

Sir,
Generalized bullous-fixed drug eruption (GBFDE), is a severe form of FDE characterized by widespread blisters and erosions, involving the whole body in addition to lesions typical of FDE. Although GBFDE is believed to have a favorable prognosis, the recent reports suggest it to have a nearly similar mortality rate like Stevens–Johnson syndrome/toxic epidermal necrolysis (SJS/TEN).[1] Cyclosporine A (CsA) that specifically targets the activity of T cells has been proposed to be beneficial in GBFDE.[2] We hereby report two cases of GBFDE who showed promising response to CsA.
Case 1
A 42-year-old male presented with multiple, discrete to confluent, and well-defined erythematous to dusky macules with superimposed bullae over the trunk and limbs for 3 days. He developed the lesions about 6 h after taking a combination pill of ibuprofen and paracetamol for joint pain. There were no associated constitutional symptoms, and general health of the patient was unaffected. Lips showed mild crusting. Genitalia had well-defined erosion over inner prepuce and glans penis. The patient also reported similar lesions of lesser severity at the same sites 2 years back, following intake of some over-the-counter analgesic for joint pain. Those lesions healed with hyperpigmentation. Based on history and clinical presentation, a diagnosis of GBFDE was made. Given the extensive involvement and risk of life-threatening consequences, rechallenge was not carried out. The patient was admitted and started on cyclosporine 5 mg/kg with excellent response and complete healing of lesions within 1 week [Figure 1a–c].
Figure 1.
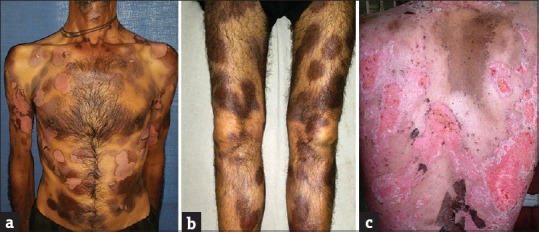
(a-c) Multipe dusky macules with superimposed ruptured bullae and healed lesions following treatment with cyclosporine
Case 2
A 24-year-old male presented with widespread dull erythema over the body with superimposed vesiculobullous lesions over trunk and limbs that developed 2 h after taking medication for common cold. He had taken multiple medicines including cetirizine, paracetamol, and pseudoephedrine. On interrogation, he admitted to have 4–5 similar episodes in the past, each of increasing severity than the previous one. On those occasions, he had taken multiple medications before the episodes for complaints of fever and cold. The patient was in good general health except for the pain and discomfort in the lesions. Lips showed erythema. The patient was treated with cyclosporine 5mg/kg with excellent response within 5-7 days. The lesions healed with hypopigmentation [Figure 2a and b]. Provocation was not carried out due to ethical reasons. The patient, however, reported after 6 months with severe burning and erythema in the healed lesions on self-administration of tablet paracetamol for fever, thereby confirming the cause of GBFDE.
Figure 2.
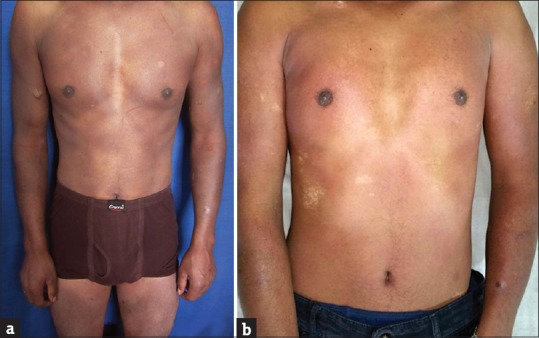
(a and b) Generalized erythema with superimposed vesiculobullous lesions and healing with hypopigmentation following treatment with cyclosporine
GBFDE, a rare variant of FDE, has been defined as widespread blisters and erosions with typical well-demarcated FDE lesions involving at least 10% of the body surface area distributed on at least 3 of 6 different anatomic sites including the head and neck, front of the trunk, back, upper extremities, lower extremities, and genitalia.[3] It has been associated with certain drugs, namely mefenamic acid, naproxen, cetirizine, ciprofloxacin, doxycycline, and nicotinic acid/laropiprant.[4]
Clinically, GBFDE may either present as generalized oval shaped, brownish livid macules with superimposed blisters or as diffuse erythema with flaccid blisters. In both forms, there are areas of intact skin between the blisters. Mucous membranes are rarely involved, and usually, there is no fever and only mild malaise. A history of a similar previous localized event regarding an FDE can often be elicited. Recurrences can lead to more widespread skin detachment, and therefore, to a more severe disease course.[5]
Histologically, GBFDE is characterized by necrotic keratinocytes that are either disseminated or present in the form of complete epidermal necrosis. The basement membrane zone shows vacuolization resulting in subepidermal blistering. The dermis usually exhibits a sparse superficial and often perivascular lymphohistiocytic infiltrate.
GBFDE can mimic SJS/TEN, staphylococcal scalded skin syndrome, and autoimmune bullous diseases such as pemphigus vulgaris and bullous pemphigoid, erythema multiforme major, and exfoliative dermatitis. In contrast to TEN, GBFDE consists of an absence or paucity of constitutional symptoms, well-demarcated blisters and erythematous patches, absence of small spots or target lesions, absence or paucity of mucosal erosions, absence or paucity of visceral complications, history of a similar eruption, and onset within hours of exposure to the associated drug. Most dermatopathologists also consider that the necrosis of the epidermis observed in GBFDE differs from that in TEN by more abundant infiltration by lymphocytes (including many regulatory T cells) and more pigment incontinence.[1] However, in the acute stage, histological differentiation may be difficult or even impossible.
The distinction between GBFDE and SJS or TEN is important because of the presumed favorable prognosis of GBFDE. However, a case–control analysis of 58 patients with GBFDE matched by age and extent of skin detachment to 170 control patients with a validated diagnosis of SJS or SJS / TEN overlap, found GBFDE to have a nearly identical mortality rate to SJS/TEN, thereby questioning the benign nature of GBFDE.[1] Therefore, the potential severity of GBFDE should not be underestimated, and it likely deserves the same symptomatic treatment and care as SJS or TEN. Severe cases of GBFDE should be regarded with the same level of urgency as cases of SJS/TEN, and the necessity of offering an effective treatment is of paramount importance.
Cyclosporine is a rapid and potent inhibitor of interleukin-2 (IL-2), and its suppressive action primarily is achieved by targeting T lymphocyte functions through IL-2, inhibition of macrophage activity, and inhibition of the activation of the Fas receptor–Fas ligand system. An additional mechanism of immunosuppression in cyclosporin A is the inhibition of CD8 T-cell activation, which in turn inhibits the apoptosis and destruction through either Fas ligand or perforin/granzyme pathway.[6]
The mechanism of FDE involves intraepidermal CD8+ T cells that are present within the lesions and play a role in local tissue destruction. Clinically, resolved FDE lesions continue to harbor a substantial number of effector memory CD8+ T-cells at the dermal-epidermal junction, which allows for recurrence of the lesions in the same location. Based on the pathogenesis of FDE, Malviya et al. proposed that cyclosporine had a superior therapeutic effect over prednisone or IVIG by specifically targeting the activity of T cells.[2] Considering the same, we also used cyclosporin in the above-mentioned cases with a favorable response in both our patients.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Lipowicz S, Sekula P, Ingen-Housz-Oro S, Liss Y, Sassolas B, Dunant A, et al. Prognosis of generalized bullous fixed drug eruption: Comparison with Stevens-Johnson syndrome and toxic epidermal necrolysis. Br J Dermatol. 2013;168:726–32. doi: 10.1111/bjd.12133. [DOI] [PubMed] [Google Scholar]
- 2.Malviya N, Cyrus N, Vandergriff T, Mauskar M. Generalized bullous fixed drug eruption treated with cyclosporine. Dermatol Online J. 2017;23 pii: 13030/qt5zw8d8 vs. [PubMed] [Google Scholar]
- 3.Cho YT, Lin JW, Chen YC, Chang CY, Hsiao CH, Chung WH, et al. Generalized bullous fixed drug eruption is distinct from Stevens-Johnson syndrome/toxic epidermal necrolysis by immunohistopathological features. J Am Acad Dermatol. 2014;70:539–48. doi: 10.1016/j.jaad.2013.11.015. [DOI] [PubMed] [Google Scholar]
- 4.Podder I, Chandra S, Das A, Gharami RC. Doxycycline induced generalized bullous fixed drug eruption. Indian J Dermatol. 2016;61:128. doi: 10.4103/0019-5154.174197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paulmann M, Mockenhaupt M. Severe drug-induced skin reactions: Clinical features, diagnosis, etiology, and therapy. J Dtsch Dermatol Ges. 2015;13:625–45. doi: 10.1111/ddg.12747. [DOI] [PubMed] [Google Scholar]
- 6.Reese D, Henning JS, Rockers K, Ladd D, Gilson R. Cyclosporine for SJS/TEN: A case series and review of the literature. Cutis. 2011;87:24–9. [PubMed] [Google Scholar]