

Abstract
Background/Aims:
Human enterocytic differentiation is altered during development, fasting, adaptation, and bariatric surgery, but its intracellular control remains unclear. We hypothesized that Schlafen 12 (SLFN12) regulates enterocyte differentiation.
Methods:
We used laser capture dissection of epithelium, qRT-PCR, and immunohistochemistry to evaluate SLFN12 expression in biopsies of control and fasting human duodenal mucosa, and viral overexpression and siRNA to trace the SLFN12 pathway in human Caco-2 and HIEC6 intestinal epithelial cells.
Results:
Fasting human duodenal mucosa expressed less SLFN12 mRNA and protein, accompanied by decreases in enterocytic markers like sucrase-isomaltase. SLFN12 overexpression increased Caco-2 sucrase-isomaltase promoter activity, mRNA, and protein independently of proliferation, and activated the SLFN12 putative promoter. SLFN12 coprecipitated Serpin B12 (SERPB12). An inactivating SLFN12 point mutation prevented both SERPB12 binding and sucrase-isomaltase induction. SERPB12 overexpression also induced sucrase-isomaltase, while reducing SERPB12 prevented the SLFN12 effect on sucrase-isomaltase. Sucrase-isomaltase induction by both SLFN12 and SERPB12 was attenuated by reducing UCHL5 or USP14, and blocked by reducing both. SERPB12 stimulated USP14 but not UCHL5 activity. SERPB12 coprecipitated USP14 but not UCHL5. Moreover, SLFN12 increased protein levels of the sucrase-isomaltase-promoter-binding transcription factor cdx2 without altering Cdx2 mRNA. This was prevented by reducing UCHL5 and USP14. We further validated this pathway in vitro and in vivo. SLFN12 or SERPB12 overexpression induced sucrase-isomaltase in human non-malignant HIEC-6 enterocytes.
Conclusions:
SLFN12 regulates human enterocytic differentiation by a pathway involving SERPB12, the deubiquitylases, and Cdx2. This pathway may be targeted to manipulate human enterocytic differentiation in mucosal atrophy, short gut or obesity.
Keywords: Schlafen 12, Serpin B12, Intestine, Epithelium, Signaling, Differentiation, UCHL5, USP14
Introduction
Enterocytic differentiation is critical to diverse processes and diseases including mucosal adaptation, fasting, lactase deficiency, and small bowel adenocarcinoma. Most dramatically, insufficient adaptation to infant short bowel syndrome causes 20–40% mortality and a prolonged course thereafter [1]. Growth factors, cytokines, matrix proteins, and physical forces all influence intestinal epithelial differentiation, and several transcription factors regulate the control of the genetic program for enterocytic differentiation [2–8]. However, these have historically been viewed as a diverse web of stimuli and signals.
Described in 1998 [9], the Schlafen (SLFN) protein superfamily remains poorly understood. Some SLFNs influence hematopoiesis [10], the immune response [11, 12], or bone biology [13], but their mechanisms of action are unclear because they lack canonical signaling motifs. The Schlafen proteins have been subclassified into short, intermediate, and long Schlafens. Although long Schlafen proteins possess a helicase domain and a nuclear targeting sequence and are therefore able to bind to nuclear DNA to modulate transcription, the short and intermediate Schlafens lack such a targeting sequence [12]. Although the Schlafen family are only loosely homologous with each other, database searches do not demonstrate overall substantial resemblance to other known proteins. Recent investigations suggest that Slfn3 influences rat enterocytic differentiation. Stimuli as diverse as TGF-beta, butyrate, and repetitive deformation each induce differentiation marker expression in IEC-6 cells by increasing Slfn3 [14]. In vivo, increasing or decreasing Slfn3 expression alters rat jejunal enterocyte expression of such differentiation markers [15]. However, Slfn3 is a rodent protein not expressed in humans, and the Schlafen superfamily is only very loosely homologous in structure and sequence, so whether any Schlafen protein might function similarly in humans is unclear. Moreover, there is no mechanistic information available about how Slfn3 or any other intermediate or short Schlafen protein function in any species except that excluding Slfn3 from the nucleus does not prevent its function [16].
Although humans do not express Slfn3, they do express one intermediate Schlafen protein, which is Schlafen 12. We therefore hypothesized that the intermediate length Schlafen 12, despite being only 40% homologous to Slfn3, might promote human enterocytic differentiation. Choosing sucrase-isomaltase expression as a marker for enterocytic differentiation, we further delineated a novel pathway by which SLFN12 acts. We showed that SLFN12 expression is markedly diminished in human duodenal epithelium during prolonged fasting in parallel with loss of enterocytic differentiation markers such as sucrase-isomaltase. We then focused on sucrase-isomaltase in more mechanistic cell culture studies in Caco-2 cells because it is an important disaccharidase in small intestinal physiology [17], well characterized as a marker of intestinal epithelial differentiation in Caco-2 cells [18–20], and important in the evaluation of intestinal adaptation in short bowel syndrome [21], while its regulation has been extensively characterized [22, 23]. Exogenous SLFN12 overexpression increases Caco-2 or non-malignant HIEC-6 sucrase-isomaltase, while reducing SLFN12 reduces Caco-2 sucrase-isomaltase. SLFN12 exerts its effects by binding to Serpin B12 (SERPB12), most likely via the SLFN12 modified ATP binding domain. SLFN12 or SERPB12 overexpression increases expression of the complementary deubiquitylases USP14 and UCHL5 in vitro, and SERPB12 stimulates USP14 deubiquitylase activity. These changes in deubiquitylase activity may in turn increase levels of transcription factors like Cdx2 that regulate enterocytic differentiation, promoting expression of enterocytic differentiation markers such as sucrase-isomaltase.
Materials and Methods
Human samples
Human duodenal mucosa was obtained by endoscopic or surgical biopsy from consenting human patients. Fasting patients had fasted at least 4 days. Control patients had fasted for 8–16 hours. Patients with intestinal pathology known or discovered on esophagogastroduodenoscopy were excluded. Most fasting patients underwent procedures for another indication such as endoscopic gastrostomy placement, while most control patients underwent esophagogastroduodenoscopy for an indication unrelated to the duodenum such as esophageal reflux. Samples were immediately placed in OCT (optimal cutting temperature) medium and frozen in liquid nitrogen. 10–15-μm sections were collected on non-charged glass slides and stored at −80°C. One set of slides was passed through an ascending alcohol series and rinsed with xylene to dehydrate the tissue and remove the alcohol. Slides were viewed under an inverted Nikon Eclipse microscope outfitted with the Arcturus Pixcell II/e Laser Capture Microdissection System and Arcview software (Arcturus, San Diego, CA). Enterocytes were lifted using CapSure LCM caps (ThermoFisher Scientific, Rockford, IL). RNA was extracted from collected cells using RNeasy Micro Kit (Qiagen, Valencia, CA) and stored at −80°C before processing as below. Another set of slides were stained using rabbit polyclonal antibody raised against a unique peptide sequence (cTRGRLYLRPELLAKRP) derived from SLFN12 (Abcam, Cambridge, MA) or goat polyclonal antibody against SI, or rabbit polyclonal villin antibody (Santa Cruz Biotechnology, Santa Cruz, CA), at 1:50 dilution in PBS followed by Vectastain Universal ABC kit detection (Vector Labs, Burlingame, CA).
Cells
Caco-2BBE cells and rat IEC6 cells (American Tissue Culture Collection, ATCC) each tested negative for mycoplasmal contamination and were maintained as described [24]. Fetal intestinal HIEC-6 cells [25] (ATCC) were maintained at 37°C with 5% CO2 in OptiMEM reduced-serum medium with 20mM HEPES, 10mM GlutaMAX, 10ng/ml Epidermal Growth Factor, and 4% fetal bovine serum, and studied within 3 passages.
DNA Plasmid constructs
We used the pEGFP-C1 expression vector from Clontech (Mountain View, CA) to generate SLFN12 construct pEGFP-C1-SLFN12. The point mutations 222 L to A (mutant 1), 233 D to A (mutant 2), 233 D to Q (mutant 3), 233 D to T (mutant 4) and 236 Y to F (mutant 5) were accomplished with the Quick Change II XL Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA). Plasmids were purified via MiniPrep (QIAGEN, Valencia, CA) prior to sequencing at the Michigan State Core Facility. After sequence confirmation, the plasmids were amplified and prepared for transient expression using the QIAGEN MaxiPrep kit (QIAGEN).
Viruses
Adv-Slfn12 was constructed by Applied Biological Materials (Richmond, BC, Canada) using a pAdeno vector, CMV promoter, and human Slfn12 insert was based on accession # NM_018042. Advenovirus amplification was as described [15, 26]. Adeno-associated viruses (AAV) were made using the AAVpro Helper Free System per manufacturer’s protocol (Takara Clontech, Mountain View, CA). Human Slfn12 and SerpinB12 inserts were synthesized through Eurofins Genomics (Louisville, KY). AAV-CMV-gene of interest plasmids were transformed into Stellar Competent Cells (Takara Clontech) and plasmid stocks were isolated using the Qiagen Plasmid Maxi kit (Qiagen). AAV particles were constructed by transfecting HEK-293T cells using the Xfect Transfection kit (Takara Clontech). AAV titers were measured using the AAVpro Titration kit for qPCR (Takara Clontech). V5SLFN12 lentivirus was assembled in HEK293T cells from pLX304 V5SLFN12 plasmid (Clone: HsCD00441704, DNASU Plasmids), lentiviral packing plasmid pCMV-dR8.2 dvpr plasmid (plasmid #8455, Addgene), and lentiviral plasmid for envelope protein pCMV-VSV-G (plasmid #8454, Addgene). Control virus substituted pLX304 empty vector (Plasmid #25890, 3rd generation lentiviral vector (LV), Empty backbone) instead of the pLX304 V5SLFN12 plasmid.
Other Materials
Dulbecco’s modified Eagle’s medium (DMEM), 0.05% Trypsin-EDTA, Lipofectamine, RNAiMAX and Plus Reagent were from Thermo Fisher (Waltham, MA), Western blot stripping reagent from Bio-Rad (Hercules, CA), and human transferrin from Roche. Antibodies to CDX2 and CDX4 were from Abcam (Cambridge, MA). Antibodies to sucrase-isomaltase, villin, USP14, UCHL5, SerpinB12, and Serpin B5 were from Santa Cruz (Dallas, TX). Rabbit monoclonal beta-acting antibody was from Cell Signaling Technology (Boston, MA). Mouse monoclonal antibody to Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was from Meridian Life Sciences, Inc (Memphis, TN). pGEX GST- vector was from GE Healthcare Bio-Sciences (Pittsburgh, PA). Antibody to V5 was from Abcam (Cambridge, MA), secondary antibodies anti-rabbit 800, anti-mouse 680, anti-mouse 680 and anti-mouse 800 were from LI-COR (Lincoln, NE). All fine reagents and Protein G-Sepharose and GST-tagged beads were from Millipore-Sigma (Billerica, MA). SensoLyte® 440 Deubiquitination Assay Kits were from ANA SPEC, (Catalog # 72204). Human recombinant USP14 (Cat# E544) and human recombinant UCHL5 (Cat#E327–025) were from Boston Biochem Inc (Cambridge, MA) and human recombinant SerpinB12 (C6-His, Catalog# CI96) was from Novoprotein (Summit, NJ). Double-stranded short interfering RNAs (siRNAs) targeting human forms of SLFN12, SerpinB12, USP14, UCHL5 and control non-targeting siRNA (NT siRNA) were from Dharmacon (Lafayette, CO).
siRNA and virus studies
70–80% confluent Caco-2 or 80–90% confluent HIEC-6 cells were transduced with 4000 viral particles/cells with Adeno-CMV control, Adeno-CMV-SLFN12 virus, Adeno-associated-ZsGreen or Adeno-associated-CMV SerpinB12 virus for two hours in 1 ml complete DMEM medium. All viruses were replication-incompetent by PCR (E1 region amplification) and direct sequencing. For lentiviral studies, 500 ul HEK293T cell viral media was added to 70–80% confluent cells with 1.5 ml opti-MEM media containing 1.5 ul polybrene at 10 ug/ml)
For siRNA studies, Caco-2 cells (300, 000 cells/well) were plated into six-well plates one day before transfection with siNT1 or siNT5 (Control, 100nM) or siUCHL5 and siUSP14 (50nM each) using RNAiMAX transfection reagent (7.5ul/well) formulated in Opti-MEM (serum-free medium) at a total volume of 300 (150+150) ul/well. The mixture was added gradually into the 1 ml of complete medium in each well. In some studies, we reduced target genes by siRNA and then transduced cells with Adeno-CMV control, Adeno-CMV-SLFN12 virus, Adeno-associated-CMV or Adeno-associated-SerpinB12 virus for two hours in 1 ml complete DMEM medium. Medium was exchanged after two hours and experiments terminated after 72 hours.
All siRNA studies were performed with at least two siRNA sequences with similar results. All viral studies were performed with at least two viral vectors, one adenoviral and one adeno-associated viral, with similar results. These have been pooled for presentation.
RNA isolation and qRT-PCR
Total RNA was isolated from Caco-2 or HIEC-6 cells using RNeasy Mini Kit, Qiashredders, DNase treatment and the QiaCube instrument per manufacturer’s protocols (Qiagen). cDNA synthesis was prepared from RNA samples using QuantiTect Reverse Transcription kit (Qiagen) or SMARTScribe Reverse Transcription kit (Takara Clontech). cDNA samples were analyzed by qPCR analysis using the BioRad CFX96 Touch Real-Time PCR Detection System and the BioRad SsoAdvanced Universal SYBR Green Supermix (BioRad Laboratories, Hercules, CA). Expression levels were determined from the threshold cycle (Ct) values using the method of 2-∆∆Ct using RPLP0 as the reference control gene. Primer design is as follows: human RPLP0 forward 5’-GCAATGTTGCCAGTGTCTG-3’, reverse 5’-GCCTTGA CC TTT TCAGCAA-3’; human Slfn12 forward 5’-ATCTGGGCTTGCAAGAGAAC-3’, reverse 5’-TTTTTGCCA GCTTCT GC TTT-3’; human sucrase-isomaltase (SI) forward 5’-GCCAGCTTATTGGGCTTTGGGTT-3’, reverse 5’-AACTGAGGAAGGTCCTGGAATGCT-3’; human SerpinB12 forward5’-GGG CTG TTG TCT CGG AAA GGT 3’, reverse 5’-AGG AGA GCA GAC CCT GCC AT-3’ human UCHL5 forward5’- CAG TGC AGT AAG GCC TGT CA-3’, reverse 5- TGG GTT CCT CTG CAA GTT GT- 3’ human USP14 forward5- GAG CTT CAG GGG AAA TGG CT- 3’, reverse 5- TCT CGG CAA ACT GTG GGA AA-3’. Primers were from IDT. PCR cycle conditions were 1 cycle of 3 minutes at 95°C, 40 cycles of 30 seconds at 95°C, 30 seconds at the annealing temperature of 60°C and 30 seconds at 72°C, and then a melt curve of 1 cycle 65°C for 30 seconds and 60 cycles 65°C for 5 seconds + 0.5°C/cycle with a ramp of 0.5°C/s and a plate read each cycle. For CDX2 expression and human SI and Vil1 expression in HIEC-6 cells, cDNA was synthesized using the SMARTScribe kit from Takara Clontech. cDNA samples were analyzed by multiplexing qPCR analysis using the BioRad CFX96 Touch Real-Time PCR Detection System and the PrimeTime Gene Expression Master Mix (IDT). Expression levels were determined from the threshold cycle (Ct) values using the method of 2-∆∆Ct using RPLP0 as the reference control gene. qPCR cycle conditions were 1 cycle of 2 minutes at 95°C, 40 cycles of 10 seconds at 95°C, and 45 seconds at the annealing temperature of 55°C. CDX2, SI (Sis), and Vil1 primer and probe combination and housekeeping gene, RPLP0, were from BioRad, and primer and probe sequences are proprietary (CDX2 Assay ID: qHsaCIP0028762, RPLP0 Assay ID: qHsaCEP0041375, Sis Assay ID: qHsaCEP0049871, Vil1 Assay ID: qHsaCIP0031545).
Flow cytometry
Caco-2 cells were stained for flow cytometry by intracellular staining using FoxP3/Transcription factor fixation/ permeabilization kit from eBioscience (San Diego, CA): anti-human SI conjugated to fluorescein (FITC) (LS-C447064) and anti-human SLFN12 conjugated to allophycocyanin (APC) (LS-C269594, LifeSpan BioSciences, Seattle, WA); and anti-human SERPINB12 conjugated to ALEXA FLUOR 594 (Bioss Antibodies, Woburn, MA). Samples were acquired on an LSR II Flow cytometer (BD, San Jose, CA) and analyzed with FlowJo software (TreeStar, Ashland OR).
Promoter activity assays
Promoter activity was assayed as previously [16]. Briefly, 104 Caco-2BBE cells/well were seeded in 96-well (0.32 cm2) plates, and co-transfected the next day with 150 ng/well of the empty vector (pEGFP-C1, Clontech Laboratories) or human SLFN12 DNA constructs (pEGFP-C1-Slfn12, pEGFP-C1-Slfn12-L222A (mutant 1), pEGFP-C1-Slfn12-D233A (mutant 2), pEGFP-C1-Slfn12-D233Q (mutant 3), pEGFP-C1-Slfn12-D233T (mutant 4), or pEGFP-C1-Slfn12-Y236F (mutant 5), with 50 ng/well of the human sucrase-isomaltase (SI) or human SLFN12 LightSwitch promoter reporter goclone (Switchgear Genomics, Carlsbad, CA). Cells in each well received a total of 150 ng of DNA with SLFN12 and SI or SLFN12 promoter reporter constructs at a 3:1 ratio. The DNA was mixed with Opti-MEM medium (10 µl/well) for 5 min, and Fugene (Switchgear Genomics) was added (3.2 µl/1.0 µg DNA). The mixture was incubated at room temperature for 20–30 minutes and then added to the cells. Forty eight hours after transfection, the experiment was terminated by adding 100ul/well (buffer + substrate) LightSwitch Luciferase assay reagent (Switchgear Genomics), then incubating for 30 minutes at room temperature in the dark. Relative Luminescence (RLU) was measured using a VERITAS Microplate Luminometer (Turner Biosystems, Sunnyvale, CA).
For promoter activity assays after combined plasmid and siRNA transfection to evaluate the effects of Cdx2 knockdown on SLFN12 induction of sucrase isomaltase promoter activity, Caco-2BBE cells (1.0 104) were seeded in 96-well (0.32 cm2) plates, and co-transfected the next day with 100 ng/well of the empty vector (pEGFP-C1, Clontech Laboratories, Inc., Mountain View, CA) or human schlafen12 DNA constructs (pEGFP-Slfn12) with 35.0 ng/well of the human Sucrase Isomaltase (SI) LightSwitch promoter reporter goclone (Switchgear Genomics, Carlsbad, CA) with non-target siRNA (siNT5) or siRNA to CDX2 (siCDX2) gene. The DNA and siRNA mixture were mixed with Opti-MEM medium (10 µl/well) for 5 min, and Dharmafect-Duo transfection reagent (Dharmacon Inc, Lafayette, CO) was added (7.5 µl/1.0 µg DNA) as per manufacturer instructions. The mixture was incubated at room temperature for 20–30 minutes and added to the cells. Sixty hours after transfection, the experiment was terminated by addition of luciferase substrate as per the manufacturer’s recommendations. Briefly, 100ul/well (buffer + substrate) LightSwitch Luciferase assay reagent (Switchgear Genomics) was added and incubated for 30 minute at room temperature in the dark. Relative Luminescence (RLU) was measured using a VERITAS Microplate Luminometer (Turner Biosystems, Sunnyvale, CA).
Deubuiquitylase activity assay
We measured USP14 or UCHL5 DUB activity in presence or absence of recombinant human SerpinB12 protein using the SensoLyte® 440 Deubiquitination Assay Kit (SensoLyte® 440 Deubiquitination Assay Kit, ANA SPEC, Catalog # 72204). A natural substrate, ubiquitin, coupled with the AMC (7-amino-4-methylcoumarin) fluorophore is used to measure the DUB activity. The human recombinant USP14 (Cat# E544) and human recombinant UCHL5 (Cat#E327–025) were from Boston Biochem (Cambridge, MA) and human recombinant SerpinB12 (C6-His, Catalog# CI96) was from Novoprotein (Summit, NJ). Upon the cleavage by a deubiquitin protease, this substrate generates the blue fluorophore that was detected at excitation/emission=354/442 nm.
Glutathione S-transferase (GST) pull-down assay
Glutathione S-transferase (GST) pull-down was used to identify potential Schlafen 12-binding partners. The GST pull-down assay was performed as previously [27] with slight modifications. Briefly, Glutathione Sepharose 4B beads were blocked with 1% bovine serum albumin for two hours at room temperature and washed with pulldown lysis buffer before being conjugated with 2.0mg recombinant GST-tagged proteins under similar conditions. Conjugated beads were incubated with lysate from GST-only-or GST-SLFN12-transfected Caco-2 cells overnight at 4°C. The beads were washed three times to remove non-specific unbound proteins. Bound proteins were eluted by adding loading buffer with sodium dodecyl sulfate (SDS) and heating at 100°C for 5 minutes before western analysis.
Co-precipitation assays
For coprecipitation of proteins, we lysed Caco-2 cells transfected with various GFP-tagged plasmids or infected lentivirus in lysis buffer (25 mM Tris-HCL PH 7.4, 150 mM NaCl, 1 mM EDTA, 0.5% Triton-X-100, sodium fluoridate, sodium pyrophosphate, sodium orthovanadate, aprotinin, leupeptin, PMSF). Equal amounts of protein assayed by BCA assay were then incubated with anti-GFP antibody and protein G sepharose beads or anti-V5 antibody conjugated to agarose beads (Abcam, Cambridge, MA) for 2 hours, washed thrice, eluted with 6X Laemmli buffer, and resolved by 10% SDS-PAGE before Western blotting.
Western blots and densitometry
Western blots were performed as previously [16] and detected by LICOR –Odyssey-Fc imaging (LI-COR Biosciences, Lincon, NE). Densitometry was conducted on exposures within the linear range.
Statistical analysis
Quantitative and semi-quantitative data were assessed for normality by Shapiro-Wilk testing and compared by two-sided t-test with Bonferroni correction for multiple comparisons and ANOVA where appropriate. Data are represented as mean+SE.
Study approval
Human studies were approved by the institutional review boards of Michigan State University, the University of North Dakota, and Sparrow Hospital. All patients gave written consent to their participation in these studies.
Results
Duodenal SLFN12 is reduced in fasting humans in parallel with enterocytic differentiation markers.
Preliminary studies compared distal duodenal SLFN12 mRNA levels in 12 human patients fasting for at least four days with 18 human patients who had been fasting for less than 24 hours. All patients underwent esophagogastroduodenoscopy for reasons unrelated to duodenal pathology. Epithelial mRNA was isolated by laser capture microscopy and q-RT-PCR. The expected decreased expression of classical enterocytic differentiation markers (Fig. 1a) including villin (38±11% of control, p<0.05), SI (34±11%, p<0.05), dipeptidyl peptidase-4 (DPP4), also known as adenosine deaminase complexing protein 2 or CD26 (cluster of differentiation 26) (49±15%, p<0.05), glucose transporter 2 (GLUT2) also known as solute carrier family 2 (facilitated glucose transporter), member 2 (SLC2A2) (28±9%, p<0.05) was accompanied by a parallel decrease in SLFN12 expression (30±10%, p<0.05) in atrophy duodenal samples. Immunolabeling demonstrated similar decreases in SLFN12, SI, and villin immunoreactivity with fasting (Fig. 1b). Interestingly, we did not observe substantial SLFN12 immunoreactivity in specimens of “intestinalized” Barrett’s esophagus (not shown). We validated the anti-SLFN12 antibody by immunostaining wild type Caco-2 cells, Caco-2 cells that overexpressed SLFN12 (induced by adenoviral SLFN12 overexpression), and Caco-2 cells in which endogenous SLFN12 had been reduced by siRNA. Rat IEC-6 cells, which do not express SLFN12, served as a negative control (Fig 1c).
Fig. 1.
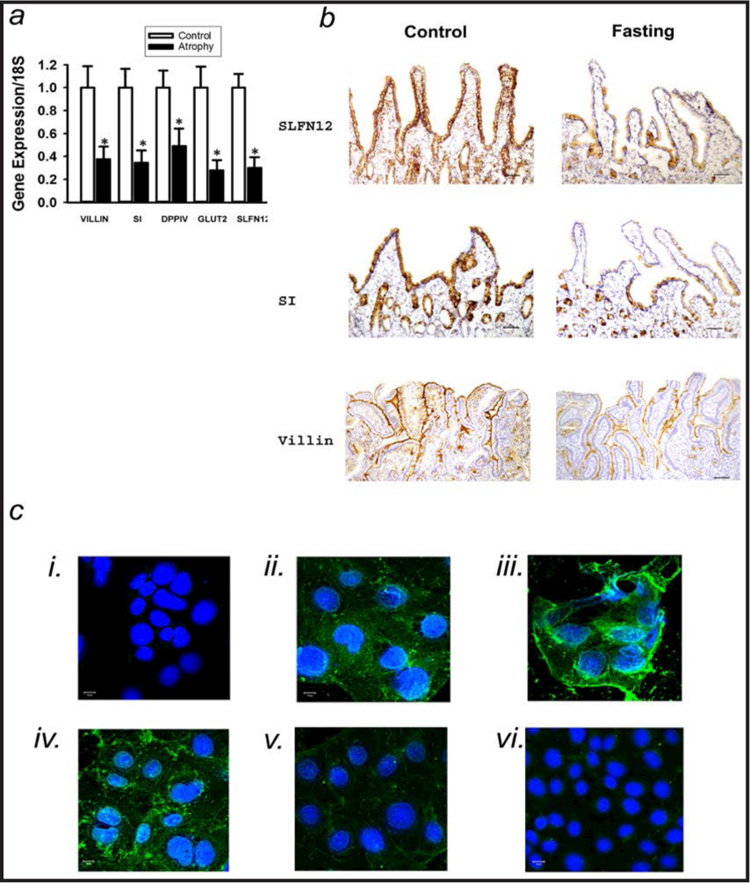
Duodenal Schlafen 12 is reduced in fasting humans in parallel with enterocyte differentiation marker expression. (a)Quantitative real-time PCR (qRT-PCR) for SLFN12, Sucrase-isomaltase (SI), Villin, DPPIV and GLUT2 from laser-captured epithelium from biopsied duodenal mucosa of control or fasting patients. Expression of SLFN12, SI, Villin, DPPIV and GLUT2 was significantly decreased in fasting patients vs. control (n=12–18,*p<0.05 by two-sided t-test with Bonferroni correction). 18S mRNA served as housekeeping control. (b) Representative SLFN12, SI, and villin stains demonstrate decreased immunoreactivity in fasting vs. control human duodenal biopsies. (Images shown are representative of 6 in each group.) (c) SLFN12 immunostaining with the same antibody for antibody validation. i) represents Caco-2 cells stained with FITC alone as a nuclear counterstain. ii) depicts wild type Caco-2 cells stained with anti-SLFN12 and the FITC counterstain. iii) shows increased SLFN12 immunoreactivity in Caco-2 cells infected with adenovirus encoding SLFN12. iv) represents wild type Caco-2 cells transfected with nontargeting NT1 siRNA, while v) represents paired Caco-2 cells similarly transfected with siRNA to SLFN12, demonstrating decreased immunoreactivity. vi) demonstrates the absence of SLFN12 immunoreactivity in rat IEC-6 cells stained with FITC and antibody to SLFN12.
Manipulating SLFN12 levels alters the expression of differentiation markers in human Caco-2 cells
We next examined the effects of SLFN12 in regulating human Caco-2BBE cell sucrase-isomaltase. Originally derived from a colon cancer, these cells express enterocytic features. Even subcloned, Caco-2BBE cells are heterogeneous, but SLFN12 and SI protein levels were highly correlated by flow cytometry within populations of Caco-2BBE cells (Fig. 2a).
Fig. 2.
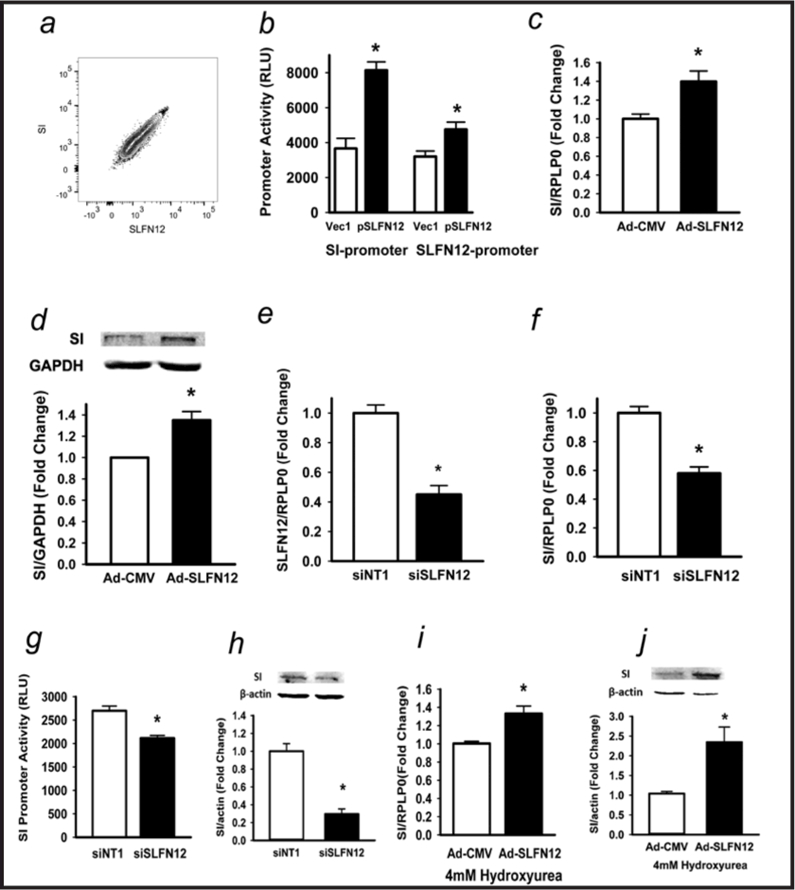
Schlafen 12 alters Caco-2 differentiation markers. (a) Flow cytometry correlating Caco-2 Slfn12 and sucrase-isomaltase (SI) protein. (b) Caco-2 cells co-transfected with human SI-promoter reporter or human SLFN12-promoter reporter constructs without or with empty vector (pEGFP-C1) or SLFN12 constructs (pEGFP-C1-SLFN12). SLFN12 enhances SI-promoter activity (n=6,*p<0.05 vs empty vector alone, left two bars) and SLFN12 promoter activity (n=6,*p<0.05 vs empty vector alone, right two bars). (c) SI expression in Caco-2 cells transduced with empty control virus (Ad-CMV) or SLFN12 adenovirus (Ad-SLFN12) for 72 hours (n=6,*p<0.05 vs empty control virus) Ribosomal Protein Lateral Stalk Subunit P1 [RPLP0] served as a control. (d) SI protein in Caco-2 cells (n=6,*p<0.05 vs empty control virus) with Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as a control. (e,f) siRNA to SLFN12 (siSLFN12) reduces SLFN12 and SI expression vs. non-targeting control siRNA (n=6,*p<0.05 vs siNT1). (g) siSLFN12 decreases Caco-2 SI promoter-driven luciferase activity vs. siNT1 in the presence of empty vector or SI promoter reporter constructs. (n=6,*p<0.05 vs siNT1). (h) siSLFN12 decreases Caco-2 SI protein levels by Western blot vs. siNT1. (n=6,*p<0.05 vs siNT1). (i) Caco-2 cells were transduced with Ad-CMV or Ad-SLFN12 in 4mM hydroxyurea. SLFN12 overexpression stimulates SI expression during complete hydroxyurea proliferative blockade (n=6,*p<0.05 vs empty control virus). (j) SLFN12 overexpression similarly increases SI protein levels after transduction with Ad-SLFN12 in 4 mM hydroxyurea vs. transduction with Ad-CMV. (n=6,*p<0.05 vs empty control virus). All statistics are by two-sided t test with Bonferroni corrections for multiple comparisons.
Exogenously increasing SLFN12 expression in subconfluent Caco-2 cells using p-CMV-EGFP-C1-SLFN12 DNA construct stimulated SI promoter activity (Fig. 2b, left bars). Interestingly, exogenous plasmid-based SLFN12 overexpression also stimulated SLFN12 promoter activity within the cells (Fig. 2b, right bars). Increasing SLFN12 expression using an adenoviral construct increased sucrase-isomaltase mRNA (Fig. 2c) and protein (Fig. 2d). Overexpressing SLFN12 using a different adeno-associated viral system yielded similar results (unpublished observations). Conversely, reducing endogenous Caco-2 SLFN12 by siRNA (n=6, p<0.05, Fig. 2e), decreased SI mRNA (n=6, p<0.05, Fig. 2f) promoter activity (n=6, p<0.05, Fig. 2g), and protein levels (Fig. 2h). Notably, SLFN12 overexpression continued to stimulate SI expression and SI protein levels when proliferation was completely blocked by 4 mM hydroxyurea (n=6, p<0.05 Fig. 2i,j).
SLFN12 acts through Serpin B12.
Caco-2 lysate was allowed to bind a GST-SLFN12 column, and LC-MS suggested SERPB12 in the subsequent eluate (not shown). This was confirmed by Western blotting of the eluate (Fig. 3a). Expression of GFP-SLFN12 in Caco-2 cells followed by immunoprecipitation with antibody to GFP coprecipitated SERPB12. (Fig. 3b) Consistent with the relatively promiscuous binding of Serpin-class proteins to other proteins, GFP alone also bound SERPB12 but to a lesser extent.
Fig. 3.
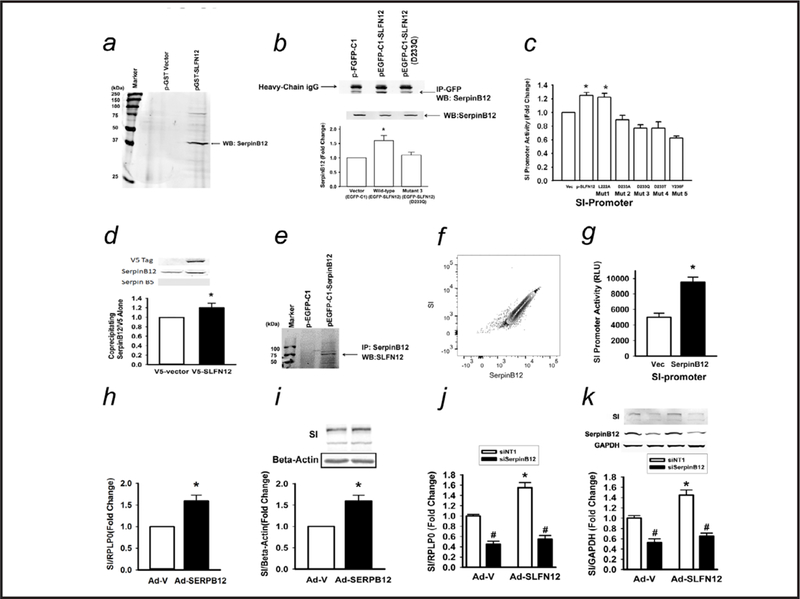
Schlafen 12 acts through SerpinB12. (a) Lysates of Caco-2 cells transfected with empty vector (GST-alone) or SLFN12 (GST-SLFN12) were purified on GST columns. (b) Coprecipitation with antibody to GFP of SerpinB12 protein from Caco-2 cells overexpressing empty vector (pEGFP-C1), SLFN12 (pEGFP-C1-SLFN12), or a SLFN12 point mutant (pEGFP-C1-SLFN12, D233Q). Constructs and lysates were immunoprecipitated with monoclonal anti-GFP and immunoblotted with polyclonal anti-SerpinB12 (n=4,*p<0.05 by ANOVA followed by two-sided t test with Bonferroni correction). (c) Point mutations to the atypical ATP-binding region of SLFN12 prevent induction of sucrase-isomaltase (SI) promoter activity. Caco-2 cells were co-transfected with human SI-promoter reporter constructs without or with empty vector (pEGFP-C1) or SLFN12 wild-type construct (pEGFP-C1-SLFN12) or SLFN12 mutants (p-EGFP-C1-SLFN12 L222A, mutant1; p-EGFP-C1-SLFN12 D233A, mutant2; p-EGFP-C1-SLFN12 D233Q, mutant3; p-EGFP-C1-SLFN12 D233T, mutant4; and p-EGFP-C1-SLFN12 Y236F, mutant5) before luciferase reporter assay (n=6,*p<0.05 by ANOVA followed by two-sided t test with Bonferroni correction). (d) Caco-2 cells were infected with a lentivirus encoding either a V5 tag (as a control) or V5-tagged SLFN12, lysed, and immunoprecipitated with antibody to the V5 tag, prior to immunoblotting for Serpin B12 (n=3, *p<0.05), V5 (not seen in control lanes because the V5 tag alone is too small and runs off the gel), and Serpin B5, which was not observed to coprecipitate although we confirmed that the Serpin B5 antibody could be used for Western blotting in these cells (not shown). (e) Caco-2 cells were transfected with empty vector (pEGFP-C1) or SerpinB12 (pEGFP-C1-SerpinB12) constructs for 72 hours. Lysates were immunoprecipitated with monoclonal anti-SerpinB12 and immunoblotted with polyclonal anti-SLFN12. (f) Flow cytometry correlating endogenous Caco-2 SerpinB12 and SI expression. (g) Caco-2 cells were co-transfected with human SI-promoter reporter construct without or with empty vector (pEGFP-C1) or SerpinB12 (pEGFP-C1-SerpinB12) and luciferase reporter activity was assayed (n=6,*p<0.05). (h) Caco-2 cells were transduced with AAV-SerpinB12 virus or empty control virus (AAV-ZsGreen) and SI mRNA was measured (n=6,*p<0.05). (i) Caco-2 cells were transduced with Ad-CMV or Ad-SerpinB12 and western blot for SI protein was performed (n=6,*p<0.05 vs empty control virus). (j) Caco-2 cells were incubated with siNT or SerpinB12 siRNA (siSerpinB12), transduced with Ad-CMV or Ad-SLFN12 for 72 hours, and SI mRNA or (k) SI protein was measured (n=6, #p<0.05, to respective siNT; *p<0.05 siNT1-Ad-SLFN12 compared to siNT-Ad-V). All statistics are by two-sided t test with Bonferroni correction, with ANOVA performed as indicated.
We have previously demonstrated that the atypical ATP-binding region of Schlafen 3 is critical to its ability to induce rat IEC-6 cell differentiation [16]. We therefore tested similar point mutations in the atypical ATP-binding region of SLFN12 for their effects on SLFN12 association with SERPB12 and on SLFN12 induction of sucrase-isomaltase promoter activity. Point mutations to the atypical ATP-binding region of SLFN12 both reduced SERPB12 co-precipitation with GFP-SLFN12 to the background levels of GFP alone (Fig. 3b) and prevented induction of sucrase-isomaltase promoter activity (Fig. 3c). We confirmed these results using a V5-tagged SLFN12 construct, again demonstrating baseline binding of SERPB12 to the V5 but again finding increased binding to the V5-tagged SLFN12 (Fig. 3d). In contrast, we did not find evidence for the binding of Serpin B5 to V5-SLFN12, suggesting the specificity of the SLFN12-SERPB12 interaction. Conversely, immunoprecipitation of SERPB12 co-precipitated endogenous wild type SLFN12 (Fig. 3e). Endogenous SERPB12 levels in heterogeneous Caco-2 cells correlated tightly with sucrase-isomaltase levels by flow cytometry (Fig. 3f). Plasmid-based overexpression of SERPB12 increased sucrase-isomaltase promoter activity (Fig. 3g) while adeno-associated viral overexpression of SERPB12 increased sucrase-isomaltase mRNA (Fig. 3h) and protein levels (Fig. 3i). Reducing SERPB12 by siRNA blocked SLFN12 effects on sucrase-isomaltase mRNA (Fig. 3j) and protein (Fig. 3k).
SLFN12 modulates sucrase-isomaltase via Serpin B12 and deubuiquitylases
SERPB12 has been predicted to bind to USP14 in hepatocellular carcinoma tissues [28], so we tested whether b-AP15, which inhibits the deubuiquitylases (DUB) UCHL5 and USP14[29] could block SLFN12 effects. Finding that it did (not shown), we next tested the more specific effects of siRNA reduction of UCHL5 and USP14. Interestingly, reducing either UCHL5 or USP14, attenuated but did not completely block the effects of SLFN12 on sucrase-isomaltase expression (not shown), while reducing both UCHL5 and USP14 together completely blocked the effect (Fig. 4a). Either SLFN12 overexpression (Fig. 4b and 4c) or SERPB12 overexpression (Fig. 4g and 4h) increased both USP14 (Fig. 4b and 4g) and UCHL5 (Fig. 4c and 4h) expression. We further confirmed that reduction of both UCHL5 and USP14 blocked the induction of SI by SLFN12 at the protein level (Fig. 4d). Effective knockdown of USP14 (Fig. 4e) and UCHL5 (Fig. 4f) was also confirmed at the protein level, as was the apparent increase in USP14 (Fig. 4e) and UCHL5 (Fig. 4f) in response to SLFN 12 overexpression.
Fig. 4.
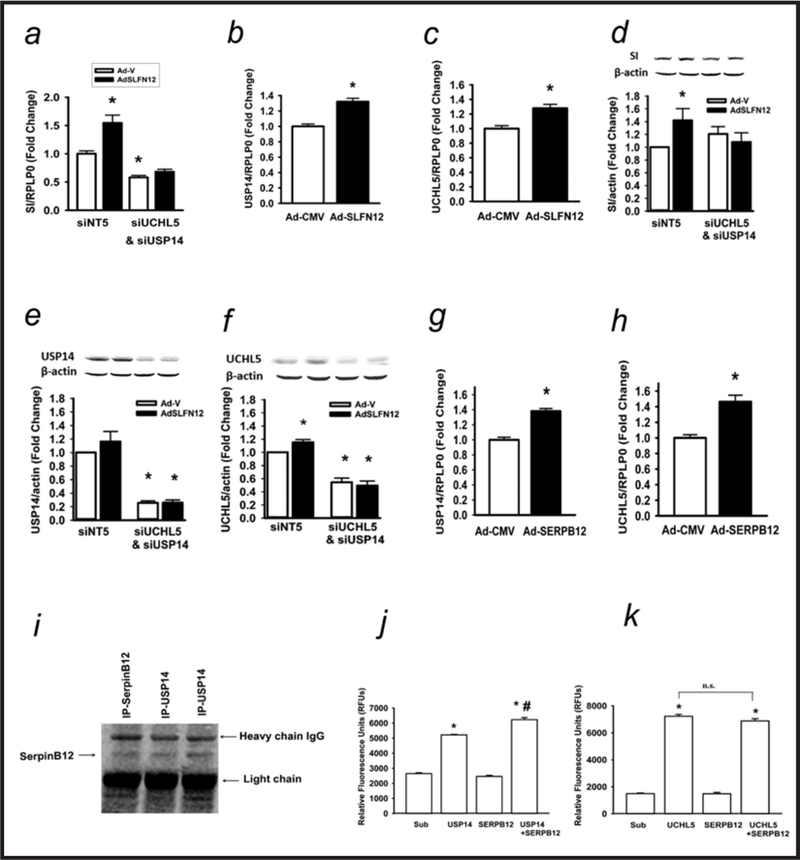
Schlafen 12 acts by modulating deubuiquitylase activity. (a) Caco-2 cells transfected with non-targeting siRNA5 (siNT5) or a combination of both USP14 (siUSP14) and UCHL5 (siUCHL5) siRNA followed by transduction with control (Ad-CMV) or SLFN12 (Ad-SLFN12) viruses for 72 hours, and SI expression was assessed (n=6,*p<0.05). (b,c) Caco-2 cells were transduced with Ad-CMV or Ad-SLFN12 for 72 hours and USP14 and UCHL5 expression was measured (n=6 each,*p<0.05). (d,e,f) Caco-2 cells were transfected with either nontargeting siRNA NT5 or combined siRNA to UCHL5 and USP15 along with either an empty vector adenovirus (Ad-V) or adenovirus encoding SLFN12 (AdSLFN12). Western blots were performed for sucraseisomaltase (d), USP14 (e), and UCHL5 (f). (g,h) Caco-2 cells were transduced with Ad-CMV or Ad-SERPB12 and USP14 and UCHL5 expression measured. (i) Caco-2 cell lysates were immunoprecipitated with monoclonal USP14 or monoclonal SerpinB12 antibodies and immunoblotted with polyclonal SerpinB12 antibody. We immunoprecipitated Serpin B12 directly as a control to confirm the correct apparent molecular weight of the co-precipitating Serpin B12. (j) Incubation with recombinant human Serpin B12 stimulated deubuiquitylase activity vs. USP14 alone (n=4,*p<0.05 to substrate (sub) or SERPB12 alone; # p<0.05 vs. USP14 alone). (k) Incubation with recombinant human Serpin B12 did not affect the UCHL5 deubuiquitylase activity vs. UCHL5 alone (n=4,*p<0.05 to substrate (Sub) or Serpin B12). All statistics are by two-sided t test with Bonferroni corrections for multiple comparisons.
SERPB12 immunoprecipitation co-precipitated USP14 (Fig. 4i), but we could not co-precipitate UCHL5 (not shown). Moreover, when we mixed purified SERPB12 with each of these two purified deubuiquitylases in vitro, SERPB12 stimulated the DUB activity of USP14 (Fig. 4j) but not UCHL5 (Fig. 4k). Since both deubiquitylases have more substantial activity when proteasome-bound in vivo than in isolation, whether SERPB12 stimulates proteasomally-bound USP14 activity in vivo or competes for the USP14 proteasomal binding site awaits further study. However, these results raise the possibility of a dual effect of the SLFN12-SerpinB12 axis on intracellular deubiquitylation, directly binding to and altering USP14 activity while indirectly increasing USP14 and UCHL5 expression.
SLFN12 DUB-dependently induces CDX2, which is required for SLFN12 induction of sucrase-isomaltase promoter activity
We selected CDX2 and CDX4 for further study as they bind the sucrase-isomaltase promoter. Infecting Caco-2BBE cells with Ad-GFP-SLFN12 increased CDX2 protein (Fig. 5a) but not CDX4 (Fig. 5b). However, CDX2 mRNA levels did not change (Fig. 5c), indicating direct regulation of protein expression. Treating the cells with siRNA to the two deubiquitylases (USP14 and UCHL5) prevented the effect of SLFN12 overexpression on CDX2 protein expression (Fig. 5d). In further studies, ≈50% reduction of CDX2 using combined siRNA (data not shown) prevented the stimulation of sucrase-isomaltase promoter activity by a SLFN12 plasmid (Fig. 5e).
Fig. 5.
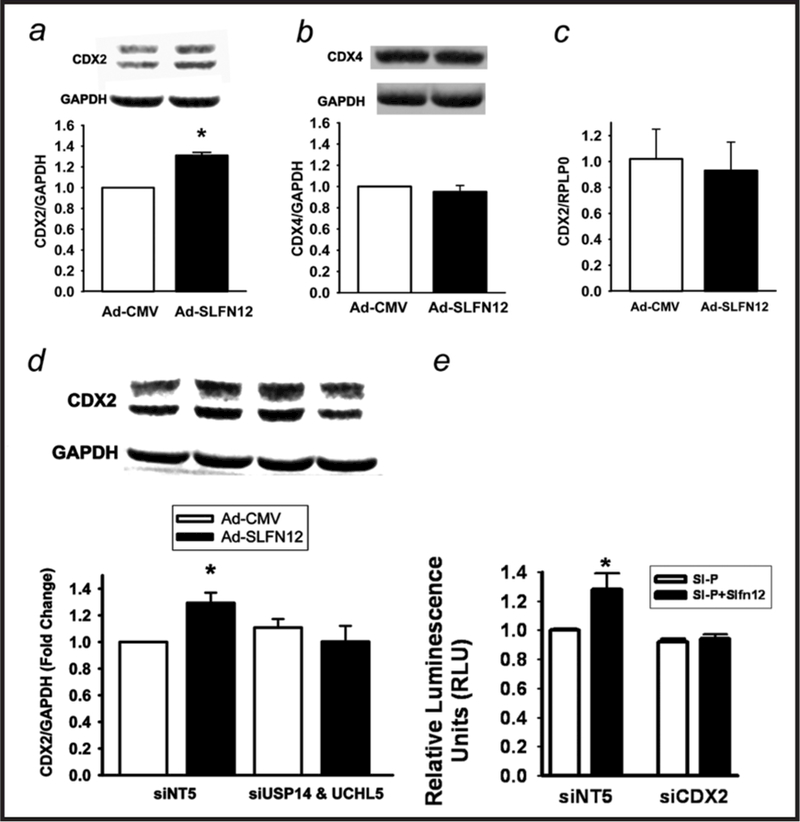
Schlafen 12 induces CDX2 transcription factor and its effects are blocked by reducing USP14 and UCHL5 deubiquitylase. (a,b) Caco-2 cells were transduced with Ad-CMV or Ad-SLFN12 for 72 hours and CDX2 or CDX4 protein was measured (n=5 for each, p>0.05). (c) Caco-2 cells were transduced with Ad-CMV or Ad-SLFN12 for 72 hours and CDX2 mRNA was assessed (n=5, p>0.05). (d) Caco-2 cells transfected with non-targeting siRNA5 (siNT5) or a combination of both USP14 (siUSP14) and UCHL5 (siUCHL5) siRNA followed by transduction with Ad-CMV or Ad-SLFN12 for 72 hours, lysis, and western blotting using CDX2 or GAPDH antibodies (n=6,*p<0.05). (e) ≈50% reduction of CDX2 by siRNA (siCDX2), data not shown prevents the induction of sucrase-isomaltase promoter activity by cotransfection with a SLFN12 plasmid (SI-P+SLFN12) in comparison to sucrase-isomaltase promoter activity after cotransfection with an empty vector control plasmid (P). In contrast, SI-P+SLFN12 induces sucrase-isomaltase promoter activity after transfection with non-targeting siRNA (siNT5). (n=6, *p<0.05). All statistics are by two-sided t test with Bonferroni corrections for multiple comparisons.
The SLFN12-Serpin B12 pathway occurs outside Caco-2 cells
Adenovirally overexpressing either SLFN12 (137.6+15.6 fold) or SERPB12 (884.3+194.8 fold) in human non-malignant fetal intestinal HIEC-6 cells stimulated sucrase-isomaltase and villin 1 mRNA expression (Fig. 6)
Fig. 6.
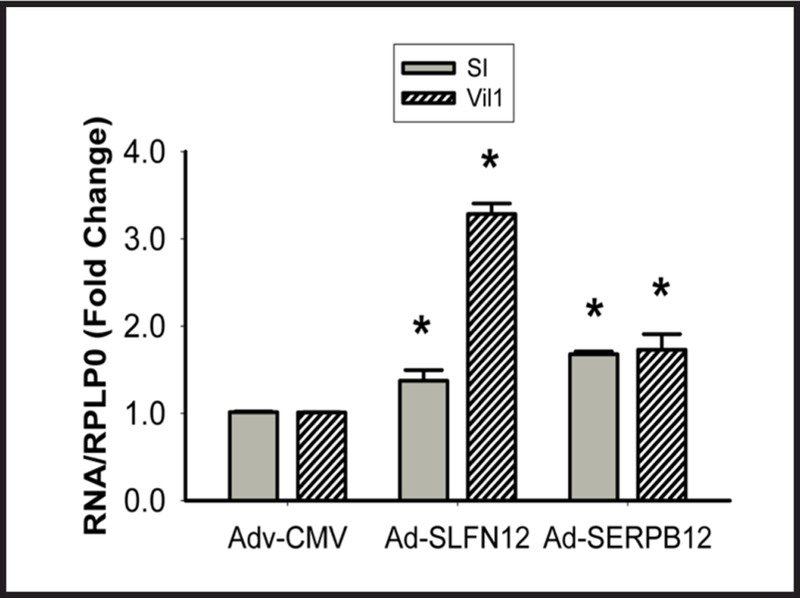
The Schlafen 12-Serpin B12 pathway regulates enterocytic sucrase-isomaltase expression in HIEC-6 cells. Infection with adenovirus expressing either SLFN12 (AdSLFN12) or SERPB12 (AdSERPB12) increases sucrase-isomaltase (SI) and villin 1 (Vil1) mRNA in HIEC-6 cells vs. adenovirus expressing only the CMV promoter (Adv-CMV) (n=6 pooled from 3 separate experiments, *p<0.05).
Discussion
Although many stimuli and transcription factors influence enterocytic differentiation, our understanding of the signaling that regulates these effects is incomplete. Our results suggest that SLFN12 critically influences human enterocytic differentiation. SLFN12 is lost during fasting as enterocytic differentiation markers are decreased, and in vitro overexpression or reduction of SLFN12 drives sucrase-isomaltase expression in Caco-2 cells, a common model for the study of human enterocytic biology, and non-malignant HIEC-6 primary human enterocytes. SLFN12 appears to act by binding to SERPB12, which in turn stimulates deubiquitylase activity, increasing Cdx2 protein. We further confirmed the role of SLFN12 and SERPB12 in this pathway in non-malignant human small intestinal HIEC-6 cells.
The Schlafens include short, intermediate, and long proteins. The short Slfn1 and Slfn2 influence T cell development [30], and Slfn2 mediates osteoclastogenesis [13]. Their subcellular mechanisms are unknown, and neither Slfn1 nor Slfn2 is expressed in humans. Humans express only SLFN1L, SLFN5, SLFN11, SLFN12, SLFN12L, SLFN13, and SLFN14. The only intermediate human Schlafens are SLFN12 and its alternate splice variant SLFN12L. Long Schlafens require a nuclear targeting sequence and a helicase domain absent in short and intermediate Schlafen proteins for their diverse nuclear actions [12]. For instance, the murine long Slfn8 impairs T cell development [31]. Human SLFN11 correlates with cancer cell responsiveness to topoisomerase inhibitors [32], and mediates retroviral resistance by inhibiting viral mRNA translation [33]. In contrast, SLFN11 inactivation has been linked in cancer cells to PARP inhibitor resistance [34]. SLFN5 also influences renal carcinoma cell motility by uncertain mechanism [35]. Both long SLFN5 and intermediate SLFN12L have been linked to intestinal metaplasia and gastric cancer [36]. In contrast to long Schlafens, SLFN12 is like rodent intermediate Slfn3, lacking a nuclear targeting sequence and localizing at least predominantly in the cytoplasm. The intermediate Schlafen proteins are poorly understood, and their subcellular mechanism is unknown. Slfn3 and SLFN12 exhibit only approximately 40% sequence homology, but the enterocytic differentiating effects of Slfn3 led us to hypothesize a similar effect for SLFN12.
Our present results suggest that SLFN12 promotes human enterocytic differentiation. Differentiation markers diminished in parallel with SLFN12 within the distal duodenal mucosa of humans fasting for four or more days. Although the subjects studied represented a heterogeneous group characterized by non-duodenal pathology, these results were statistically significant and suggestive. Although it would be impossible to prove that all phenotypic characteristics of a human enterocyte require SLFN12, these results certainly suggest that some may. These SLFN12 effects may seem modest, but they are consistent in magnitude with changes in mucosal atrophy that others have observed in vivo [37, 38] and the effects of differentiating stimuli on Caco-2 cells in vitro [39–43]. Since many markers of enterocytic differentiation are enzymes or transport proteins, small changes in such proteins can catalyze greater downstream effects.
Caco-2BBE cells, originally derived from human colon cancer, resemble human enterocytes, and are commonly used to model enterocyte biology. Caco-2BBE cells are imperfect, and ultimately cannot change from colon cancer to a non-malignant enterocyte. However, our cell culture work suggests that SLFN12 expression drives SI promoter activity and expression in Caco-2 and HIEC-6 cells, just as SLFN3 does in intact rodents. The induction of SLFN12 promoter activity by exogenous SLFN12 overexpression suggests a feedback loop that contributes to the speed with which enterocytes differentiate in vivo as they migrate from crypt to villus tip or in response to dietary changes or other stimuli. Cytosolic SLFN12 cannot bind the nuclear SLFN12 promoter directly. More likely, the cascade modulating deubiquitylation described here alters levels of transcription factors that influence the SLFN12 promoter. This awaits delineation.
Intermediate-length schlafens such as SLFN12 lack obvious signaling motifs and are structurally homologous only with other schlafens, motivating the question of how SLFN12 acts. The activity of rodent Slfn3 resides in its N-terminal half, and point mutations in the central domain prevent Slfn3 promotion of rat enterocyte differentiation [16]. However, this does not illuminate the downstream mechanism by which Slfn3 or any other intermediate or short Schlafen protein acts. We demonstrate here that similar mutations not only inactivate SLFN12 but also prevent SLFN12 binding to SERPB12. Furthermore, SLFN12 effects are inhibited by reducing SERPB12 with siRNA and mimicked by SERPB12 overexpression. Taken together, all these observations suggest that SLFN12 acts via its interaction with SERPB12.
The mechanistic importance of this interaction between SLFN12 and Serpin B12 awaits further exploration. Although a ternary complex has not been demonstrated between SLFN12, Serpin B12, and USP14, such a complex may exist transiently. SLFN12 could present Serpin B12 to USP14 or facilitate their interaction. Alternatively, SLFN12 binding to Serpin B12 could alter the Serpin B12 conformation so that it binds to a chaperone protein, or Serpin B12 subcellular localization could be altered to facilitate subsequent Serpin B12 interaction with USP14. Although it might be tempting to suggest that the central domain of SLFN12 is involved in this mechanism, this domain seems to more closely resemble an AlbA_2 domain that typically interacts with nucleotides, rather than the divergent AAA ATPase-like domain that is suggested by misannotation in some databases. This makes a direct catalytic modification of Serpin B12 by Schlafen12 unlikely. The confusion regarding the identity of this domain in the literature may have arisen due InterPro (http://www.ebi.ac.uk/interpro/entry/IPR007421) and other sources describing this region as a “schlafen AAA domain”; however, the so-called schlafen AAA domain is actually identified in proteins by InterPro using an AlbA_2 DNA binding domain signature (http://pfam.xfam.org/family/PF04326). The prior AAA confusion may have arisen because of remote sequence similarity in part of the AlbA_2 domains with part of some AAA domains (J. Chen and L. A. Kuhn, manuscript in preparation). Thus, whether SLFN12 acts with regard to Serpin B12 as a scaffolding, linking, or targeting molecule, or in some other fashion, awaits further study.
The serpins are themselves a complex superfamily. Serpins have highly conserved overall structures despite wide variability in specifics, exhibiting N-terminal helical domains and C-terminal beta-pleated sheets with conjoining hinges. They classically bind irreversibly and block proteases (hence SERine Protease INhibitors), but not all serpins do so [44]. Although most serpins are specific for serine proteases, some inhibit cysteine proteases or both cysteine and serine proteases, and some do not inhibit proteases at all [45] but bind other proteins without suppressing their function as chaperones or transport proteins [46]. B clade Serpins (also called ovalbumin serpins) are diverse in their targets. SERPB12 even binds GFP, although not as effectively as it binds SLFN12. The function of SERPB12 is unknown, although other Clade B serpins may act intracellularly. For instance, maspin (SerpinB5) is a tumor suppressor gene that modulates breast cancer cell adhesion and invasion and is lost in invasive breast cancers [47] Other members of this family include ovalbumin itself, PAI2, and elastase inhibitor. SERPB12, also called yukopin, targets both trypsin and plasmin, but seems likely to have other effects as well [45].
Our present results suggest that the SLFN12-Serpin B12 axis stimulates USP14 deubuiquitylase activity. This, in turn, represents a key mechanism by which these proteins influence enterocytic differentiation since the SLFN12 effects are blocked by pharmacologic inhibition or molecular reduction of the deubuiquitylases USP14 and UCHL5. USP14 and UCHL5 are cysteine proteases associated with the 19S regulatory subunit of the proteasome, with overlapping and sometimes complementary roles. Because of their slightly different deubiquitinating profiles, USP14 can either rescue proteins from proteasomal degradation by removing the polyubiquitin chain that facilitates proteasomal interaction or simply recycle the ubiquitin from a protein already commited to proteasomal interaction while UCHL5 more frequently facilitates proteasomal degradation [48–50]. However, these two structurally distinct deubiquitylases have overlapping and partially redundant functions so that they can compensate for each other’s activity [51, 52], as we have observed here. This could explain why it was necessary to reduce both USP14 and UCHL5 to block the downstream effects of the Schlafen-Serpin-USP14 pathway we postulate here. Alternatively, it is possible that, as reported by Fang in hepatocellular carcinoma tissues [28], SERPB12 also interacts with UCHL5 directly in this pathway even though we were unable to co-precipitate the two proteins here. The further downstream elements of modulating deubiquitylation await conclusive elucidation, but changes in DUB activity might modulate the proteasomal degradation of proteins such as transcription factors such as Cdx2 to regulate nuclear gene expression. This possibility awaits further study, but could help explain how Slfn3 [16] (and perhaps by hypothesized extension SLFN12) might act within the cytosol to ultimately regulate gene expression.
Although we have studied the effects of SLFN12 on enterocytic differentiation, these results may have broader implications for the mechanisms by which intermediate Schlafen proteins influence other cell types. SLFN12 has previously been reported to alter the phenotype of other cells, including prostatic cancer cells [53] and activated T cells [54, 55], and to inhibit dendritic cell differentiation [11]. Slfn4, also intermediate length, regulates myelopoiesis [56]. However, the mechanisms by which all these effects occur are unknown. Whether these effects also occur via SERPB12 and downstream influences on deubuiquitylation awaits study. For instance, SLFN12 was recently reported to interact directly with phosphodiesterase 3A in cancer cells, including stromal tumors, and to drive cancer chemotherapeutic resistance to phosphodiesterase 3 inhibitors [57, 58], but this interaction does not appear to be important in non-malignant cells [59, 60].
Most attention has focused on increasing mucosal mass in short gut with enterocytic mitogens like glutamine and teduglutide that may actually decrease enterocytic differentiation [61, 62], and on decreasing available intestinal mucosal mass by Roux-en-Y intestinal bypass in morbid obesity. Our results raise the possibility that enterocytic differentiation can be manipulated directly independently of proliferation to enhance or reduce enterocyte nutrient absorption and digestion. While further work is required to more fully define the pathway by which SLFN12 works, SLFN12 may be an important mediator of human enterocytic differentiation. Schlafen 12 and the pathways by which it acts may represent important targets for altering human small intestinal functional capacity in settings as diverse as prolonged fasting, mucosal atrophy during total parenteral nutrition, short gut syndrome, and morbid obesity, while the pathway we have described could have broader applicability to the modulation of other cells’ phenotype by SLFN12 and even other intermediate Schlafens.
Acknowledgements
This work was funded by NIHRO1DK096137 (MDB). Flow cytometry was supported in part by IDeA Awards P20GM103442 and P20GM113123. Assistance by Pam Haan and Dr. Harvey Bumpers is gratefully acknowledged.
Footnotes
Disclosure Statement
The authors declare that they have no competing interests.
References
- 1.Wales PW, Christison-Lagay ER: Short bowel syndrome: epidemiology and etiology. Semin Pediatr Surg 2010;19:3–9. [DOI] [PubMed] [Google Scholar]
- 2.Basson MD: In vitro evidence for matrix regulation of intestinal epithelial biology during mucosal healing. Life Sci 2001;69:3005–3018. [DOI] [PubMed] [Google Scholar]
- 3.Basson MD, Li GD, Hong F, Han O, Sumpio BE: Amplitude-dependent modulation of brush border enzymes and proliferation by cyclic strain in human intestinal Caco-2 monolayers. J Cell Physiol 1996;168:476–488. [DOI] [PubMed] [Google Scholar]
- 4.Crawley SW, Mooseker MS, Tyska MJ: Shaping the intestinal brush border. J Cell Biol 2014;207:441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Emenaker NJ, Basson MD: Short chain fatty acids inhibit human (SW1116) colon cancer cell invasion by reducing urokinase plasminogen activator activity and stimulating TIMP-1 and TIMP-2 activities, rather than via MMP modulation. J Surg Res 1998;76:41–46. [DOI] [PubMed] [Google Scholar]
- 6.Gayer CP, Basson MD: The effects of mechanical forces on intestinal physiology and pathology. Cell Signal 2009;21:1237–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang CZ, Yu T, Chen QK: DNA Methylation Dynamics During Differentiation, Proliferation, and Tumorigenesis in the Intestinal Tract. Stem Cells Dev 2015;24:2733–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rubin DC, Levin MS: Mechanisms of intestinal adaptation. Best Pract Res Clin Gastroenterol 2016;30:237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwarz DA, Katayama CD, Hedrick SM: Schlafen, a new family of growth regulatory genes that affect thymocyte development. Immunity 1998;9:657–668. [DOI] [PubMed] [Google Scholar]
- 10.Fletcher SJ, Johnson B, Lowe GC, Bem D, Drake S, Lordkipanidze M, Guiu IS, Dawood B, Rivera J, Simpson MA, Daly ME, Motwani J, Collins PW, Watson SP, Morgan NV, Genotyping UK, Phenotyping of Platelets study g: SLFN14 mutations underlie thrombocytopenia with excessive bleeding and platelet secretion defects. J Clin Invest 2015;125:3600–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puck A, Aigner R, Modak M, Cejka P, Blaas D, Stockl J: Expression and regulation of Schlafen (SLFN) family members in primary human monocytes, monocyte-derived dendritic cells and T cells. Results Immunol 2015;5:23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mavrommatis E, Fish EN, Platanias LC: The schlafen family of proteins and their regulation by interferons. J Interferon Cytokine Res 2013;33:206–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee NK, Choi HK, Yoo HJ, Shin J, Lee SY: RANKL-induced schlafen2 is a positive regulator of osteoclastogenesis. Cell Signal 2008;20:2302–2308. [DOI] [PubMed] [Google Scholar]
- 14.Yuan L, Yu Y, Sanders MA, Majumdar AP, Basson MD: Schlafen 3 induction by cyclic strain regulates intestinal epithelial differentiation. Am J Physiol Gastrointest Liver Physiol 2010;298:G994–G1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kovalenko PL, Yuan L, Sun K, Kunovska L, Seregin S, Amalfitano A, Basson MD: Regulation of epithelial differentiation in rat intestine by intraluminal delivery of an adenoviral vector or silencing RNA coding for Schlafen 3 PLoS One 2013;8:e79745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chaturvedi L, Sun K, Walsh MF, Kuhn LA, Basson MD: The P-loop region of Schlafen 3 acts within the cytosol to induce differentiation of human Caco-2 intestinal epithelial cells. Biochim Biophys Acta 2014;1843:3029–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gericke B, Amiri M, Naim HY: The multiple roles of sucrase-isomaltase in the intestinal physiology. Mol Cell Pediatr 2016;3:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noda S, Yamada A, Tanabe R, Nakaoka K, Hosoi T, Goseki-Sone M: Menaquinone-4 (vitamin K2) up-regulates expression of human intestinal alkaline phosphatase in Caco-2 cells. Nutr Res 2016;36:1269–1276. [DOI] [PubMed] [Google Scholar]
- 19.Roostaee A, Guezguez A, Beausejour M, Simoneau A, Vachon PH, Levy E, Beaulieu JF: Histone deacetylase inhibition impairs normal intestinal cell proliferation and promotes specific gene expression. J Cell Biochem 2015;116:2695–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sambuy Y, De Angelis I, Ranaldi G, Scarino ML, Stammati A, Zucco F: The Caco-2 cell line as a model of the intestinal barrier: influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol Toxicol 2005;21:1–26. [DOI] [PubMed] [Google Scholar]
- 21.Piena-Spoel M, Sharman-Koendjbiharie M, Yamanouchi T, Tibboel D: “Gut-feeling” or evidence-based approaches in the evaluation and treatment of human short-bowel syndrome. Pediatr Surg Int 2000;16:155–164. [DOI] [PubMed] [Google Scholar]
- 22.Traber PG: Control of gene expression in intestinal epithelial cells. Philos Trans R Soc Lond B Biol Sci 1998;353:911–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Inoue S, Honma K, Mochizuki K, Goda T: Induction of histone H3K4 methylation at the promoter, enhancer, and transcribed regions of the Si and Sglt1 genes in rat jejunum in response to a high-starch/low-fat diet. Nutrition 2015;31:366–372. [DOI] [PubMed] [Google Scholar]
- 24.Walsh MF, Ampasala DR, Hatfield J, Vander Heide R, Suer S, Rishi AK, Basson MD: Transforming growth factor-beta stimulates intestinal epithelial focal adhesion kinase synthesis via Smad- and p38-dependent mechanisms. Am J Pathol 2008;173:385–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perreault N, Beaulieu JF: Use of the dissociating enzyme thermolysin to generate viable human normal intestinal epithelial cell cultures. Exp Cell Res 1996;224:354–364. [DOI] [PubMed] [Google Scholar]
- 26.Seregin SS, Aldhamen YA, Appledorn DM, Hartman ZC, Schuldt NJ, Scott J, Godbehere S, Jiang H, Frank MM, Amalfitano A: Adenovirus capsid-display of the retro-oriented human complement inhibitor DAF reduces Ad vector-triggered immune responses in vitro and in vivo. Blood 2010;116:1669–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Basson MD, Zeng B, Wang S: Akt1 binds focal adhesion kinase via the Akt1 kinase domain independently of the pleckstrin homology domain. J Physiol Pharmacol 2015;66:701–709. [PubMed] [Google Scholar]
- 28.Fang Y, Mu J, Ma Y, Ma D, Fu D, Shen X: The interaction between ubiquitin C-terminal hydrolase 37 and glucose-regulated protein 78 in hepatocellular carcinoma. Mol Cell Biochem 2012;359:59–66. [DOI] [PubMed] [Google Scholar]
- 29.Tian Z, D’Arcy P, Wang X, Ray A, Tai YT, Hu Y, Carrasco RD, Richardson P, Linder S, Chauhan D, Anderson KC: A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood 2014;123:706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahmadi S, Veinotte LL: Effect of Schlafen 2 on natural killer and T cell development from common T/natural killer progenitors. Pak J Biol Sci 2011;14:1002–1010. [DOI] [PubMed] [Google Scholar]
- 31.Geserick P, Kaiser F, Klemm U, Kaufmann SH, Zerrahn J: Modulation of T cell development and activation by novel members of the Schlafen (slfn) gene family harbouring an RNA helicase-like motif. Int Immunol 2004;16:1535–1548. [DOI] [PubMed] [Google Scholar]
- 32.Tian L, Song S, Liu X, Wang Y, Xu X, Hu Y, Xu J: Schlafen-11 sensitizes colorectal carcinoma cells to irinotecan. Anticancer Drugs 2014;25:1175–1181. [DOI] [PubMed] [Google Scholar]
- 33.Lin YZ, Sun LK, Zhu DT, Hu Z, Wang XF, Du C, Wang YH, Wang XJ, Zhou JH: Equine schlafen 11 restricts the production of equine infectious anemia virus via a codon usage-dependent mechanism. Virology 2016;495:112–121. [DOI] [PubMed] [Google Scholar]
- 34.Murai J, Feng Y, Yu GK, Ru Y, Tang SW, Shen Y, Pommier Y: Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget 2016;7:76534–76550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sassano A, Mavrommatis E, Arslan AD, Kroczynska B, Beauchamp EM, Khuon S, Chew TL, Green KJ, Munshi HG, Verma AK, Platanias LC: Human Schlafen 5 (SLFN5) Is a Regulator of Motility and Invasiveness of Renal Cell Carcinoma Cells. Mol Cell Biol 2015;35:2684–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merchant JL, Ding L: Hedgehog Signaling Links Chronic Inflammation to Gastric Cancer Precursor Lesions. Cell Mol Gastroenterol Hepatol 2017;3:201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song J, Wolf SE, Wu XW, Finnerty CC, Gauglitz GG, Herndon DN, Jeschke MG: Starvation-induced proximal gut mucosal atrophy diminished with aging. JPEN J Parenter Enteral Nutr 2009;33:411–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolf SE, Debroy MA, Ikeda H, Jeschke M, Matin S, Rajaraman S, Ko TC, Englander EW, Norman JG, Thompson JC: Increased small bowel epithelial turnover in interleukin-1 receptor knockout mice. Ann Surg 2000;232:42–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Couto MR, Goncalves P, Catarino TA, Martel F: The Effect of Inflammatory Status on Butyrate and Folate Uptake by Tumoral (Caco-2) and Non-Tumoral (IEC-6) Intestinal Epithelial Cells. Cell J 2017;19:96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shao Y, Wolf PG, Guo S, Guo Y, Gaskins HR, Zhang B: Zinc enhances intestinal epithelial barrier function through the PI3K/AKT/mTOR signaling pathway in Caco-2 cells. J Nutr Biochem 2017;43:18–26. [DOI] [PubMed] [Google Scholar]
- 41.Sun X, Yang Q, Rogers CJ, Du M, Zhu MJ: AMPK improves gut epithelial differentiation and barrier function via regulating Cdx2 expression. Cell Death Differ 2017;24:819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van De Walle J, Hendrickx A, Romier B, Larondelle Y, Schneider YJ: Inflammatory parameters in Caco-2 cells: effect of stimuli nature, concentration, combination and cell differentiation. Toxicol In vitro 2010;24:1441–1449. [DOI] [PubMed] [Google Scholar]
- 43.Wang Q, Zhou Y, Rychahou P, Fan TW, Lane AN, Weiss HL, Evers BM: Ketogenesis contributes to intestinal cell differentiation. Cell Death Differ 2017;24:458–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huntington JA: Serpin structure, function and dysfunction. J Thromb Haemost 2011;9:26–34. [DOI] [PubMed] [Google Scholar]
- 45.Izuhara K, Ohta S, Kanaji S, Shiraishi H, Arima K: Recent progress in understanding the diversity of the human ov-serpin/clade B serpin family. Cell Mol Life Sci 2008;65:2541–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Silverman GA, Whisstock JC, Bottomley SP, Huntington JA, Kaiserman D, Luke CJ, Pak SC, Reichhart JM, Bird PI: Serpins flex their muscle: I. Putting the clamps on proteolysis in diverse biological systems. J Biol Chem 2010;285:24299–24305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Endsley MP, Zhang M: Investigating maspin in breast cancer progression using mouse models. Methods Enzymol 2011;499:149–165. [DOI] [PubMed] [Google Scholar]
- 48.Selvaraju K, Mazurkiewicz M, Wang X, Gullbo J, Linder S, D’Arcy P: Inhibition of proteasome deubiquitinase activity: a strategy to overcome resistance to conventional proteasome inhibitors? Drug Resist Updat 2015;21–22:20–29. [DOI] [PubMed]
- 49.Lee BH, Lu Y, Prado MA, Shi Y, Tian G, Sun S, Elsasser S, Gygi SP, King RW, Finley D: USP14 deubiquitinates proteasome-bound substrates that are ubiquitinated at multiple sites. Nature 2016;532:398–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ristic G, Tsou WL, Todi SV: An optimal ubiquitin-proteasome pathway in the nervous system: the role of deubiquitinating enzymes. Front Mol Neurosci 2014;7:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koulich E, Li X, DeMartino GN: Relative structural and functional roles of multiple deubiquitylating proteins associated with mammalian 26S proteasome. Mol Biol Cell 2008;19:1072–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.D’Arcy P, Brnjic S, Olofsson MH, Fryknas M, Lindsten K, De Cesare M, Perego P, Sadeghi B, Hassan M, Larsson R, Linder S: Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat Med 2011;17:1636–1640. [DOI] [PubMed] [Google Scholar]
- 53.Kovalenko PL, Basson MD: Schlafen 12 expression modulates prostate cancer cell differentiation. J Surg Res 2014;190:177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Puck A, Hopf S, Modak M, Majdic O, Cejka P, Bluml S, Schmetterer K, Arnold-Schrauf C, Gerwien JG, Frederiksen KS, Thell E, Leitner J, Steinberger P, Aigner R, Seyerl-Jiresch M, Zlabinger GJ, Stockl J: The soluble cytoplasmic tail of CD45 (ct-CD45) in human plasma contributes to keep T cells in a quiescent state. Eur J Immunol 2017;47:193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andersson E, Eldfors S, Edgren H, Ellonen P, Vakeva L, Ranki A, Mustjoki S: Novel TBL1XR1, EPHA7 and SLFN12 mutations in a Sezary syndrome patient discovered by whole exome sequencing. Exp Dermatol 2014;23:366–368. [DOI] [PubMed] [Google Scholar]
- 56.van Zuylen WJ, Garceau V, Idris A, Schroder K, Irvine KM, Lattin JE, Ovchinnikov DA, Perkins AC, Cook AD, Hamilton JA, Hertzog PJ, Stacey KJ, Kellie S, Hume DA, Sweet MJ: Macrophage activation and differentiation signals regulate schlafen-4 gene expression: evidence for Schlafen-4 as a modulator of myelopoiesis. PLoS One 2011;6:e15723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Waal L, Lewis TA, Rees MG, Tsherniak A, Wu X, Choi PS, Gechijian L, Hartigan C, Faloon PW, Hickey MJ, Tolliday N, Carr SA, Clemons PA, Munoz B, Wagner BK, Shamji AF, Koehler AN, Schenone M, Burgin AB, Schreiber SL, Greulich H, Meyerson M: Identification of cancer-cytotoxic modulators of PDE3A by predictive chemogenomics. Nat Chem Biol 2016;12:102–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nazir M, Senkowski W, Nyberg F, Blom K, Edqvist PH, Jarvius M, Andersson C, Gustafsson MG, Nygren P, Larsson R, Fryknas M: Targeting tumor cells based on Phosphodiesterase 3A expression. Exp Cell Res 2017;361:308–315. [DOI] [PubMed] [Google Scholar]
- 59.Vandenberghe P, Hague P, Hockman SC, Manganiello VC, Demetter P, Erneux C, Vanderwinden JM: Phosphodiesterase 3A: a new player in development of interstitial cells of Cajal and a prospective target in gastrointestinal stromal tumors (GIST). Oncotarget 2017;8:41026–41043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Corradini E, Klaasse G, Leurs U, Heck AJ, Martin NI, Scholten A: Charting the interactome of PDE3A in human cells using an IBMX based chemical proteomics approach. Mol Biosyst 2015;11:2786–2797. [DOI] [PubMed] [Google Scholar]
- 61.Chaturvedi LS, Basson MD: Glucagonlike peptide 2 analogue teduglutide: stimulation of proliferation but reduction of differentiation in human Caco-2 intestinal epithelial cells. JAMA Surg 2013;148:1037–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Turowski GA, Rashid Z, Hong F, Madri JA, Basson MD: Glutamine modulates phenotype and stimulates proliferation in human colon cancer cell lines. Cancer Res 1994;54:5974–5980. [PubMed] [Google Scholar]