3β) in migrating astrocytes and during epithelial cell death 16, 17. aPKC forms a complex with active form of GSK3β, whereas aPKC activity is required for GSK3β inactivation, leading to the stabilization of microtubules and MTOC reorientation. In the cardiovascular system, it has been shown that PAR‐3 regulates sprouting behavior of endothelial cells (ECs) during angiogenesis 18; meanwhile, the role of PAR‐3 in endothelial polarization in living organisms remains elusive.
In light of these observations, we investigate the role of endothelial PAR‐3 in EC polarization. EC‐specific inducible PAR‐3 loss‐of‐function mice exhibit compromised endothelial polarity to the flow axis in a flow‐rate‐dependent manner but do not show overt effects on apical–basal polarity. Shear stress controls the spatio‐temporal antagonism of the PAR‐3/aPKClambda/iota (aPKCλ), one of two isoforms of aPKC, complex versus the GSK3β/aPKCλ complex through the RhoA/Rho‐kinase pathway, resulting in spatially controlled microtubule stabilization in the direction of flow. Moreover, vascular inflammatory responses are increased by regulating NF‐κB, a key regulator of inflammation, nuclear localization downstream of GSK3β in PAR‐3 loss‐of‐function conditions. Importantly, pharmacological suppression of GSK3β restored increased NF‐κB nuclear localization but not endothelial polarity to the flow axis. Our results indicate an unexpected relationship between endothelial polarity to flow and vascular inflammation downstream of PAR‐3.
Results
PAR‐3 controls endothelial polarity toward the direction of flow in a shear rate dependent manner but is not essential for apical–basal polarization in the growing vasculature
To gain more insight into the role of PAR‐3 in endothelial polarity, we first observed the effect of PAR‐3 on shear stress‐dependent endothelial polarization in response to blood flow. EC nuclei and Golgi, visualized by immunostaining with ERG1/2/3, a marker of EC nucleus 19 and GM130 or GOLPH4 antibodies, respectively, showed that Golgi was regularly found in the upstream side of the nuclei in vessels at postnatal day 6 (P6) retina (Fig 1A). This was particularly obvious in ECs in the major vessels, including the artery and vein (Fig 1A). In contrast, Golgi orientation was often reversed in ECs of Pard3, the gene encoding PAR‐3, EC‐specific inducible KO mice (Pard3 iΔEC) (Fig 1A). To further analyze the misorientation of Golgi toward the flow direction throughout the entire retinal vasculature, we employed a computational approach to calculate the efficiency of endothelial adaptation to flow by measuring the angle between EC axial polarity vectors (EC nucleus‐to‐Golgi axis) and the predicted blood flow vectors 10, 20, 21. All of the vascular beds in the retina were categorized into four groups: artery, vein, capillary, and sprouting front (Appendix Fig S1A); polarization of ECs toward the flow direction determined by Polarity Index was measured (Fig 1B). While the majority of the ECs showed polarized Golgi orientation in each vascular bed (Appendix Fig S1B), endothelial Golgi orientation against flow in Pard3 iΔEC mutants was significantly disrupted in the artery, vein, and capillaries of the vascular plexus, but not in the sprouting front (Fig 1C). Interestingly, correlative analysis of wall shear stress (WSS) and EC polarization in the capillary vascular bed of control and Pard3 iΔEC mice showed that EC polarity is compromised in low‐to‐medium WSS regions, but not at high levels of WSS in the retina (Fig 1D).
Figure 1. PAR‐3 is important for the establishment of endothelial polarity toward the flow axis.
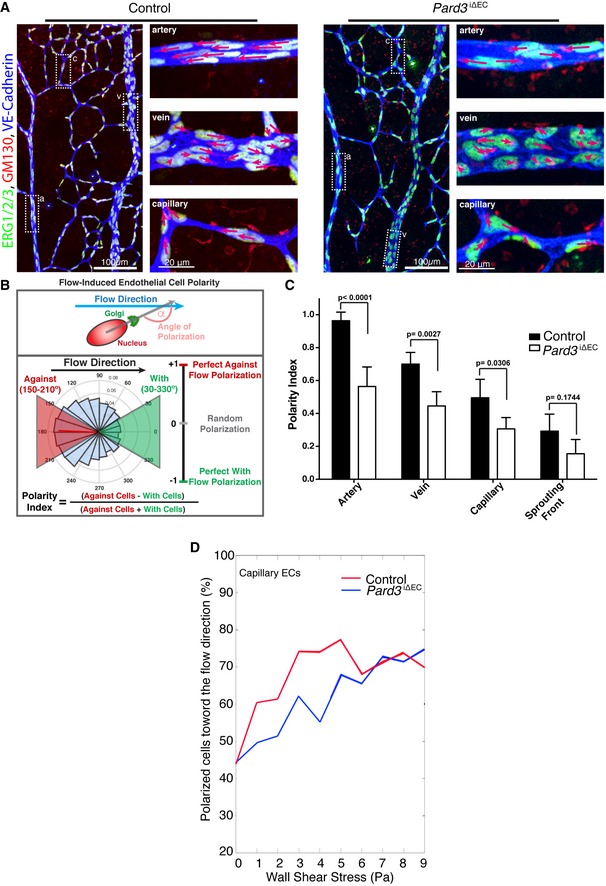
- Endothelial axial polarity phenotype in control and Pard3 iΔEC P6 mouse retinal vasculature. Left panels show a large field of view of corresponding retinal vasculature. Right panels show Golgi orientation of ECs in designated areas, corresponding to highlighted areas in left panel (a: artery; v: vein; c: capillary). Green: ERG1/2/3; red: GM130; blue: VE‐cadherin. Arrows are drawn from the center of EC nuclei to the Golgi. Scale bars: 100 μm (left columns, lower magnification) and 20 μm (right columns, higher magnification).
- Schema for the Polarity Index calculation. The angle of polarization (α) was calculated from the flow direction and the angle of the nucleus‐to‐Golgi vector.
- Analysis of the endothelial Polarity Index, relative to predicted blood flow for each specific vascular bed (n = 3 mice). Data are presented as mean ± SD. P‐values are indicated in the figure (Student's t‐test).
- Correlative analysis of wall shear stress and EC polarization in the capillary vascular bed.
PAR‐3 controls cell‐to‐cell contact formation and thereby epithelial polarization 22. To investigate the role of PAR‐3 in EC polarity in the growing vasculature, we next examined EC‐EC junction and apical–basal polarity markers in P6 mouse retinal vasculature with immunostaining. Vascular endothelial (VE)‐cadherin establishes a homophilic complex at the joint point and forms an adherens junction, which defines apical–basal polarity 3. Podocalyxin is widely expressed on the apical surface of lumenized vascular endothelial cells, whereas collagen type IV is secreted by the basal region of the endothelium 23, 24. While Pard3 iΔEC mutants showed the expected sprouting defects as previously reported 18 (Fig EV1A–C), immunostaining within anti‐VE‐cadherin antibody in Pard3 iΔEC retina did not reveal any clear defects (Fig EV1A). Three‐dimensional reconstituted images of the retinal vasculature stained with anti‐VE‐cadherin, anti‐Podocalyxin, and anti‐collagen type IV antibodies further confirmed equivalent apical–basal polarization between control and Pard3 iΔEC mice (Fig EV1D). These results suggest that PAR‐3 is important for endothelial polarity to the flow axis in vivo at low‐to‐moderate but not at high levels of shear stress nor apical–basal polarization during angiogenesis.
Figure EV1. PAR‐3 KO does not exhibit overt defects on adherens junction formation and apical–basal polarization in the retinal vasculature.
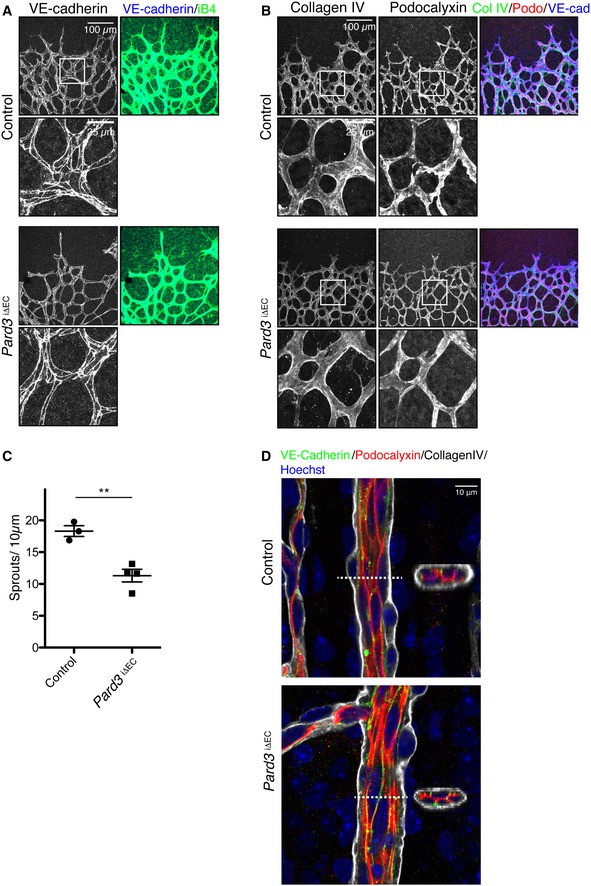
- Staining of control and Pard3 iΔEC P6 mice retina with adherens junction marker (VE‐cadherin) and endothelial cell marker isolectin‐B4 (iB4).
- Staining of control and Pard3 iΔEC P6 retina with basement membrane marker (collagen IV), apical membrane marker (podocalyxin), and isolectin‐B4 (iB4).
- Quantification of the number of sprouts/10 μm in angiogenic front. Data are presented as mean ± SEM (n = 3 retinas). Difference **P < 0.01, analyzed by Student's t‐test.
- Staining of control and Pard3 iΔEC P6 retina artery with basement membrane marker (collagen IV), apical membrane marker (podocalyxin), adherens junction marker (VE‐cadherin), and nuclei (Hoechst 33342, Sigma). 3D‐reconstituted cross‐section images from the region indicated with white dashed lines are shown in the images.
PAR‐3 controls endothelial polarity to the flow axis and inflammatory responses in established vessels
The inner curvature of the aorta is an area where ECs experience low‐to‐moderate WSS and disturbed flow, and which is prone to atheroma plaque formation 6. To examine whether the role of PAR‐3 in endothelial polarity toward the flow axis is restricted only to developmental processes, we next investigated endothelial polarity toward the flow axis in the aortic arch in the established vessels of adult Pard3 iΔEC mice. En face confocal microscopy analysis of ECs in the inner curvature to sidewall at the proximal arch showed a gradient of morphological diversity (Figs 2A and EV2A). To quantify endothelial polarity toward the flow axis at the arterial arch, we measured the angle of the EC axial polarity vector with the proximal–distal axis and a Polarity Index was calculated (Figs EV2B and 1B). To evaluate EC orientation, we measured the angle between the primary axis of the best fitting ellipse to the EC shape and the orthogonal axis of the direction of flow (Fig EV2C). In control mice, ECs showed roundish shape at the inner curvature, whereas those at the sidewall were aligned and showed a polarized morphology (Fig 2A–C). Consistent with our observation in the retinal vasculature (Fig 1), endothelial Golgi in the sidewall exhibited polarized localization against the flow direction; however, ECs in the inner curvature did not (Fig 2A–C). At the marginal zone, the region in between the sidewall and the inner curvature, endothelial orientation, and Golgi axis were also polarized even though cell shapes were roundish (Fig 2A–C). In Pard3 iΔEC mice, Golgi orientation was significantly compromised only in ECs at the marginal zone, whereas cell elongation and orientation were not affected (Fig 2A–C). These observations further suggest that PAR‐3 controls endothelial polarity toward the flow axis in response to moderate shear stress.
Figure 2. Loss of endothelial PAR‐3 disrupts regional axial cell polarity and increases pro‐inflammatory response of ECs.
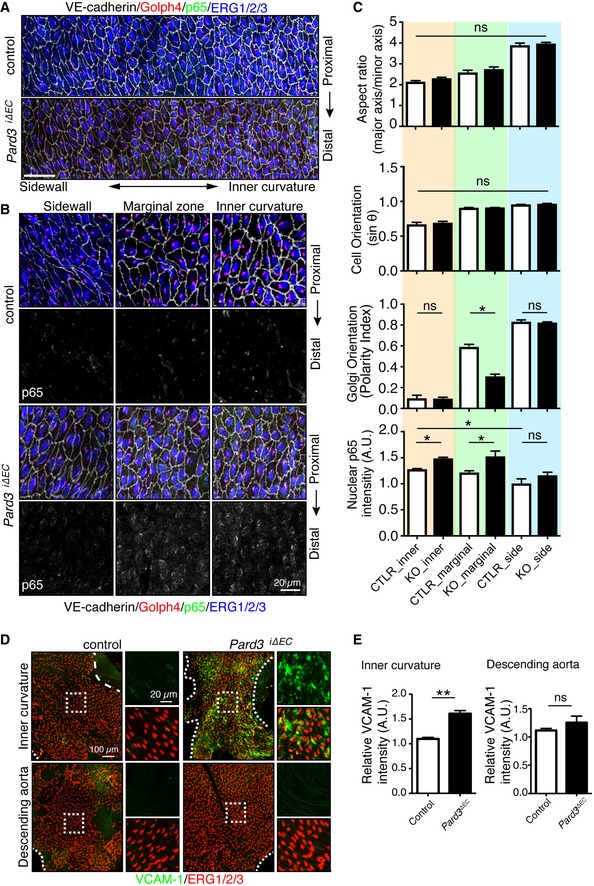
- En face confocal microscopy of the aortic arch from the inner curvature to sidewall, stained with anti‐VE‐cadherin (gray), GOLPH4 (red), NF‐κB p65 subunit (green), and ERG1/2/3 (blue) from P56 male mice. Axis of the aorta is indicated on right side of images.
- Higher magnification images from (A). Lower panels show p65 signal (gray).
- Regional quantification of the cell elongation, cell orientation, Golgi orientation (Polarity Index), and nuclear p65 intensity of ECs.
- En face confocal microscopy of the inner curvature of the aortic arch and descending aorta from P56 control and Pard3 iΔEC mice. VCAM‐1 is green, and EC nuclei (ERG1/2/3) are red.
- Quantification of the relative intensity of VCAM‐1 signal in the ECs of inner curvature of the aorta and descending aorta from control and Pard3 iΔEC mice.
Figure EV2. Analysis of ECs in the aorta.
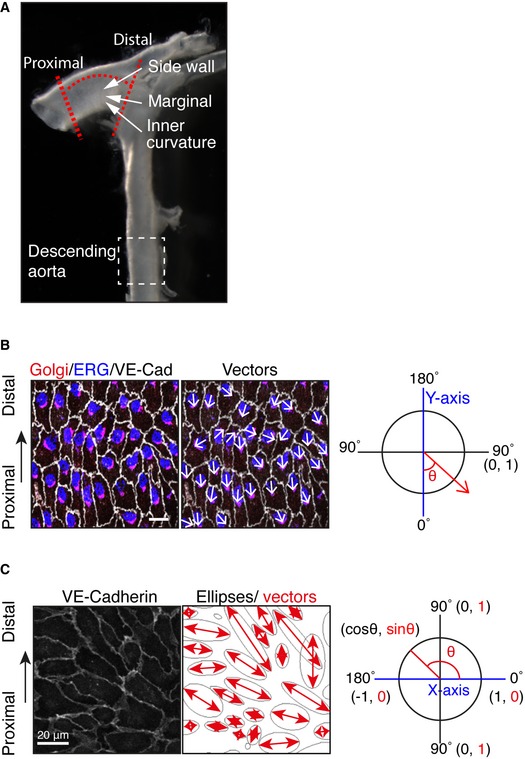
- To examine the effect of loss of PAR‐3, the highlighted region of the aorta within the red dotted lines was dissected and analyzed.
- To quantify endothelial Golgi orientation toward flow, the angle between the axial polarity vector and the proximal–distal vector (−1 to 1) was analyzed. White arrows indicate the axial polarity vectors from each EC.
- Left panel shows the VE‐cadherin signal of aorta ECs. Right panel shows the ellipse fitted to each of the EC, generated by ImageJ software. Red arrows indicate the vectors of the major axis of each ellipse. Distribution of the sin(θ) of the angle between the axial polarity vector and the proximal–distal vector was analyzed (0 to 1).
The inner curvature of the aorta is an area prone to atherosclerosis. Disrupted endothelial polarity toward the flow axis is considered to be pro‐inflammatory, which increases the susceptibility for atherosclerotic plaque formation 25, 26, 27. To clarify the relationship between endothelial polarity toward the flow axis and endothelial inflammatory responses, we examined the NF‐κB complex nuclear localization, a key regulator of inflammation. p65, a major component of the NF‐κB complex, immunoreactivity was gradually increased in the ECs from the sidewall to the inner curvature in both control and Pard3 iΔEC mice. Suggestive of a molecular connection, p65 staining intensity inversely related to an increased endothelial polarization (Fig 2B and C). However, significantly nuclear‐localized p65 protein was found at the marginal and the inner curvature in Pard3 iΔEC aortas but control mice showed diffuse localization (Fig 2B and C). On the other hand, Golgi orientation at the inner curvature was not affected in mutants (Fig 2B and C). Active NF‐κB induces expression of vascular cell adhesion molecule‐1 (VCAM‐1), controlling the adhesion of leukocytes to the endothelium 28. In accordance, Pard3 iΔEC mice showed a dramatic increase in VCAM‐1 protein expression in the inner curvature of the aorta compared to control mice (Fig 2D and E). Interestingly, ECs of the descending aorta in Pard3 iΔEC mice did not show VCAM‐1 upregulation, where ECs are subjected to high shear stress (Fig 2D and E), confirming the specific requirement of PAR‐3 signaling in low‐to‐moderate WSS regions. These results were further confirmed with primary cultured aortic ECs. When ECs from control mice were subjected to flow, the VCAM‐1 mRNA level was downregulated. However, this was compromised in ECs isolated from PAR‐3 KO mice (Appendix Fig S2). Taken together, these results suggest that PAR‐3 simultaneously negatively regulates NF‐κB activation and positively controls Golgi orientation in response to low‐to‐moderate blood flow.
Atherosclerosis formation was increased in PAR‐3 EC‐specific inducible KO mice
As disrupted endothelial polarity toward the flow axis and inflammation is linked to atherosclerosis, we hypothesized that Pard3 iΔEC mice would be more susceptible to the development of atherosclerotic lesions. Thus, we next bred Pard3 iΔEC mice with apolipoprotein (Apo) E KO mice, a well‐established model to study atherosclerosis. To induce PAR‐3 gene knockout, tamoxifen was injected daily from P42 to P46, then control and Pard3 iΔEC mice were placed on high‐fat diet. To analyze the effect of PAR‐3 KO on the onset of atherosclerotic plaque formation, mice were sacrificed after 10 weeks of high‐fat diet feeding. Then, aortas were collected and stained with Oil Red O to highlight lipid accumulation. Control ApoE −/−mutant animals showed Oil Red O staining at the inner curvature and branching points of aortic arches but not in the descending aorta (Fig 3A and B). However, under these conditions, Pard3 iΔEC/ApoE −/− mutants showed a significant expansion of the region stained with Oil Red O (Fig 3A and B), while neither serum cholesterol levels nor body weight was affected (Fig 3C and D). To further confirm the role of PAR‐3 in atherosclerosis formation, en face staining of the aortic arch was performed with an antibody against MOMA‐2, a marker of monocytes/macrophages. MOMA‐2‐positive area was increased in Pard3 iΔEC/ApoE −/− mice in the neointima of aortic arch when compared to control mice, indicating an increase in macrophage infiltration in endothelial‐specific Pard3 loss‐of‐function mice (Fig 3E and F). Thus, PAR‐3 inhibits atherosclerosis onset by blocking endothelial inflammation.
Figure 3. Loss of endothelial PAR‐3 accelerates regional atherosclerosis development.
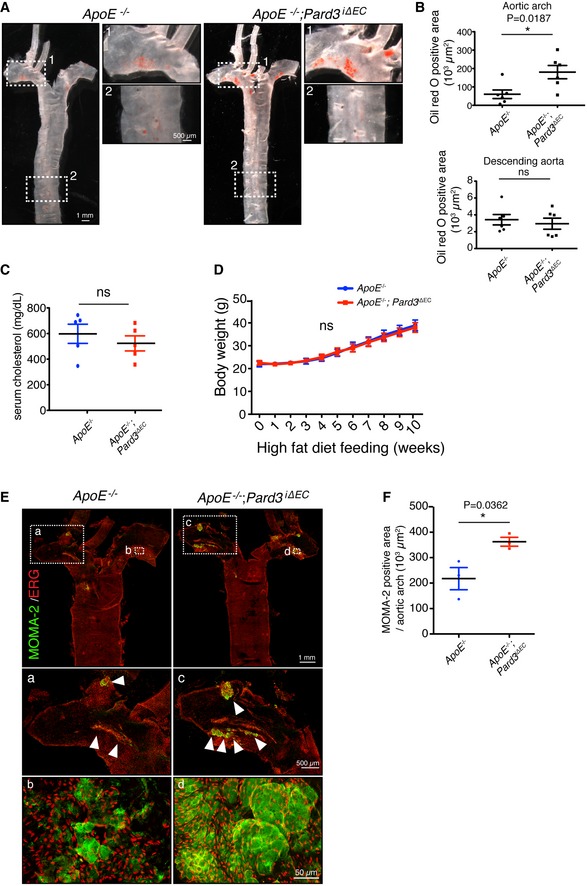
- Representative aorta of mice fed with high‐fat diet for 10 weeks (18‐week‐old male mice) stained en face with Oil Red O. Smaller panels show higher magnification images from the aortic arch (1) and descending aorta (2).
- Quantification of Oil Red O‐positive regions in aortic arch (upper panel) and descending aorta (lower panel).
- Quantification of serum cholesterol level of control (ApoE −/−) and ApoE −/−; Pard3 iΔEC animals after 10 weeks of high‐fat diet feeding.
- Body weights of control (ApoE −/−) and ApoE −/−; Pard3 iΔEC animals after 10 weeks of high‐fat diet feeding.
- Representative aorta of mice fed with high‐fat diet for 10 weeks (18‐week‐old male mice) stained en face with monocyte/macrophage marker (MOMA‐2, green) and EC marker (EGR, red). White arrowheads indicate MOMA‐2‐positive areas. Smaller panels (a, b, c, d) show higher magnification images from the aortic arch.
- Quantification of MOMA‐2‐positive areas in the aortic arch.
The antagonism between the PAR‐3/aPKC complex and the aPKC/GSK3β complex regulates GSK3β activation
To gain mechanistic insight into the role of PAR‐3 in endothelial polarity toward the flow axis in response to shear stress, we established an in vitro culture system. We confirmed efficient knocked down (KD) of PAR‐3 in HUVECs with two different siRNAs (siPAR‐3#1 and #2) (Appendix Fig S3A), and these siRNAs were used to examine the function of PAR‐3 in flow‐mediated polarity establishment. Confluent HUVECs were seeded in flow chambers coated with fibronectin and exposed to a range of shear stress. Consistent with the in vivo observations, Golgi polarization was compromised in PAR‐3 KD cells in the presence of low‐to‐moderate flow but not when exposed to high flow (Fig EV3A–C). Moreover, ECs isolated from Pard3 iΔEC and control mice aorta were subjected to the same range of shear stress. We confirmed the flow‐rate‐dependent compromised Golgi orientation in Pard3 iΔEC aortic ECs (Appendix Fig S3B). However, the amount of ECs isolated from mice was limited. Thus, we decided to use HUVEC for further analysis.
Figure EV3. PAR‐3 regulates EC Golgi reorientation toward flow in vitro .
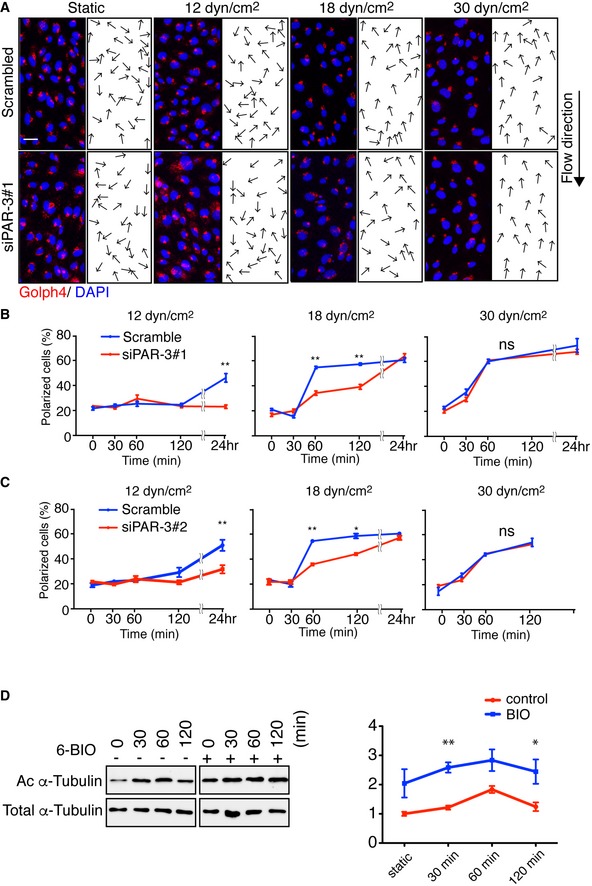
-
ARepresentative images of flow chamber‐cultured ECs transfected with control (Scrambled) or PARD3‐specific siRNA (siPAR‐3#1) and exposed to the indicated value of shear stress for 60 min. The cells were stained for nuclei (DAPI, blue) and Golgi apparatus (GOLPH4, red). Black arrows indicate corresponding axial polarity vectors. Flow direction is indicated on the right. Scale bar, 35 μm.
-
B, CAxial polarity of ECs treated with control (Scrambled) or PARD3‐specific siRNAs (siPAR‐3#1, B; or siPAR‐3#2, C) in response to 12, 18, and 30 dyn/cm2 laminar flow for the indicated time.
-
CWestern blotting of EC lysates treated with 18 dyn/cm2 laminar flow for indicated times, with 1 μM of 6BIO (+) or control (−) containing growth medium. Upper panels show the blot of acetylated α‐tubulin (Ac α‐tubulin), and lower panels show total tubulin. Quantitation of relative intensity of Ac α‐tubulin is shown.
GSK3β is a ubiquitously expressed and constitutively active protein kinase, which was implicated in cytoskeletal reorganization, a number of chronic diseases and inflammation 29, 30. Phosphorylation of GSK3β at serine 9 (S9) residue downregulates its catalytic activity 29, 30. aPKC, a member of the PAR polarity complex, forms a complex with unphosphorylated active form of GSK3β, and S9 phosphorylation dissociates the complex 16. Additionally, the role of GSK3β on microtubules stabilization and Golgi polarization under flow has been shown 31. The dual role of PAR‐3 in polarity and inflammation prompted us to investigate the link between GSK3β and the PAR polarity complex in the context of endothelial flow response. However, the role of PAR‐3 in aPKC/GSK3β complex dynamics is unclear. Thus, we first examined the effect of flow on GSK3β activity. Phosphorylation of GSK3β at S9 was increased in 30 min under 18 dyn/cm2 flow and was sustained for 120 min; meanwhile, it was compromised in PAR‐3 KD cells (Fig 4A and Appendix Fig S3C). Under these conditions, tubulin acetylation, a marker of stabilized microtubules, was increased in a time‐course‐dependent manner in control ECs but not in PAR‐3 KD cells in response to flow (Fig 4A). PAR‐3 KD did not affect GSK3β S9 phosphorylation in low‐ and high‐flow conditions (Appendix Fig S3D and E). Overexpression of PAR‐3 with aPKCλ HEK293 cells resulted in increased phosphorylation of GSK3β at S9 in a PAR‐3 dose‐dependent manner (Fig 4B). Conversely, treatment with GSK3β inhibitor, 6BIO, in cultured ECs resulted in an increased amount of acetylated tubulin both with and without flow (Fig EV3D). These results suggest that PAR‐3 controls microtubule stability via inhibition of GSK3β in a dose‐dependent manner. The previous report showed that PAR‐3 does not form a complex with GSK3β16; therefore, we hypothesize that the dose‐dependent effect of PAR‐3 on GSK3β phosphorylation is due to antagonism between the aPKC/PAR‐3 and the aPKC/GSK3β complexes (Fig 4C). To test this hypothesis, PAR‐3, GSK3β, and aPKCλ were transiently overexpressed in HEK293 cells and immunoprecipitated with PAR‐3. Consistent with previous reports, aPKCλ, but not GSK3β, was co‐immunoprecipitated with PAR‐3 (Appendix Fig S4A) 16. Furthermore, aPKCλ, but not PAR‐3, was co‐immunoprecipitated with GSK3β (Appendix Fig S4B). These results support our hypothesis of two different protein complexes. PAR‐3/aPKCλ complex formation has been shown to be controlled by RhoA/Rho‐kinase pathway 32. To test if Rho‐kinase could influence differential PAR‐3/aPKC/GSK3β complex formation, we treated HEK293 cells with Rho‐kinase inhibitor, Y‐27632, and examined complex formation by immunoprecipitation. The amount of aPKCλ co‐precipitated with PAR‐3 was increased by 1.5 times compared to control after treatment with the Rho‐kinase inhibitor (Appendix Fig S4A). Under these conditions, the amount of aPKCλ‐GSK3β co‐precipitation was decreased by approximately 50% compared to control (Appendix Fig S4B). Thus, Rho‐kinase activity favors aPKCλ/GSK3β complex over PAR‐3/aPKCλ complex. Previous reports have shown that RhoA/Rho‐kinase activity in ECs is affected by flow 33, 34. To further confirm this complex formation, we first immunoprecipitated aPKCλ in HUVECs and observed that both PAR‐3 and GSK3β were co‐precipitated (Fig 4D). Rho‐kinase inhibitor treatment increased the amount of co‐precipitated PAR‐3, whereas the amount of GSK3β was decreased (Fig 4D). Finally, we investigated the effect of laminar flow on the complex formation. When HUVECs were placed under moderate flow (18 dyn/cm2), PAR‐3/aPKCλ complex formation was induced (Fig 4E). In contrast, the amount of the aPKCλ/GSK3β complex was reduced (Fig 4E). These results suggest that blood flow could control the antagonism between the PAR‐3/aPKCλ and aPKCλ/GSK3β complexes through RhoA activity (Fig 4F).
Figure 4. Balance between the PAR‐3/aPKCλ complex versus the aPKCλ/GSK3β complex modulates GSK3β activity.
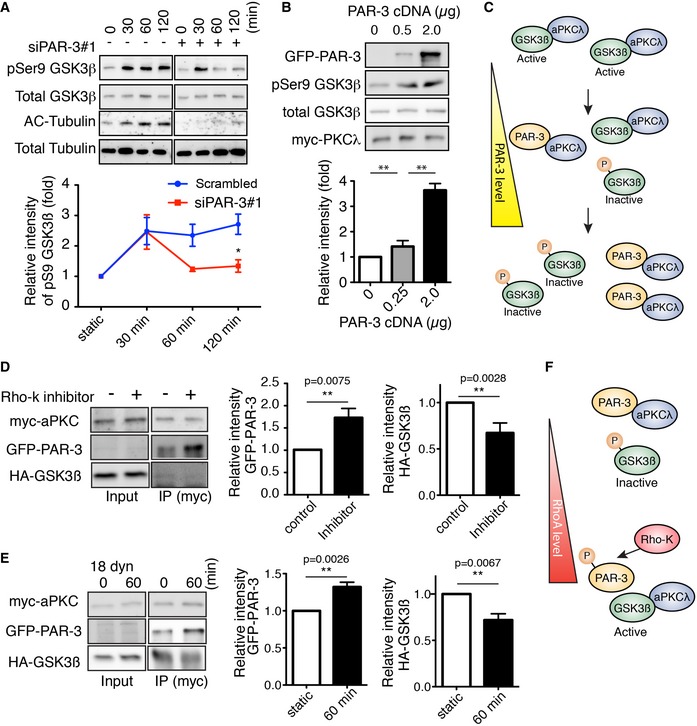
- Western blotting of ECs transfected with control scrambled siRNA (indicated as “‐” in the figure) or siPAR‐3#1 and exposed to 18 dyn/cm2 laminar flow.
- HEK293 cells were transfected with different amounts of GFP‐PAR‐3 cDNA with HA‐GSK3β and myc‐aPKCλ and were analyzed by Western blotting.
- Model for PAR‐3‐mediated GSK3β inactivation. The balance between PAR‐3/aPKCλ complex versus the aPKCλ/GSK3β complex controls GSK3β activity.
- HUVECs were transfected with indicated cDNAs and were treated with or without Rho‐kinase inhibitor (Y‐27632, 20 μM). After immunoprecipitation with indicated antibodies, samples were analyzed by Western blotting.
- HUVECs were transfected with indicated cDNAs, seeded in the flow chamber, and exposed to 18 dyn/cm2 laminar flow for 60 min. After immunoprecipitation with anti‐myc antibody, samples were analyzed with indicated antibodies by Western blotting. Relative intensity of each signal was statistically analyzed.
- RhoA/Rho‐kinase pathway controls GSK3β activity by modulating balance between the PAR‐3/aPKCλ complex versus the aPKCλ/GSK3β complex.
Spatio‐temporal antagonism between the PAR‐3/aPKC complex and aPKC/GSK3β complex regulated by RhoA controls microtubule stabilization under flow
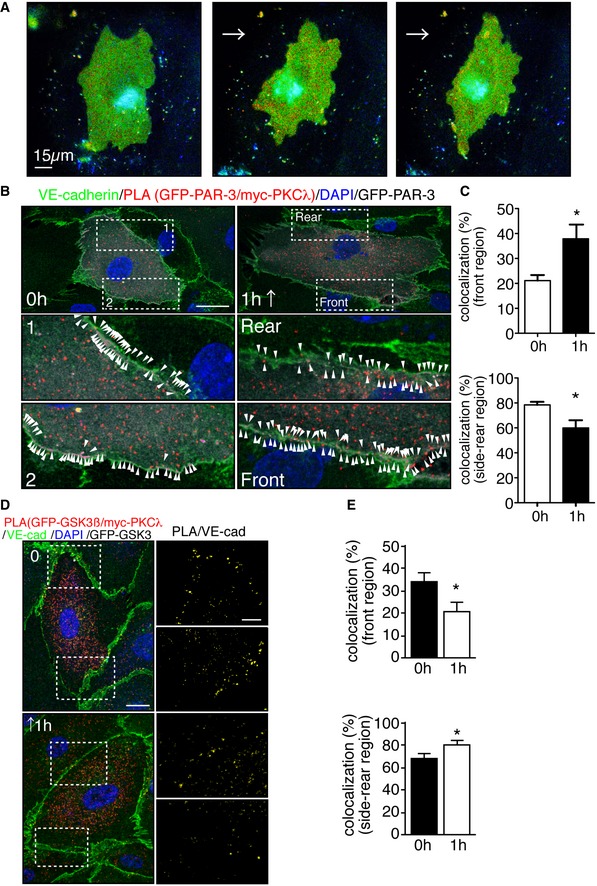
Interestingly, blood flow has been shown to spatio‐temporally restricted Rac1 activity in cultured ECs 33, 35, which is known to antagonize RhoA activity 11. We next examined spatio‐temporal RhoA activation in ECs under flow. A lentiviral vector containing RhoA Fluorescence resonance energy transfer (FRET)‐biosensor, Raichu‐RhoA, was used to infect ECs 36. Infected HUVECs were subjected to moderate flow. Remarkably, we observed restricted FRET signal at the rear region of ECs, distal to the flow direction (Fig 5A and B, and Movie EV1). Conversely, low flow failed to induce spatially restricted RhoA activation (Fig EV4A and Movie EV2). Given the spatial distribution of RhoA activation under flow, we next investigated the spatial distribution of PAR‐3/aPKCλ and aPKCλ/GSK3β complexes in ECs exposed to shear stress. HUVECs were treated with flow, and spatial distribution of endogenous PAR‐3/aPKCλ was examined via the proximity ligation assay (PLA). The aPKCλ/PAR‐3 complex was observed more frequently in the front region of ECs than the rear region under flow and treatment of ECs with the Rho‐kinase inhibitor equalized the distribution of this protein complex within the cells (Fig 5C and D). Conversely, the aPKC/GSK3β complex was observed more frequently in the rear region of EC, which was equalized by Rho‐kinase inhibitor treatment (Fig 5E and F). To further confirm the effect of moderate flow on the distribution of these protein complexes, HUVECs were transfected with GFP‐PAR‐3/myc‐aPKCλ or GFP‐GSK3β/myc‐aPKCλ and subjected to flow. Consistently, the GFP‐PAR‐3/myc‐aPKCλ complex was observed more frequently in the front region of ECs than the rear region under flow and GFP‐GSK3β/myc‐aPKCλ was more abundant at the rear region of ECs (Fig EV4B–E). Concurrently, acetylated tubulin accumulated in the front region of HUVECs (Fig 5G and H). Altogether, these data depict that flow‐dependent RhoA/Rho‐kinase activity determines the spatial distribution of PAR‐3/aPKCλ and aPKCλ/GSK3β complexes, which in turn delimits GSK3β activity and microtubule stability in ECs exposed to moderate blood flow.
Figure 5. Spatio‐temporal antagonism of the PAR‐3/aPKCλ complex versus the aPKCλ/GSK3β complex controls microtubule stabilization under flow.
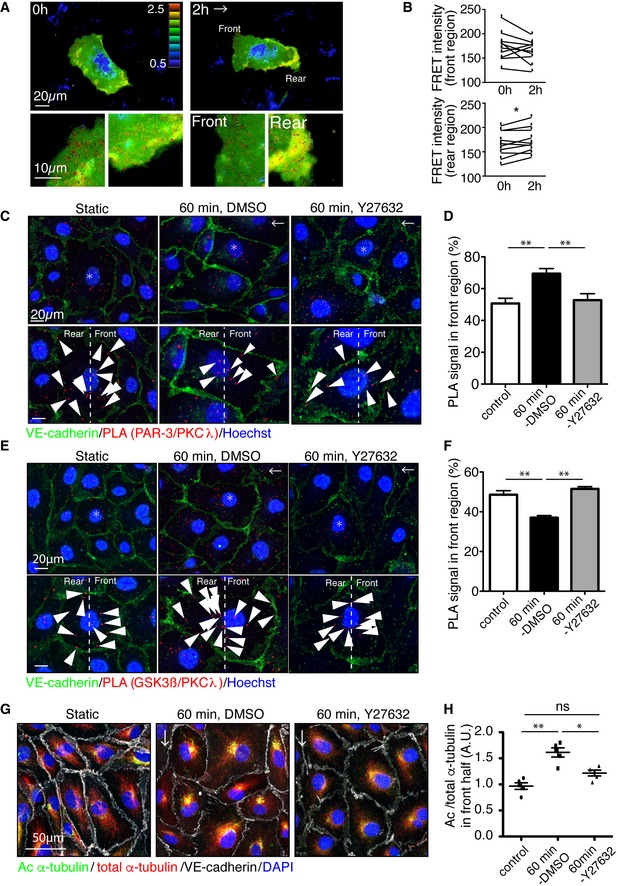
-
ARepresentative time‐lapse images of ECs transfected with RhoA biosensor under 18 dyn/cm2 laminar flow. FRET/CFP ratio is represented in IMD mode.
-
BStatistical analysis of FRET intensity of EC front and rear region.
-
C–FPLA analysis of Par3 and aPKCλ (C, D) or GSK3β and aPKCλ (E, F) in EC in static and treated with 18 dyn/cm2 laminar flow for 60 min. Arrowheads indicate PLA signal. (D, F) Quantification of the PLA images. Percentile of the PLA signal in the front half of the ECs toward flow was calculated.
-
GAcetylated and total α‐tubulin in ECs under 18 dyn/cm2 flow, with or without Rho‐kinase inhibitor (Y‐27632, 20 μM).
-
HQuantification of regional Ac/total α‐tubulin ratio in the front and rear half of each cells.
Figure EV4. Spatio‐temporal antagonism of the PAR‐3/aPKCλ complex versus the aPKCλ/GSK3β complex controls microtubule stabilization under flow.
- Representative time‐lapse images of ECs transfected with RhoA biosensor. FRET/CFP ratio is represented in IMD mode. Cells were subjected to laminar flow of 12 dyn/cm2 flow for 2 h. Scale bar, 15 μm.
- PLA in ECs expressing GFP‐PAR‐3 and myc‐PKCλ in static and under 18 dyn/cm2 flow for 1 h. Arrowheads indicate PAR‐3/aPKCλ PLA signal colocalized with EC junction. Scale bar, 20 μm.
- Quantification of the images shown in (B).
- Representative images of PLA in ECs expressing GFP‐GSK3β and myc‐PKCλ in static (0 h) and after 1 h subjected to 18 dyn/cm2 flow. EC junction (VE‐cadherin) is green, nuclear stain (DAPI) is blue, and GFP‐GSK3β is gray. Right panels show higher magnification images of the indicated areas. Yellow signals indicate PLA signal colocalized with EC junction. Scale bars, 10 μm (left panel) and 20 μm (right panel).
- Quantification of percentile of the PLA signals in the front and rear region of the ECs under static and 1‐h treatment with flow.
PAR‐3 controls NF‐κB nuclear localization via GSK3β in a manner independent of EC polarity toward the flow axis
GSK3β has also been shown to control endothelial inflammatory responses via modulation of NF‐κB activity 37. Consistently, GSK3β inhibition results in reduced vascular inflammation in vivo 38. To understand the relationship between the PAR complex and GSK3β activity in response to flow, we examined the effect of GSK3β inhibition on flow‐mediated Golgi polarization and on NF‐κB complex nuclear localization. Treatment of ECs with 6BIO normalized p65 nuclear accumulation in PAR‐3‐deficient ECs (Appendix Fig S4A and B), but did not have additional effects on Golgi polarization against the flow direction (Appendix Fig S4C and D). Next, ECs isolated from Pard3 KO and control mice were incubated with 6BIO in the presence of flow. In 30 to 60 min, nuclear localization of p65 was increased, which was further enhanced in ECs isolated from PAR‐3 KO mice aorta. Treatment of ECs with 6BIO normalized p65 nuclear accumulation in PAR‐3‐deficient ECs (Fig EV5).
Figure EV5. PAR‐3 controls nuclear localization of p65 in aortic endothelial cells in a GSK3β activity‐dependent manner.
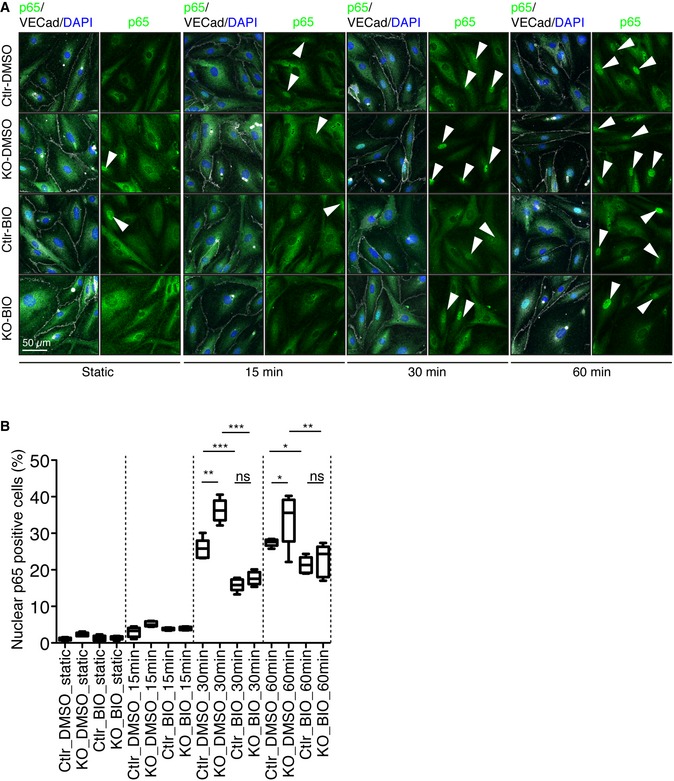
- Representative images of control and Pard3KO ECs treated with control (DMSO) or GSK3β inhibitor (6‐BIO, 1 μM) under static conditions or after subjected to 12 dyn/cm2 flow for the indicated times. EC junction (VE‐cadherin) is gray, NF‐κB p65 subunit (p65) is green, and nuclear stain (DAPI) is blue. Scale bar, 50 μm. White arrowheads indicate p65‐positive nuclei.
- Quantitative analysis of the percentile of p65‐positive ECs. Data are shown as box and whisker plots. The box spans the interquartile range, horizontal central values are median, and the whiskers extend to the highest and lowest observations (n = 50 cells); statistical significance (*P < 0.05, **P < 0.01, ***P < 0.001) was evaluated by one‐way ANOVA and Tukey's multiple comparison post hoc analysis.
Finally, we examined the effect of GSK3β inhibition in EC polarity toward the flow axis and inflammation in vivo. CHIR99021, a specific inhibitor for GSK3β, was injected into Pard3 iΔEC mice, followed by en face confocal microscopy analysis of aortic ECs. In control mice, Golgi orientation in the marginal zone was compromised by the inhibitor injection. Consistent with the in vitro observations, compromised endothelial polarity toward the flow axis in Pard3 iΔEC mice was not enhanced by the inhibitor injection (Fig 6A and B), Under these conditions, increased endothelial nuclear localization of p65 in Pard3 iΔEC mice at the inner curvature and the marginal zone of the aorta was rescued by the inhibitor injection (Fig 6A and C). These observations indicate that PAR‐3 is essential for flow‐dependent polarity toward the flow axis, but is not necessary for an anti‐inflammatory response.
Figure 6. GSK3β indirectly controls EC polarity toward the flow axis and anti‐inflammatory effects in vivo .
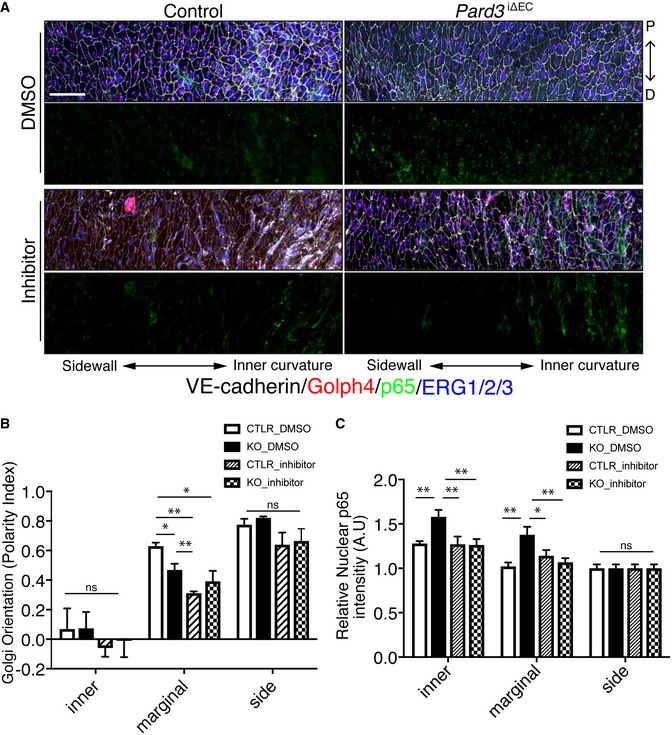
-
AEn face confocal microscopy of the aortic arch from the inner curvature to sidewall of control and mutant mice treated with GSK3β inhibitor. Scale bar: 50 μm.
-
B, CRegional quantification of Golgi orientation (Polarity Index, B) and nuclear p65 intensity (C) of ECs. Data are presented as mean ± SEM (n = 5 mice, n = 50 cells from each region). Differences: *P < 0.05, **P < 0.01, analyzed with two‐way ANOVA with Tukey's multiple comparison post hoc analysis.
Discussion
Establishment of cell polarity is essential for tissue formation and homeostasis, whereas disrupted cell polarity is often seen in disease conditions. Although loss of cell polarity was previously considered a by‐product of abnormal cell accumulation, a body of evidence has shown a causative role in tumor initiation and progression of attenuated cell polarity 39. Loss of PAR‐3 results in impaired apical–basal polarization in epithelial cells and promotes tumorigenesis in breast cancer 40, 41. In contrast, PAR‐6 and aPKC are often overexpressed in various cancers 1. In the cardiovascular system, ECs possess apical–basal polarity and polarity toward the flow axis. Shear stress induces endothelial cytoskeletal reorganization, cell shape elongation, and alignment toward the flow direction in vitro and in vivo. Conversely, ECs have a roundish and unpolarized shape when exposed to a low rate of flow or disturbed flow and therefore are subjected to low shear stress 6. Endothelial PAR‐3 did not seem to be critical for the establishment of apical–basal polarity and cell–cell junction formation (Fig EV1). However, PAR‐3 controlled cytoskeletal remodeling and Golgi reorientation in a flow‐rate‐dependent manner, suggesting its role in endothelial sensitization toward shear stress. Compromised EC polarity toward the flow axis was assumed to be a hallmark of the pro‐inflammatory condition based on a number of clinical observations 6, 42. Although these previous observations suggest a causative link of EC polarity toward the flow axis to anti‐inflammatory responses, this relationship has not been investigated in living organisms. Here, we have shown that ECs at the sidewall of the aorta have an elongated and aligned shape with oriented Golgi; meanwhile, ECs at the inner curvature have roundish shapes with unpolarized Golgi. On the other hand, ECs at the marginal zone in between the sidewall and inner curvature did not have elongated and oriented shapes, but EC Golgi were polarized toward the proximal–distal axis of the aorta (Figs 2 and 7). Blood flow around the marginal zone might be slower than that around the sidewall; however, flow over time is directed. Thus, Golgi orientation is the more sensitive marker than cell shape and alignment not only in the culture system, but also in the aorta. NF‐κB pathway is a key regulator of endothelial pro‐inflammatory response. Strong shear stress triggers anti‐inflammatory response by excluding NF‐κB from the EC nucleus, whereas low and pulsative flow activate NF‐κB DNA binding 43, 44. In control animals, immunoreactivity of a p65 antibody was observed in the ECs of the inner curvature and was gradually decreased to the sidewall of the aortic arch. Under these conditions, Pard3 iΔEC mice showed significantly elevated NF‐κB expression and nuclear localization at both the inner curvature and the marginal zone of the aorta, while mislocalization of Golgi was only observed at the marginal zone (Fig 2A). Additionally, a previous study showed that disturbed flow increases p65 expression level via JNK1 activation 45 and that loss of PAR‐3 activates JNK1 in tumor cells 46. These reports suggest that in Pard3 iΔEC mice aorta nuclear p65 translocation is controlled by PAR‐3/GSK3β axis, whereas p65 protein upregulation may be mediated by the PAR‐3/JNK1 pathway. These observations further support our conclusion that PAR‐3 is the sensitizer of ECs to shear stress and indicate that elevated NF‐κB nuclear localization occurs in a manner independent of EC polarity toward the flow axis, represented by Golgi orientation. Consistent with this, Pard3 iΔEC mice had increased VCAM‐1 expression at the inner curvature of the aortic arch but not at the descending aorta and higher levels of atherosclerotic plaque formation in the ApoE‐deficient background (Fig 3). Using the flow chamber system, we found that flow‐induced S9 phosphorylation of GSK3β was not sustained and, therefore, GSK3β is overactivated in PAR‐3 loss of function. Under these conditions, Golgi misorientation in PAR‐3 KD cultured ECs was not restored by GSK3β inhibitor; meanwhile, increased NF‐κB p65 subunit nuclear localization in PAR‐3 KD ECs and PAR‐3 KO ECs was rescued by GSK3β inhibitor (Fig EV5 and Appendix Fig S5). Furthermore, these observations were supported in the in vivo model (Fig 6). Taken together, our data suggest that spatio‐temporally regulated GSK3β activation is important for cytoskeletal reorganization, but spatial information within the individual ECs was not critical for vascular inflammatory responses (Fig 7). Cytoskeletal reorganization is a dynamic process and is important for motile behavior of cells. Consistently, vessel pruning, which is achieved by directed cell migration 10, was increased in P6 retinal vasculature of EC‐specific PAR‐3 KO mice 18. This may, therefore, be linked to defective EC Golgi orientation in ECs.
Figure 7. Discrete regulation of axial cell polarity and inflammation by the balance between the PAR‐3/aPKCλ and aPKCλ/GSK3β complexes.
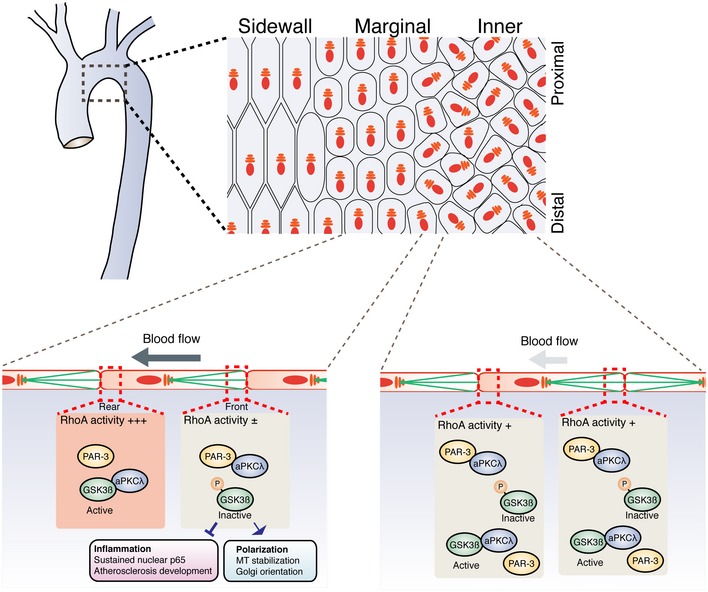
Laminar flow discretely controls endothelial polarity toward the flow axis and vascular inflammatory responses. In the aortic arch, EC Golgi at the sidewall and marginal regions are polarized toward the flow direction. On the other hand, ECs at the inner curvature are not polarized toward the flow direction and have a high risk for atherosclerotic plaque formation. ECs at the marginal region and the inner curvature in Pard3 iΔEC mice showed misoriented Golgi, while those at the sidewall do not. Middle flow restricted RhoA activation to the rear region of ECs, resulting in the non‐uniform distribution of the PAR‐3/aPKCλ and aPKCλ/GSK3β complexes along the flow direction. In the front region, aPKCλ forms a complex with PAR‐3, and hence, the free inactive form of GSK3β is increased. This GSK3β inactivation mediates both EC planar cell polarization and pro‐inflammatory responses. While spatial distribution of GSK3β activation would be important of EC polarity toward the flow axis, local activation of GSK3β is not essential for vascular inflammation.
In this study, we found that PAR‐3 modulates GSK3β activity via its interaction with aPKCλ. Laminar flow‐mediated phosphorylation of the S9 residue of GSK3β in 30 min, and this phosphorylation was sustained for 120 min (Fig 4A). GSK3β phosphorylation at S9 was increased in 30 min in PAR‐3 KD ECs under flow, while it was not sustained for 120 min (Fig 4A). These observations may suggest that the responsible kinase for S9 could be different in a time‐course‐dependent manner. Consistently, many protein kinases, including PKC, AKT, and PKA, could phosphorylate and control GSK3β activity 47. Interestingly, it has been shown that GSK3β phosphorylates aPKC and promotes its degradation via the ubiquitin system 48. The balance between the aPKC/GSK3β and the aPKC/PAR‐3 complexes might be involved in a positive or negative feedback signaling of GSK3β, therefore acting as a signaling hub for cellular responses.
An increasing body of evidence highlights the clinical impact of Rho‐kinase inhibitors and GSK3β inhibitors on the treatment of many diseases, including cardiovascular dysfunction 49. Abnormal RhoA/Rho‐kinase activation contributes to smooth muscle contraction observed in vasospasm, hypertension, or pulmonary hypertension. In contrast, inhibition of Rho‐kinase by a Rho‐kinase inhibitor, fasudil, decreases phosphorylation of MLC and thereby leading to vasodilation 50, 51, 52, 53. Additionally, long‐term treatment with fasudil improves vascular inflammation, remodeling, and atherosclerosis 54. RhoA/Rho‐kinase induces actomyosin reorganization and indirectly controls gene transcription via serum response factor (SRF) and myocardin‐related transcription factor (MRTF) 55, 56. GSK3β is also involved in a wide variety of disease states including inflammation, but to generate a kinase activity‐specific inhibitor is challenging 57. Our results suggest that the cell polarity complexes control key regulators of inflammatory response by modulating GSK3β activity downstream of Rho‐kinase. Recently, an integrin‐YAP/TAZ signaling cascade, which controls cell proliferation, was identified as a target of statins in the context of atherosclerosis formation 58, 59. Interestingly, RhoA regulates YAP/TAZ activation via cytoskeletal reorganization 60, and here, we show RhoA controls the antagonism between PAR‐3/aPKCλ and aPKCλ/GSK3β complexes. Thus, it is tempting to speculate that regulation of RhoA by blood flow is atheroprotective. Genetic association studies linking polarity complexes with inflammatory disease such as atherosclerosis have not been reported; therefore, our findings might stimulate numerous future studies and could be useful for the development of new therapeutic interventions.
Materials and Methods
Mouse genetics
Experiments involving animals were conducted in accordance with institutional guidelines and laws, and following the protocols approved by the local animal ethics committees and authorities (Regierungspraesidium Darmstadt, B2/1073 and B2/1122, and Universidade de Lisboa). Chd5‐CreERT2 transgenic mice were bred onto a background of animals containing a loxP‐flanked Pard3 18. Cre activity in male and female neonatal mice was induced by intra‐peritoneal (ip) injection of tamoxifen (T5648, Sigma‐Aldrich, MO) from P1 to P3 according to the protocol as previously described 18. The phenotype of the mutant mice was analyzed at P6, and tamoxifen‐injected Chd5‐CreERT2‐negative littermates were used as controls. The knockout efficiency was confirmed as previously reported 18. To induce Cre activity in adult male mice, ip injection of tamoxifen was performed from P42 to P46, and the phenotype was analyzed at P56.
For atherosclerosis analysis, B6.129P2‐Apoetm1Unc/J 61 mice were bred with Pard3 flox/flox; Chd5‐CreERT2 mice. Cre activity in male mice was induced by ip injection of tamoxifen from P42 to P46. Both control and Pard3‐deficient mice in ApoE background were maintained on a mixture of normal chow and high‐fat diet (E15126‐34, ssniff EF R/M with 30% Fat; ssniff Spezialdiäten, Germany) for 1 week after the injection; then, they were fed high‐fat diet for 10 weeks. Littermates without Chd5‐CreERT2 allele were used as control (ApoE −/−, Fig 2).
For rescue experiments, Pard3 flox/flox; Chd5‐CreERT2 male mice were used. Cre activity was induced by ip injection of tamoxifen from P42 to P46. 50 mg/kg of CHIR99021 (InSolution GSK‐3 Inhibitor XVI, CHIR99021, 361571) or DMSO was ip injected at P56. Twelve hours after injection, the mice were deeply anesthetized and sacrificed with trans‐cardiac perfusion and the aorta was dissected.
Retina staining
For retina staining, eyes of P6 mice were prefixed in 2% PFA for 10 min at room temperature and retinas were dissected in PBS. Eyeballs were further dissected, and the retinas were fixed in 4% PFA for 10 min at room temperature, permeabilized, and blocked in 1% BSA (Sigma, A4378) and 0.3% Triton X‐100 overnight at 4°C with gentle rocking. Next, they were washed three times in Pblec buffer (1 mM CaCl2, 1 mM MgCl2, 1 mM MnCl2, and 1% Triton X‐100 in PBS) and incubated with biotinylated isolectin B4, a marker of ECs 62 (Vector, B‐1205, Griffonia simplicifolia lectin I, 1:50) and primary antibodies, overnight at 4°C with gentle rocking VE‐cadherin (AF938, R&D systems); Golgi (GM130α, 560066, BD Pharmingen, Franklin Lakes, NJ; Anti‐GOLPH4 antibody, ab28049, Abcam, Cambridge, UK); collagen type IV (Anti‐Collagen Antibody, Type IV, AB756P, Merck Millipore); Mouse Podocalyxin Antibody (AF1556, R&D systems); and nucleus (Hoechst 33342, Sigma‐Aldrich, MO). Retinas were washed five times with 0.5% BSA and 0.15% Triton X‐100 and incubated with Alexa Fluor‐coupled streptavidin (Invitrogen, 1:100) and the corresponding Alexa Fluor‐coupled secondary antibody (Invitrogen, 1:500) in blocking buffer for 2 h at room temperature and mounted using Fluoromount‐G (SouthernBiotech, 0100‐01).
Cell lines and primary cultures
Pooled human umbilical vein endothelial cells (HUVECs) were purchased from Pelobiotech (Frankfurt, Germany) and used between passages 2 and 4. Mouse aortic ECs were isolated according to a previous report with modifications. 63 Three Pard3 flox/flox; Chd5‐CreERT2 mice at age P56 were deeply anesthetized with Ketamine/Xylazine injection, and the aortas were harvested. The aortas were incubated with Dispase II (4 U/ml, D4693, Sigma) for 60 min at 37°C, and ECs were dissociated by flushing the inner lumen of the aortas with 5 ml of growth medium (EGM‐2, CC‐3162, Lonza, Basel, Switzerland or Endothelial Cell Growth Medium 2, C‐22011, PromoCell, Germany). The ECs were isolated with rat anti‐VE‐Cadherin antibody (555289, BD Biosciences) and sheep anti‐rat IgG‐coated Dynabeads (11035, Thermo Fisher Scientific; Waltham, MA). To induce gene KO, the ECs were incubated with growth medium containing 1 μM 4‐hydroxytamoxifen (H6278, Sigma) or PBS (control) for 48 h prior to flow treatment. ECs were cultured in EGM‐2 medium (CC‐3162, Lonza; Basel, Switzerland). HEK293 cells were obtained from the American Type Culture Collection (LGC Standards, Molsheim Cedex, France) and cultured in DMEM (D6546, Sigma) with 10% fetal bovine serum (FBS; Biochrom GmbH, Berlin, Germany), 2 mM l‐glutamine (25030‐024, Gibco, Invitrogen, Life Technologies; Darmstadt, Germany), 100 IU/ml penicillin, and 100 μg/ml streptomycin.
Gene knockdown strategies
Oligonucleotide siRNA duplexes were purchased from GE Healthcare Dharmacon, Inc. Individual ON‐TARGETplus PARD3 (J‐015602‐05 5′‐AAGCAUGGAUUUAGGUAUA‐3′; J‐015602‐06 5′‐AGACUAAACUCAAUACAGU‐3′; J‐015602‐07 5′‐CGAUAAAGACAGACUGGUA‐3′; J‐015602‐08 5′‐GAUGGCGACCUUCGAAAUA‐3′) or scrambled control (D‐001810‐01 ON‐TARGETplus Non‐targeting siRNA#1, 5′‐UGGUUUACAUGUCGACUAA‐3′) was transfected into HUVECs using oligofectamine (12252011, Thermo Fisher) according to the manufacturer's instructions. Briefly, 4 × 100,000 HUVECs were seeded onto 60‐mm dish and cultured with growth medium (EGM‐2, 2% FBS) for 12 h. Thirty minutes before transfection, cells were washed once with PBS and incubated with Opti‐MEM. Scrambled or siPARD3s were diluted with Opti‐MEM, mixed with oligofectamine, and incubated for 30 min at room temperature. siRNA mixture was added onto the dish at final concentration of 100 nM, incubated for 6 h at 37°C, and added twice the volume of the growth medium containing 6% FBS (final concentration: 2% FBS). After 12 h, medium was changed to normal growth medium (2% FBS). Cells were analyzed 48 h post‐transfection. J‐015602‐08 and J‐015602‐06 were used in this study as siPAR‐3#1 and siPAR‐3#2, respectively.
Cell culture and microfluidic chamber experiments
All flow applying experiments were performed with ibidi pump system, a combination of pump, fluidic unit, and controlling software (10902; ibidi, Germany) with ibidi perfusion chamber slides (μ‐Slide I 0.4 Luer ibiTreat, 80176 and μ‐Slide IV 0.4 ibiTreat, 80606; ibidi, Germany) with perfusion set RED (10962; ibidi). Chambers were coated with fibronectin (human plasma, 354008, Corning, NY) with 1.5 μg/cm2 in PBS for 1 h at 37°C. Twelve hours post‐transfection, 2 × 100,000 HUVECs were plated in the ibidi slides (μ‐Slide I) and maintained in growth medium. Local shear stress was calculated using pump controlling software (ibidi) with viscosity 0.07.
Golgi orientation and EC polarity analysis
Cells were exposed to shear stress for 0, 30, 60, 120 min, or 24 h using growth media, then fixed using 4% PFA for 10 min, and washed three times in PBS. Cells were stained for EC junction (VE‐cadherin, AF938, R&D systems), Golgi (GM130, 560066, BD Pharmingen, Franklin Lakes, NJ; Anti‐GOLPH4 antibody, ab28049, Abcam, Cambridge, UK), and nucleus (DAPI, 1/10,000, Sigma‐Aldrich, MO). Orientation of the Golgi was evaluated by looking at the angle of the vector from the center of nucleus toward the center of Golgi compared to flow direction using ImageJ software (NIH, MD). For quantification, the angles were converted to cos(θ) using Excel software (Microsoft, DC; Figs 2, 6, EV2 and EV3). To analyze the polarization, the ECs whose Golgi was within 90° of angle toward flow (−45° to 45°) were defined as “polarized”. For each experiment, three fields containing more than 90 cells have been analyzed.
Cell shape analysis
Using ImageJ software (NIH, MD), EC junction images (VE‐cadherin) were converted from 8‐bit images to the thresholded images. From these images, the shape of ECs was extracted and ellipses that fit to the cell shape were generated using “analyze particles” command. The angle of the ellipses toward the axis of aorta (in vivo, Figs 2 and EV2) and the aspect ratio (ratio of the major axis length to the minor axis length of ellipses) were calculated by ImageJ. For quantification, the angle of ellipses was converted to sin(θ) using Excel software (Microsoft, DC).
Immunoprecipitation from cultured cells
HEK cells were seeded into 60‐mm plate, incubated in DMEM/10%FBS for 12 h, and transfected with pEGFP‐PAR‐3, pCAGGS‐myc‐aPKCλ, pCAGGS‐HA‐GSK3β, and pCAGGS‐FLAG‐GSK3β or control empty vectors with PEI max (Polysciences, PA). HUVECs were seeded in 10‐cm plate, incubated in growth medium (2% FBS) for 12 h, and transfected with jetPEI‐HUVEC (108‐05N, Polyplus, Illkirch, FRANCE) with 12 μg DNA and 36 μl of jetPEI/10‐cm dish. Cells were maintained in the growth medium for 12 h after transfection and proceed to further treatments. For flow treatment, cDNA‐transfected ECs were seeded in four μ‐Slide I 0.4 coated with fibronectin, 2 × 100,000 cells each slide. For inhibitor treatment, cells were starved with serum (12 h for HEK cells and 4 h for HUVECs) and incubated with fresh growth medium for 30 min with control solvent or Rho‐k inhibitor (20 μM Y‐27632, 688002, Merck Millipore, MA). Cells were washed once with ice‐cold PBS and harvested with lysis buffer (50 mM Tris–HCl, pH 7.4; 1% NP‐40; 150 mM NaCl; Protease inhibitor cocktail, P2714, Sigma; Phosphatase Inhibitor Cocktail Set V, 524629, Merck Millipore). HUVECs seeded in four flow chambers were harvested with 300 μl lysis buffer using 1‐ml syringe and Serial Connector tube 10830, ibidi). All harvesting procedure was performed on ice with buffers containing protease/phosphatase inhibitors and EDTA to prevent unexpected stimulation. Cell lysates were sonicated with a Bioruptor (Diagenode, NJ, USA), centrifuged for 15 min, and protein concentration of the supernatant was determined using a BCA protein assay (Pierce BCA Protein Assay Kit, 23225, Thermo). The normalized supernatants were mixed with 1 μg of anti‐GFP antibody and 10 μl bed volume of Dynabeads Protein A (10001D, Thermo Fisher; GFP immunoprecipitation), 15 μl bed volume of anti‐FLAG‐M2 magnetic beads (M8823, SIGMA; Flag immunoprecipitation), or 10 μl bed volume of anti‐c‐Myc magnetic beads (88842, Thermo Fisher; myc immunoprecipitation), and incubated for 1 h at 4°C with rotation. Beads were washed with lysis buffer five times, and the immunoprecipitates were boiled with 1× SDS sample buffer (50 mM Tris–HCl pH 6.8, 2% SDS, 10% glycerol, 1% β‐mercaptoethanol, 12.5 mM EDTA, and 0.02% bromophenol blue) for 15 min and analyzed by Western blotting using specific antibodies.
Western blot analysis of cultured cell lysates
HEK cells were rinsed once with ice‐cold PBS, harvested with 1× SDS sampling buffer (50 mM Tris–HCl pH 6.8, 2% SDS, 10% glycerol, 1% β‐mercaptoethanol, 12.5 mM EDTA, 0.02% bromophenol blue), and boiled at 95°C for 10 min. To prepare HUVEC cell lysate from flow chambers, cells were rinsed once with ice‐cold PBS and harvested with 100 μl of 1× SDS sample buffer without β‐mercaptoethanol and bromophenol blue. The lysates were sonicated with a Bioruptor (Diagenode, NJ, USA) to reduce viscosity, and protein concentration was determined using a BCA protein assay kit (Pierce BCA Protein Assay Kit). The lysates were then supplemented with β‐mercaptoethanol and bromophenol blue and boiled. Protein concentration was normalized with 1xSDS buffer. Western blot analysis was carried out according to standard laboratory practices.
Staining of tissues and cells
For en face aorta staining, whole aortas were fixed in 4% PFA for 10 min on ice. Aortas were permeabilized and blocked in 1% BSA and 1% Triton X‐100 in PBS buffer overnight at 4°C with gentle rocking and incubated with indicated antibodies. For multiple labeling using rabbit‐derived antibodies, blocking of rabbit IgG with anti‐rabbit IgG F(ab′)2 fragment from donkey (NA9340V, GE Healthcare, UK; 1/200 dilution) was performed. First, aortas were incubated with anti‐VE‐cadherin (BD Biosciences, 555289, 1/300) and anti‐NF‐κB p65 (8242, Cell Signaling Technology, MA) antibodies, followed with Alexa Fluor‐coupled secondary antibody (1/500). Next, free rabbit IgG was blocked with anti‐rabbit IgG F(ab′)2 fragment with overnight incubation at 4°C with gentle rocking, followed with post‐fixation with 4% PFA for 10 min at RT. This procedure was repeated twice, with anti‐GOLPH4 antibody (Abcam, ab28049; 1/300) and anti‐ERG 1/2/3 antibody (ab92513, 1/500). The aortas were dissected and flat‐mounted using Fluoromount‐G.
Staining of cells in the perfusion chamber was performed according to the company's instruction. Briefly, 2 × 100,000 HUVECs were seeded in the μ‐Slide I 0.4 Luer (ibidi) and incubated for 24 h before flow treatment. Cells within the chamber were fixed with 4% PFA for 10 min at room temperature, washed with PBS three times, and permeabilized with 0.1% Triton X‐100 for 10 min. The cells were incubated with specific primary and secondary antibodies diluted with antibody buffer (0.1% BSA/PBS).
Quantification of atherosclerosis in the aorta
Aortas of 18‐week‐old mice were fixed with 4% PFA for 30 min at RT, followed with 60% isopropanol for 5 min, and stained with 3% Oil Red O for 15 min. For en face immunostaining, aortas from control (ApoE −/−) and KO (ApoE −/−; Pard3 iΔEC) were fixed with 4% PFA for 10 min at RT and permeabilized and blocked in 1% BSA and 1% Triton X‐100 in PBS buffer overnight at 4°C with gentle rocking, The aortas were dissected and incubated with anti‐MOMA‐2 (MAB1852; Merck Millipore) and ERG1/2/3 antibody, followed by Alexa‐conjugated secondary antibodies. Volocity quantitation (PerkinElmer, MA) software was used to measure surface coverage of the atherosclerotic lesions.
Serum cholesterol measured
Mice were anesthetized with i.p. injection of Ketamin/Xylazin. Blood was collected from the superior vena cava and left at room temperature for 30 min. The blood was centrifuged at 2,000 g for 15 min at 4°C, and plasma (supernatant) was collected. Total cholesterol was measured using a colorimetric kit (Cholesterol Fluorometric Assay Kit, 10007640, Germany).
Lentivirus production
HEK293T cells were transfected with pPB‐bsr2‐Raichu‐1237x, pCMV‐VSV‐G‐RSV‐Rev, and psPAX2 with PEI Max. Cells were maintained for 48 h at 37°C, and the supernatant was harvested and concentrated with Lenti‐X concentration according to manufactures instruction (Clontech, 631231). All viral experiments were performed in P2 laboratory in Kyoto University Graduate School of Medicine (Japan), following the protocols approved by the local ethics committees.
Time‐lapse FRET imaging
HUVECs were infected with the aforementioned recombinant lentivirus. After 72 h, 200,000 cells were seeded into the perfusion chamber (μ‐slide I 0.4 Luer; ibidi). Cells were maintained for 24 h and treated with 18 dyn/cm2 of laminar flow under the microscope. Cells were imaged with an inverted microscope (IX71 or IX81; Olympus, Tokyo, Japan) equipped with a 60× objective lens (Olympus), a cooled CCD camera (CoolSNAP HQ or CoolSNAP K4; Roper Scientific, Tucson, AZ), an LED illumination system (CoolLED precisExcite; Molecular Devices, Sunnyvale, CA), an IX2‐ZDC laser‐based autofocusing system (Olympus), and an MD‐XY30100T‐Meta automatically programmable XY stage (SIGMA KOKI, Tokyo, Japan). The following filters used for the dual‐emission imaging studies were obtained from Omega Optical (Brattleboro, VT): an XF1071 (440AF21) excitation filter, an XF2034 (455DRLP) dichroic mirror, and two emission filters (XF3075 for CFP and XF3079 for YFP). After background subtraction, FRET/CFP ratio images were created with MetaMorph software (Universal Imaging, West Chester, PA) and represented by the intensity‐modulated display mode. In the intensity‐modulated display mode, eight colors from red to blue are used to represent the FRET/CFP ratio, with the intensity of each color indicating the mean intensity of FRET and CFP. For the quantification, the peripheral region of the cell (10 pixels from the edge) was extracted and divided into four parts of 90° toward the direction of flow. FRET and CFP intensities were analyzed for each area, and the results were exported to Excel software (Microsoft Corporation, Redmond, WA). The ratio of raw FRET/CFP value versus the reference value was defined as the normalized FRET/CFP value.
Proximity ligation assay
PLA reaction was performed using Duolink in situ orange starter kit mouse/rabbit (DUO92102, Sigma) according to the company instruction. To analyze the complex formation of endogenous proteins, 8 × 10,000 HUVECs were seeded into the perfusion chamber (μ‐slide IV 0.4; ibidi) coated with fibronectin and incubated for 24 h at 37°C in growth medium. Cells were treated with 18 dyn/cm2 laminar flow for 60 min, fixed with 4% PFA for 10 min at room temperature, and permeabilized with 0.1% Triton X‐100 for 10 min. aPKCλ/PAR‐3 complex formation was analyzed with anti‐PAR‐3 antibody (kindly gifted from Dr. Kaibuchi, rabbit polyclonal) and anti‐aPKCλ antibody (mouse monoclonal, 610207, BD Biosciences, CA). GSK3β/aPKCλ complex formation was analyzed with anti‐GSK3β antibody (rabbit monoclonal, clone D5C5Z, Cell Signaling #12456) and anti‐aPKCλ antibody (mouse monoclonal, 610207). For exogenous protein analysis, HUVECs were transfected with pCAGGS‐myc‐PKCλ and pEGFP‐Par3 or pEGFP‐GSK3β (Fig EV4) using jetPEI‐HUVEC (Polyplus, 108‐05N). Transfected ECs were stained with anti‐GFP‐antibody conjugated with Alexa Fluor 488 (rabbit polyclonal, A‐21311, Thermo Fisher) and anti‐myc antibody (mouse monoclonal, clone 9E10, sc‐40, Santa Cruz Biotechnology, TX). To analyze the protein complex formation on the cell–cell contact site, the colocalization image between PLA (GFP‐PAR‐3/myc‐PKCλ, Fig EV4B and C; GFP‐ GSK3β/myc‐PKCλ, Fig EV4D and E) and EC junction (VE‐cadherin) was generated by Volocity quantitation module (PerkinElmer, MA). The image was divided into four parts of 90° toward the direction of flow, and percentile of colocalized signal in each part was quantified with ImageJ software (NIH, MA).
Quantification of nuclear‐localized p65 signal
En face microscope images of aorta from 8W control or Pard3 iΔEC mice were analyzed with Volocity quantitation software (PerkinElmer). The ROI of EC nuclei (ERG 1/2/3) signal were extracted, and the intensity of p65 that colocalize with EC nuclei was counted. The signal of ERG 1/2/3 was used to normalize each image.
For in vitro analysis, mouse aortic ECs were seeded into the perfusion chamber (μ‐slide I Luer 0.4, fibronectin coated; 3 × 100,000 cells). Cells were exposed to 12 dyn/cm2 of laminar flow for 0, 15, 30, and 60 min, fixed with 4% PFA. Cells were permeabilized with 0.1% Triton X‐100 and incubated with anti‐p65 (Cell Signaling Technology, 8242) and anti‐VE‐cadherin, followed with secondary antibody conjugated with Alexa Fluor dyes. Nuclei were visualized with DAPI. For inhibitor experiment, growth medium containing 1 μM BIO (Sigma, B1686) was used.
RNA extraction and qPCR analysis
For analysis of messenger RNA expression levels, total RNA was isolated from ECs using the Quick RNA‐Mini Kit (Zymo Research; Freiburg, Germany) and 0.5–1 mg per reaction was used to generate cDNA with the Superscript VILO cDNA Synthesis Kit (11755050, Thermo Fisher). qPCR was carried out using a StepOnePlus real‐time PCR machine using TaqMan Gene Expression Assays (VCAM‐1, Mm01320970_m1, and GAPDH, Mm99999915_g1 as internal control) with TaqMan Fast Advanced Master mix (4444556, Thermo Fisher).
Polarity network analysis
Given the complexity and technical aspects of the PolNet Analysis, the full details related to the methodology are published in a separate manuscript 64. We present here a brief description of our methodology.
First, the plexus was manually segmented using Adobe Photoshop (San Jose, CA), producing a binary mask which was subsequently skeletonized using a Voronoi diagram‐based method (http://uk.mathworks.com/matlabcentral/fileexchange/27543-skeletonization-using-voronoi). The local vessel diameters were calculated using maximum inscribed circles at multiple positions along each vessel segment, and this information was used to construct a 3D model of the plexus 20, code available at https://github.com/UCL/BernabeuInterface2014. The surface defined by this model was used as the input to a Lattice Boltzmann Computational Fluid Dynamics solver, HemeLB (https://github.com/UCL/hemelb), run on a High Performance Computing Cluster. The raw fluorescence images were processed in MATLAB using the built‐in “Ginput” function to add points corresponding to the nucleus and Golgi of each cell and recording their locations. These positions defined a vector with magnitude and angle describing the spatial relationship between the points. Pairs of points were recorded for each cell, one at the center of the nucleus and one at the center of the Golgi, defining a vector with a magnitude and angle describing the spatial relationship between the points. The WSS values from the flow simulation were recorded at the positions of the cell nuclei, with the WSS at each point described by a vector giving the magnitude and angle of the applied shear stress. Each plexus was subdivided into artery, vein, capillary, and sprouting front regions, and each cell was assigned to one of these vascular beds. The angular distributions were compared using the Kuiper test (the circular statistics equivalent of the Kolmogorov–Smirnov test) with each comparison yielding a P‐value indicating the likelihood that the two samples are drawn from the same underlying angular distribution. The calculation was performed using the Circular Statistics Toolbox from MATLAB's FileExchange 65. In addition, we binned the angular data according to WSS magnitude to plot the proportion of cells within 45° of being anti‐aligned with the flow as a function of WSS. We calculated the scalar product of the two vectors, given by magnitude(cell polarity)*magnitude(WSS)*cos(θ) which combines information about the length and relative angles of the vectors. By plotting the scalar product versus WSS, we were able to extract a gradient corresponding to magnitude(cell)*cos(θ), i.e. the projection of the cell polarity vector onto the axis defined by the WSS vector. A larger negative gradient corresponds to a larger polarization effect for a given WSS.
Quantification
Data are based on at least three independent experiments or three mutant and control animals for each stage and result shown. The data are presented as mean ± SEM or box and whisker plots. All statistical analyses were carried out using Prism software (Graph Pad, CA). A P < 0.05 was considered significant.
Author contributions
THik and MN designed the study. Golgi orientation in the retinal vasculature was analyzed by PB, CAF, and HG. RhoA FRET analysis was done in Kyoto University under the supervision by MM. RPB and SO contributed experimental reagents and insights. All other experiments have performed by THik, FM, MR, AP, ML, TK, and THir. CAF and MN wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Review Process File
Acknowledgements
Funding for this project was mainly provided by the Excellence Cluster Cardio‐Pulmonary System, the German Research Foundation and Deutsche Forschungsgemeinschaft (NA 1195/3‐1). FM, ML, and MN were supported by GRK2213. THik was supported by EMBO short‐term fellowship (EMBO ASTF 607‐2015). CAF and PB were supported by FCT investigator (IF/00412/2012), EC‐ERC Starting Grant (AXIAL.EC‐679368), FCT grant (EXPL/BEX‐BCM/2258/2013), H2020‐TWINN‐2015 (ReTuBi‐692322), and Portugal 2020 Program (LISBOA‐01‐0145‐FEDER‐00739). The authors acknowledge Dr. Miguel Bernabeu‐Llinares, Dr. Anna Pezzarossa and the HemeLB development team, and the UCL Research Software Development Team (RSD@UCL) for their contribution to this work.
EMBO Reports (2018) 19: e45253
References
- 1. Aranda V, Nolan ME, Muthuswamy SK (2008) Par complex in cancer: a regulator of normal cell polarity joins the dark side. Oncogene 27: 6878–6887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Adams RH, Alitalo K (2007) Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 8: 464–478 [DOI] [PubMed] [Google Scholar]
- 3. Herbert SP, Stainier DY (2011) Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat Rev Mol Cell Biol 12: 551–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Potente M, Gerhardt H, Carmeliet P (2011) Basic and therapeutic aspects of angiogenesis. Cell 146: 873–887 [DOI] [PubMed] [Google Scholar]
- 5. Hahn C, Schwartz MA (2009) Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol 10: 53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chiu JJ, Chien S (2011) Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev 91: 327–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rogers KA, McKee NH, Kalnins VI (1985) Preferential orientation of centrioles toward the heart in endothelial cells of major blood vessels is reestablished after reversal of a segment. Proc Natl Acad Sci USA 82: 3272–3276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Malek AM, Izumo S (1996) Mechanism of endothelial cell shape change and cytoskeletal remodeling in response to fluid shear stress. J Cell Sci 109(Pt 4): 713–726 [DOI] [PubMed] [Google Scholar]
- 9. Coan DE, Wechezak AR, Viggers RF, Sauvage LR (1993) Effect of shear stress upon localization of the Golgi apparatus and microtubule organizing center in isolated cultured endothelial cells. J Cell Sci 104(Pt 4): 1145–1153 [DOI] [PubMed] [Google Scholar]
- 10. Franco CA, Jones ML, Bernabeu MO, Geudens I, Mathivet T, Rosa A, Lopes FM, Lima AP, Ragab A, Collins RT et al (2015) Dynamic endothelial cell rearrangements drive developmental vessel regression. PLoS Biol 13: e1002125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Iden S, Collard JG (2008) Crosstalk between small GTPases and polarity proteins in cell polarization. Nat Rev Mol Cell Biol 9: 846–859 [DOI] [PubMed] [Google Scholar]
- 12. Suzuki A, Ohno S (2006) The PAR‐aPKC system: lessons in polarity. J Cell Sci 119: 979–987 [DOI] [PubMed] [Google Scholar]
- 13. Joberty G, Petersen C, Gao L, Macara IG (2000) The cell‐polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol 2: 531–539 [DOI] [PubMed] [Google Scholar]
- 14. Nishimura T, Yamaguchi T, Kato K, Yoshizawa M, Nabeshima Y, Ohno S, Hoshino M, Kaibuchi K (2005) PAR‐6‐PAR‐3 mediates Cdc42‐induced Rac activation through the Rac GEFs STEF/Tiam1. Nat Cell Biol 7: 270–277 [DOI] [PubMed] [Google Scholar]
- 15. Chen X, Macara IG (2005) Par‐3 controls tight junction assembly through the Rac exchange factor Tiam1. Nat Cell Biol 7: 262–269 [DOI] [PubMed] [Google Scholar]
- 16. Etienne‐Manneville S, Hall A (2003) Cdc42 regulates GSK‐3beta and adenomatous polyposis coli to control cell polarity. Nature 421: 753–756 [DOI] [PubMed] [Google Scholar]
- 17. Kim M, Datta A, Brakeman P, Yu W, Mostov KE (2007) Polarity proteins PAR6 and aPKC regulate cell death through GSK‐3beta in 3D epithelial morphogenesis. J Cell Sci 120: 2309–2317 [DOI] [PubMed] [Google Scholar]
- 18. Nakayama M, Nakayama A, van Lessen M, Yamamoto H, Hoffmann S, Drexler HC, Itoh N, Hirose T, Breier G, Vestweber D et al (2013) Spatial regulation of VEGF receptor endocytosis in angiogenesis. Nat Cell Biol 15: 249–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Randi AM, Sperone A, Dryden NH, Birdsey GM (2009) Regulation of angiogenesis by ETS transcription factors. Biochem Soc Trans 37: 1248–1253 [DOI] [PubMed] [Google Scholar]
- 20. Bernabeu MO, Jones ML, Nielsen JH, Kruger T, Nash RW, Groen D, Schmieschek S, Hetherington J, Gerhardt H, Franco CA et al (2014) Computer simulations reveal complex distribution of haemodynamic forces in a mouse retina model of angiogenesis. J R Soc Interface 11: pii: 20140543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Franco CA, Jones ML, Bernabeu MO, Vion AC, Barbacena P, Fan J, Mathivet T, Fonseca CG, Ragab A, Yamaguchi TP et al (2016) Non‐canonical Wnt signalling modulates the endothelial shear stress flow sensor in vascular remodelling. Elife 5: e07727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rodriguez‐Boulan E, Macara IG (2014) Organization and execution of the epithelial polarity programme. Nat Rev Mol Cell Biol 15: 225–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Strilic B, Kucera T, Eglinger J, Hughes MR, McNagny KM, Tsukita S, Dejana E, Ferrara N, Lammert E (2009) The molecular basis of vascular lumen formation in the developing mouse aorta. Dev Cell 17: 505–515 [DOI] [PubMed] [Google Scholar]
- 24. Baluk P, Morikawa S, Haskell A, Mancuso M, McDonald DM (2003) Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am J Pathol 163: 1801–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lawrence T (2009) The nuclear factor NF‐kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 1: a001651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tzima E, Irani‐Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA (2005) A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437: 426–431 [DOI] [PubMed] [Google Scholar]
- 27. Gareus R, Kotsaki E, Xanthoulea S, van der Made I, Gijbels MJ, Kardakaris R, Polykratis A, Kollias G, de Winther MP, Pasparakis M (2008) Endothelial cell‐specific NF‐kappaB inhibition protects mice from atherosclerosis. Cell Metab 8: 372–383 [DOI] [PubMed] [Google Scholar]
- 28. Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T (1995) Transcriptional regulation of endothelial cell adhesion molecules: NF‐kappa B and cytokine‐inducible enhancers. FASEB J 9: 899–909 [PubMed] [Google Scholar]
- 29. Cohen P, Frame S (2001) The renaissance of GSK3. Nat Rev Mol Cell Biol 2: 769–776 [DOI] [PubMed] [Google Scholar]
- 30. Jope RS, Yuskaitis CJ, Beurel E (2007) Glycogen synthase kinase‐3 (GSK3): inflammation, diseases, and therapeutics. Neurochem Res 32: 577–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McCue S, Dajnowiec D, Xu F, Zhang M, Jackson MR, Langille BL (2006) Shear stress regulates forward and reverse planar cell polarity of vascular endothelium in vivo and in vitro . Circ Res 98: 939–946 [DOI] [PubMed] [Google Scholar]
- 32. Nakayama M, Goto TM, Sugimoto M, Nishimura T, Shinagawa T, Ohno S, Amano M, Kaibuchi K (2008) Rho‐kinase phosphorylates PAR‐3 and disrupts PAR complex formation. Dev Cell 14: 205–215 [DOI] [PubMed] [Google Scholar]
- 33. Wojciak‐Stothard B, Ridley AJ (2003) Shear stress‐induced endothelial cell polarization is mediated by Rho and Rac but not Cdc42 or PI 3‐kinases. J Cell Biol 161: 429–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tzima E, del Pozo MA, Shattil SJ, Chien S, Schwartz MA (2001) Activation of integrins in endothelial cells by fluid shear stress mediates Rho‐dependent cytoskeletal alignment. EMBO J 20: 4639–4647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu Y, Collins C, Kiosses WB, Murray AM, Joshi M, Shepherd TR, Fuentes EJ, Tzima E (2013) A novel pathway spatiotemporally activates Rac1 and redox signaling in response to fluid shear stress. J Cell Biol 201: 863–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yoshizaki H, Ohba Y, Kurokawa K, Itoh RE, Nakamura T, Mochizuki N, Nagashima K, Matsuda M (2003) Activity of Rho‐family GTPases during cell division as visualized with FRET‐based probes. J Cell Biol 162: 223–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eto M, Kouroedov A, Cosentino F, Luscher TF (2005) Glycogen synthase kinase‐3 mediates endothelial cell activation by tumor necrosis factor‐alpha. Circulation 112: 1316–1322 [DOI] [PubMed] [Google Scholar]
- 38. Choi SE, Jang HJ, Kang Y, Jung JG, Han SJ, Kim HJ, Kim DJ, Lee KW (2010) Atherosclerosis induced by a high‐fat diet is alleviated by lithium chloride via reduction of VCAM expression in ApoE‐deficient mice. Vascul Pharmacol 53: 264–272 [DOI] [PubMed] [Google Scholar]
- 39. Lee M, Vasioukhin V (2008) Cell polarity and cancer–cell and tissue polarity as a non‐canonical tumor suppressor. J Cell Sci 121: 1141–1150 [DOI] [PubMed] [Google Scholar]
- 40. McCaffrey LM, Montalbano J, Mihai C, Macara IG (2012) Loss of the Par3 polarity protein promotes breast tumorigenesis and metastasis. Cancer Cell 22: 601–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xue B, Krishnamurthy K, Allred DC, Muthuswamy SK (2013) Loss of Par3 promotes breast cancer metastasis by compromising cell‐cell cohesion. Nat Cell Biol 15: 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chatzizisis YS, Coskun AU, Jonas M, Edelman ER, Feldman CL, Stone PH (2007) Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: molecular, cellular, and vascular behavior. J Am Coll Cardiol 49: 2379–2393 [DOI] [PubMed] [Google Scholar]
- 43. Mohan S, Mohan N, Sprague EA (1997) Differential activation of NF‐kappa B in human aortic endothelial cells conditioned to specific flow environments. Am J Physiol 273: C572–C578 [DOI] [PubMed] [Google Scholar]
- 44. Wang C, Baker BM, Chen CS, Schwartz MA (2013) Endothelial cell sensing of flow direction. Arterioscler Thromb Vasc Biol 33: 2130–2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cuhlmann S, Van der Heiden K, Saliba D, Tremoleda JL, Khalil M, Zakkar M, Chaudhury H, le Luong A, Mason JC, Udalova I et al (2011) Disturbed blood flow induces RelA expression via c‐Jun N‐terminal kinase 1: a novel mode of NF‐kappaB regulation that promotes arterial inflammation. Circ Res 108: 950–959 [DOI] [PubMed] [Google Scholar]
- 46. Archibald A, Mihai C, Macara IG, McCaffrey L (2015) Oncogenic suppression of apoptosis uncovers a Rac1/JNK proliferation pathway activated by loss of Par3. Oncogene 34: 3199–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Sokolosky M, Abrams SL, Montalto G, D'Assoro AB, Libra M, Nicoletti F et al (2014) GSK‐3 as potential target for therapeutic intervention in cancer. Oncotarget 5: 2881–2911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Colosimo PF, Liu X, Kaplan NA, Tolwinski NS (2010) GSK3beta affects apical‐basal polarity and cell‐cell adhesion by regulating aPKC levels. Dev Dyn 239: 115–125 [DOI] [PubMed] [Google Scholar]
- 49. Shimokawa H, Takeshita A (2005) Rho‐kinase is an important therapeutic target in cardiovascular medicine. Arterioscler Thromb Vasc Biol 25: 1767–1775 [DOI] [PubMed] [Google Scholar]
- 50. Nakayama M, Amano M, Katsumi A, Kaneko T, Kawabata S, Takefuji M, Kaibuchi K (2005) Rho‐kinase and myosin II activities are required for cell type and environment specific migration. Genes Cells 10: 107–117 [DOI] [PubMed] [Google Scholar]
- 51. Shimokawa H, Seto M, Katsumata N, Amano M, Kozai T, Yamawaki T, Kuwata K, Kandabashi T, Egashira K, Ikegaki I et al (1999) Rho‐kinase‐mediated pathway induces enhanced myosin light chain phosphorylations in a swine model of coronary artery spasm. Cardiovasc Res 43: 1029–1039 [DOI] [PubMed] [Google Scholar]
- 52. Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M et al (1997) Calcium sensitization of smooth muscle mediated by a Rho‐associated protein kinase in hypertension. Nature 389: 990–994 [DOI] [PubMed] [Google Scholar]
- 53. Nagaoka T, Morio Y, Casanova N, Bauer N, Gebb S, McMurtry I, Oka M (2004) Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 287: L665–L672 [DOI] [PubMed] [Google Scholar]
- 54. Shimokawa H, Morishige K, Miyata K, Kandabashi T, Eto Y, Ikegaki I, Asano T, Kaibuchi K, Takeshita A (2001) Long‐term inhibition of Rho‐kinase induces a regression of arteriosclerotic coronary lesions in a porcine model in vivo . Cardiovasc Res 51: 169–177 [DOI] [PubMed] [Google Scholar]
- 55. Liu HW, Halayko AJ, Fernandes DJ, Harmon GS, McCauley JA, Kocieniewski P, McConville J, Fu Y, Forsythe SM, Kogut P et al (2003) The RhoA/Rho kinase pathway regulates nuclear localization of serum response factor. Am J Respir Cell Mol Biol 29: 39–47 [DOI] [PubMed] [Google Scholar]
- 56. Olson EN, Nordheim A (2010) Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol 11: 353–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pandey MK, DeGrado TR (2016) Glycogen synthase kinase‐3 (GSK‐3)‐targeted therapy and imaging. Theranostics 6: 571–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio R, Piazza S et al (2014) Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol 16: 357–366 [DOI] [PubMed] [Google Scholar]
- 59. Wang L, Luo JY, Li B, Tian XY, Chen LJ, Huang Y, Liu J, Deng D, Lau CW, Wan S et al (2016) Integrin‐YAP/TAZ‐JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 540: 579–582 [DOI] [PubMed] [Google Scholar]
- 60. Low BC, Pan CQ, Shivashankar GV, Bershadsky A, Sudol M, Sheetz M (2014) YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS Lett 588: 2663–2670 [DOI] [PubMed] [Google Scholar]
- 61. Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N (1992) Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci USA 89: 4471–4475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ismail JA, Poppa V, Kemper LE, Scatena M, Giachelli CM, Coffin JD, Murry CE (2003) Immunohistologic labeling of murine endothelium. Cardiovasc Pathol 12: 82–90 [DOI] [PubMed] [Google Scholar]
- 63. Kobayashi M, Inoue K, Warabi E, Minami T, Kodama T (2005) A simple method of isolating mouse aortic endothelial cells. J Atheroscler Thromb 12: 138–142 [DOI] [PubMed] [Google Scholar]
- 64. Bernabeu MO, Jones ML, Nash RW, Pezzarossa A, Coveney PV, Gerhardt H, Franco CA (2018) PolNet: a tool to quantify network‐level cell polarity and blood flow in vascular remodeling. Biophys J 114: 2052–2058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Berens P (2009) CircStat: a MATLAB toolbox for circular statistics. J Stat Softw 31: 1–21 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Review Process File