

Abstract
Liver damage is typically inferred from serum measurements of cytoplasmic liver enzymes. DNA molecules released from dying hepatocytes are an alternative biomarker, unexplored so far, potentially allowing for quantitative assessment of liver cell death. Here we describe a method for detecting acute hepatocyte death, based on quantification of circulating, cell-free DNA (cfDNA) fragments carrying hepatocyte-specific methylation patterns. We identified 3 genomic loci that are unmethylated specifically in hepatocytes, and used bisulfite conversion, PCR, and massively parallel sequencing to quantify the concentration of hepatocyte-derived DNA in mixed samples. Healthy donors had, on average, 30 hepatocyte genomes/ml plasma, reflective of basal cell turnover in the liver. We identified elevations of hepatocyte cfDNA in patients shortly after liver transplantation, during acute rejection of an established liver transplant, and also in healthy individuals after partial hepatectomy. Furthermore, patients with sepsis had high levels of hepatocyte cfDNA, which correlated with levels of liver enzymes aspartate aminotransferase (AST) and alanine aminotransferase (ALT). Duchenne muscular dystrophy patients, in which elevated AST and ALT derive from damaged muscle rather than liver, did not have elevated hepatocyte cfDNA. We conclude that measurements of hepatocyte-derived cfDNA can provide specific and sensitive information on hepatocyte death, for monitoring human liver dynamics, disease, and toxicity.
Keywords: Hepatology, Transplantation
Keywords: Diagnostics, Epigenetics
A method for detecting human hepatocyte death using methylation patterns of cell-free DNA circulating in plasma.
Introduction
The clinical standard to assess liver damage is serum measurement of alanine aminotransferase (ALT) and aspartate aminotransferase (AST), reflecting hepatocyte injury leading to release of these enzymes into the blood. While in extensive clinical use, these tests do have important limitations. First, the enzymes are not absolutely hepatocyte specific. AST is expressed in cardiac and skeletal muscle, kidney, brain, pancreas, lung, leukocytes, and erythrocytes (1), while ALT is primarily present in the liver and kidneys, with low amounts in the heart and skeletal muscle (2). Second, liver enzymes do not always reflect the full burden of disease and in various hepatic pathologies have been shown to be insufficient (3–6). A possible reason for this is that the enzymes could be released from dying as well as reversibly injured cells, and therefore do not indicate the exact nature of damage to the liver, nor the number of injured cells. Third, ALT and AST have long half-lives (approximately 47 and 17 hours, respectively) (2), which limits the ability to monitor rapid changes in liver damage.
Short-lived fragments of DNA released from dying cells are emerging as a valuable biomarker. Cell-free DNA (cfDNA) analysis is used as a liquid biopsy to detect DNA derived from fetal cells, cancer cells, or transplanted organs, based on genetic differences between the host and the tissue of interest (7–11). We and others have recently used tissue-specific DNA methylation patterns to determine the tissue origins of cfDNA, allowing the identification of cell death in specific tissues even if they carry a genome that is identical to that of the host (12–14). Here we describe the application of this approach for the detection of cfDNA derived from hepatocytes. We characterized hepatocyte-specific DNA methylation patterns, and used these as specific biomarkers to detect hepatocyte-derived cfDNA in the plasma of healthy individuals and in people with an injured liver. Our results support the potential utility of hepatocyte-specific DNA methylation markers for minimally invasive, specific and sensitive detection and monitoring of hepatocellular death.
Results
Identification of hepatocyte-specific DNA methylation markers.
By comparing publicly available human tissue methylomes we selected 3 genomic loci, adjacent to the ITIH4, IGF2R, and VTN genes, which were unmethylated in the liver compared with other tissues and cell types (Figure 1A and Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.120687DS1). Moreover, 2 of the markers (IGF2R and ITIH4) were methylated in cholangiocytes, indicating cell-type specificity (Supplemental Figure 1). Experiments with genomic DNA extracted from a panel of human tissues indicated that the sequence of each of these loci includes 4–7 cytosine-guanine dinucleotides (CpG sites), which are fully unmethylated in hepatocytes and methylated in all other tissues (Figure 1B and Methods). We then spiked human liver DNA into human leukocyte DNA in different proportions and quantified unmethylated ITIH4, IGF2R, and VTN molecules. We found excellent correlation between the measured methylation signal and the input material, and were able to detect a liver signal even when liver DNA was diluted 1:1,000 in leukocyte DNA (Figure 1C). Thus, we have defined short sequences of DNA, whose methylation status constitutes an epigenetic signature unique to hepatocytes relative to blood cells and other tissues, allowing detection of hepatocyte-derived DNA in mixed samples.
Figure 1. Liver-specific markers.

(A) Structure of the 3 loci (adjacent to the ITIH4, IGF2R, and VTN genes) used as hepatocyte biomarkers. Lollipops represent CpG sites. Red indicates CpG sites represented in the Infinium HumanMethylation450K BeadChip. Arrows mark positions of PCR primers. Markers are defined by the methylation status of CpG sites between primers. (B) Methylation status of ITIH4, IGF2R, and VTN in DNA from multiple tissues. Shown is the percentage of molecules in which all CpG sites were unmethylated. (C) Spike-in experiments. Human liver DNA was mixed with human leukocyte DNA in the indicated proportions (0 to 20%), and the percentage of fully unmethylated hepatocyte markers was determined.
Hepatocyte-derived cfDNA in healthy individuals.
We measured the frequency of unmethylated ITIH4, IGF2R, and VTN molecules in plasma, reasoning that the presence of hepatocyte-derived genomic DNA fragments would reflect the rate of hepatocyte death in an individual. We prepared cfDNA from plasma samples, and performed bisulfite conversion, PCR amplification, and massively parallel sequencing, as previously described, to assess how many copies of each marker were present in plasma (12). Healthy individuals had on average 30 hepatocyte genomes/ml plasma, reflective of basal liver turnover (Figure 2, A and B and Supplemental Figure 2, A and B).
Figure 2. Liver-derived cfDNA in healthy individuals.

(A) Concentration in genome equivalents (Geq)/ml of hepatocyte-derived cfDNA in the plasma of healthy donors. Green, red, and blue indicate the estimation for VTN, ITIH4, and IGF2R markers, respectively. The concentration was measured by multiplying the fraction of hepatocyte cfDNA by the concentration of total cfDNA (Supplemental Figure 2). (B) Estimation of the concentration of hepatocyte-derived cfDNA in the plasma of healthy donors, averaging the values for all markers. Each data point represents one individual donor. Dashed line indicates average + 2 standard deviations. (C) Estimation of the concentration of hepatocyte-derived cfDNA in the plasma of healthy donors (n = 12) at 3 time points. T0, after a 12-hour fast; T30, half an hour after a meal; T120, two hours after a meal. (D–G) Lack of correlation between hepatocyte cfDNA in healthy donors and ALT levels (D), AST levels (E), BMI (F), and age (G) of the same donors.
To assess how dynamic the levels of hepatocyte-derived cfDNA are in healthy individuals, we measured hepatocyte-derived cfDNA as a function of fasting and food intake. Twelve healthy individuals were sampled after a 12-hour fast, and 30 minutes and 2 hours after a meal. Individual donors had distinct levels of hepatocyte cfDNA, and these levels did not differ between the time points (Figure 2C and Supplemental Figure 2, C and D), suggesting that the level of hepatocyte cfDNA is an individual, relatively stable trait. More studies will be required to substantiate and understand this interesting phenomenon. The baseline measurements of AST and ALT did not correlate with baseline hepatocyte cfDNA. Furthermore, we found no correlation between basal hepatocyte cfDNA and body mass index or age (Figure 2D).
Hepatocyte cfDNA following liver transplantation.
To validate the hepatocyte cfDNA assay in a setting where massive hepatocyte death is expected, we examined serum samples from 18 patients that underwent liver transplantation. We recorded low levels of hepatocyte cfDNA before transplantation and a very strong signal shortly after reperfusion of the transplant, which declined dramatically in the days that followed (Figure 3A and Supplemental Figure 3). Hepatocyte cfDNA correlated with the liver enzymes ALT and AST, providing another level of validation to the cfDNA assay (Figure 3B).
Figure 3. Hepatocyte cfDNA in liver transplant recipients.

(A) Hepatocyte-derived DNA in the serum of 18 liver transplant recipients. Each patient was sampled at 4 time points as indicated. Graph shows the average values of the 3 liver markers in each sample. Dashed line indicates average + 2 standard deviations of healthy controls. (B) Pearson’s correlation between hepatocyte cfDNA levels and circulating liver enzymes in liver transplant recipients. (C) Hepatocyte-derived DNA in the serum of 6 liver transplantation patients who had an episode of graft rejection. Each patient was examined at the 6 time points indicated. Graph shows the average values of the fraction of the 3 liver markers in each sample. Each line represents the values for 1 patient.
We further examined serum samples from 6 liver transplant recipients that were sampled periodically after transplantation, including episodes of acute rejection. All 6 sets of samples showed a similar pattern of hepatocyte cfDNA: low levels before the procedure, and high levels following transplantation, which declined to baseline by day 45 after transplantation. In 5 of 6 patients the hepatocyte cfDNA signal was elevated during biopsy-proven rejection; strikingly, in 3 patients we recorded elevated hepatocyte cfDNA prior to clinically detectable rejection (Figure 3C).
Hepatocyte-derived cfDNA following liver donation.
Next, we examined the levels of hepatocyte cfDNA after partial hepatectomy. We examined 14 sets of serum samples from healthy individuals who donated part of their liver. Patients were sampled prior to partial hepatectomy, and 12, 30, and 95 days after, where liver regeneration should have been complete. We observed abnormally high hepatocyte cfDNA 12 days after surgery, which later declined (Figure 4 and Supplemental Figure 4). Strikingly, by day 12 after surgery the levels of ALT and AST were already back within the normal range, but still above the baseline of each individual. These findings suggest that hepatocyte cfDNA measurement might be a more sensitive indicator of liver damage than enzyme measurements, at least under some circumstances.
Figure 4. Identification of hepatocyte-derived cfDNA after partial hepatectomy.
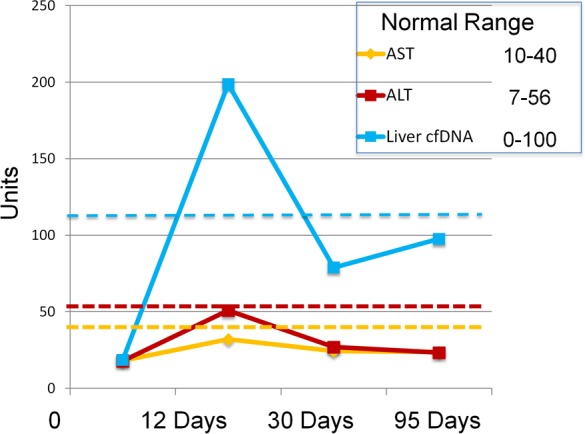
Hepatocyte-derived DNA in the serum of 14 liver donors after partial hepatectomy. Each patient was sampled at 4 time points as indicated. Graph shows the average values of the 3 liver markers in each sample (genome equivalents/ml) as well as AST and ALT values. Dashed line indicates average + 2 standard deviations of healthy controls.
Hepatocyte-derived cfDNA in sepsis patients.
Sepsis often involves massive liver damage, in addition to cell death in multiple additional tissues. To examine how liver damage in this complex setting is reflected in hepatocyte-derived cfDNA, we analyzed 56 plasma samples obtained during sepsis. The amount of hepatocyte-derived cfDNA was significantly higher in sepsis patients compared with healthy controls (Figure 5 and Supplemental Figure 5). Hepatocyte cfDNA values were in good correlation with the levels of liver enzymes ALT and AST measured in the same patients (Figure 5), supporting specificity of hepatocyte cfDNA measurements even in the background of extensive cell death in multiple tissues.
Figure 5. identification of liver-derived cfDNA in sepsis.

(A) Hepatocyte cfDNA in the plasma of patients with sepsis. P value determined by 2-tailed Mann-Whitney test. (B) Pearson’s correlation between hepatocyte cfDNA levels and circulating AST and ALT in septic patients.
Hepatocyte-derived cfDNA in Duchenne muscular dystrophy patients.
Patients with Duchenne muscular dystrophy (DMD) and other muscle dystrophies often present with high levels of ALT and AST, which are derived from damaged muscle rather than from the liver (15–17). We examined whether our hepatocyte-specific methylation markers are elevated in this setting. We tested plasma samples of 10 DMD patients who had elevated levels of ALT and AST. In all samples the concentration of hepatocyte-derived cfDNA was in the normal range (Figure 6), consistent with a non-liver origin of AST and ALT and further supporting specificity of the hepatocyte cfDNA assay.
Figure 6. Identification of hepatocyte-derived cfDNA in Duchenne muscular dystrophy (DMD).

Hepatocyte-derived cfDNA and transaminases in the plasma of 10 DMD patients. Dashed line indicates cutoff for healthy individuals.
A digital droplet PCR assay for measurement of hepatocyte cfDNA.
The experiments described above were performed using massively parallel sequencing, requiring access to a next generation sequencer and the application of a bioinformatics pipeline to interpret sequencing results. To simplify the method, we developed a PCR-based version of the assay that alleviates the need for sequencing. We designed primers for digital droplet PCR (ddPCR) after bisulfite conversion of cfDNA, and probes that recognize blocks of unmethylated CpGs in the amplified marker regions. We focused on the VTN and IGF2R markers, which had multiple CpGs in close proximity.
ddPCR using both amplicons showed no signal in leukocyte DNA and a strong signal in hepatocyte DNA (Figure 7A). We then examined 6 sets of plasma samples from 6 patients before and after liver transplantation (the same samples used in Figure 2). Similar to the sequencing results, the ddPCR assay revealed a strong and transient elevation of hepatocyte cfDNA in plasma shortly after transplantation, which declined thereafter, strongly suggesting validity of the assay (Figure 7B).
Figure 7. Digital droplet PCR for the identification of liver-derived cfDNA.

(A) Hepatocyte and leukocyte DNA examined using ddPCR. (B) Hepatocyte-derived DNA in the plasma of 6 liver transplant recipients. Each patient was sampled at 4 time points as indicated. Graph shows the average values of the 2 liver markers in each sample.
Discussion
We described here a highly specific, sensitive, and quantitative assay for the detection of human hepatocyte cfDNA, reflecting hepatocyte death in healthy individuals as well as patients with liver disease. We characterized 3 genomic loci that are unmethylated specifically in hepatocytes, compared with a large panel of human tissues and cell types, and used these as biomarkers. We note that the selection of these markers was rather arbitrary. All 3 loci were unmethylated in hepatocytes, highly methylated in other tissues and cell types, and each had multiple adjacent CpG sites that behaved similarly within the same molecule. The latter feature (also termed methylation haplotype block; refs. 12, 18) increases the signal-to-noise ratio between hepatocyte and non-hepatocyte DNA. Importantly, our computational analysis indicates that the genome contains hundreds of additional sequences harboring clusters of adjacent CpG sites with hepatocyte-specific methylation patterns. Such loci could also be used as hepatocyte biomarkers. Multiplexing several such markers can in principle increase the sensitivity of the assay without compromising specificity, potentially far exceeding current methods.
Our experiments with serum and plasma samples were designed to provide proof of concept that hepatocyte cfDNA can be detected, and is reflective of liver damage based on clinical parameters or circulating liver enzymes. Indeed, the assay was able to detect the expected elevation in hepatocyte cfDNA shortly after liver transplantation, during acute rejection of a liver transplant, after partial hepatectomy, and in patients with sepsis. We note 2 insights into liver dynamics that can already be gained from the data. First, the detection of hepatocyte cfDNA in plasma suggests that at least some of the DNA from dying hepatocytes is cleared through blood, and not through other routes such as bile secretion or complete digestion by Kupffer cells. Second, different healthy individuals had typical levels of hepatocyte cfDNA, which did not change following overnight fast or after a meal, raising the possibility that baseline hepatocyte turnover is a personal, stable trait.
Measurements of hepatocyte cfDNA have several distinct features compared with standard enzyme assays, which may find utility in research and eventually in clinical settings. First, hepatocyte specificity of the cfDNA assay may prove useful in ambiguous conditions, where damage may have happened in tissues that express classic liver markers; one example for such a situation is DMD. Second, elevation of serum AST/ALT indicates liver damage that could result from either cell death or a transient leak. The cfDNA signal, involving cleavage and release of genomic DNA, is more likely to reflect actual hepatocyte death. Third, it is hard to infer the number of damaged cells from levels of circulating liver enzymes; in contrast, the cfDNA assay, counting individual DNA molecules in plasma, can in principle report on the exact number of dying hepatocytes. Lastly, the short half-life of cfDNA (estimated at 15–60 minutes) (19) compared with the slow clearance of liver enzymes provides a near real-time indication of liver damage, which can be important in dynamic settings.
The hepatocyte cfDNA assay may find utility in many areas of liver biology and disease. For example, determination of hepatocyte death in non-alcoholic steatohepatitis (NASH) at present requires a liver biopsy, which can theoretically be replaced by cfDNA (3, 4, 6). Another important case is autoimmune hepatitis, in which measurements of liver enzymes often fail to disclose ongoing liver damage (5). In these situations, liver disease may silently progress to cirrhosis without marked elevations in either AST or ALT. Elevations of hepatocyte cfDNA may also prove informative for early detection of liver transplant rejection, for assessing hepatotoxicity of drugs, for diagnosis and monitoring of hepatocellular carcinoma, and for studies of human liver dynamics in physiological settings. Testing these possibilities will require large, blinded clinical trials, made possible with the biomarkers and methods described here. We also note that in pathologies involving damage to multiple cell types, combining cfDNA measurements of hepatocytes and other cells, for example specific immune cell types or cholangiocytes, may increase the diagnostic power of the cfDNA approach.
Methods
Study approval.
All clinical studies were approved by the ethics committees of the University of Pennsylvania and Hadassah Medical Center.
Biomarkers.
Tissue-specific methylation biomarkers were selected after a comparison of publicly available genome-wide DNA methylation data sets generated using the Illumina Infinium HumanMethylation450K BeadChip array. The comparison included in addition the methylome of human cholangiocytes, generated locally by sorting dissociated liver tissue. The analysis of the cholangiocyte methylome (and cholangiocyte cfDNA biomarkers) will be reported elsewhere.
Sample preparation and DNA processing.
Blood samples were collected in EDTA-containing plasma-preparation tubes and centrifuged for 10 minutes at 1,500 g and 4°C. The supernatant was transferred to a fresh 15-ml conical tube without disturbing the cellular layer and centrifuged again for 10 minutes at 3,000 g and 4°C. The supernatant was collected and stored at –80°C.
Cell-free DNA was extracted from 1–4 ml of plasma using the QIAsymphony liquid-handling robot (Qiagen) and treated with bisulfite (Zymo Research). DNA concentration was measured using Qbit double-strand molecular probes (Invitrogen). Bisulfite-treated DNA was PCR amplified using primers specific for bisulfite-treated DNA but independent of methylation status at monitored CpG sites.
Primers were barcoded, allowing the mixing of samples from different individuals when sequencing products. Sequencing was performed on PCR products using MiSeq Reagent Kit v2 (MiSeq, Illumina method) or NextSeq 500/550 v2 sequencing reagent kits. Sequenced reads were separated by barcode, aligned to the target sequence, and analyzed using custom scripts written and implemented in MATLAB (MathWorks). Reads were quality filtered based on Illumina quality scores. Reads were identified by having at least 80% similarity to target sequences and containing all the expected CpGs in the sequence. CpGs were considered methylated if “CG” was read and were considered unmethylated if “TG” was read. The efficiency of bisulfite conversion was assessed by analyzing the methylation of non-CpG cytosines.
ddPCR.
We established a procedure for ddPCR, in which bisulfite-treated cfDNA is interrogated using methylation-sensitive TaqMan probes. The limited length of probes (up to 30 bp) dictated that they could cover only 2 to 4 informative CpG sites. In the IGF2R locus 4 CpGs were covered. However, in the VTN = locus, only 2 CpGs were covered by a probe predicting a relatively high frequency of noise (positive droplets) in DNA from non-liver tissue. In the sequencing-based assay we addressed this problem by documenting the methylation status of 5 adjacent cytosines (Figure 1A), which greatly increased specificity. To implement this concept in the ddPCR platform we designed 2 TaqMan probes, each recognizing lack of methylation in a different cluster of cytosines (each containing 2 CpG sites) within the same amplified 100-bp fragment from the VTN locus. Each probe was labeled with a different fluorophore, such that we could identify droplets in which both probes found a target. Such droplets would be interpreted as containing a VTN cfDNA fragment in which all 4 targeted cytosines were unmethylated. This resulted in a ddPCR assay with the improved specificity afforded by interrogating multiple cytosines on the same DNA molecule.
Each 20-μl volume reaction mix consisted of ddPCR Supermix for Probes (No dUTP) (Bio-Rad), 900 nM primer, 250 nM probe, and 2 μl of sample. The mixture and droplet generation oil were loaded onto a droplet generator (Bio-Rad). Droplets were transferred to a 96-well PCR plate and sealed. The PCR was run on a thermal cycler as follows: 10 minutes of activation at 95°C, 47 cycles of a 2-step amplification protocol (30 seconds at 94°C denaturation and 60 seconds at 56°C), and a 10-minute inactivation step at 98°C. The PCR plate was transferred to a QX100 Droplet Reader (Bio-Rad), and products were analyzed with QuantaSoft (Bio-Rad) software. Discrimination between droplets that contained the target (positives) and those that did not (negatives) was achieved by applying a fluorescence amplitude threshold based on the amplitude of reads from the negative template control.
Statistics.
To assess the significance of differences between groups, we used a 2-tailed Mann–Whitney test. P less than 0.05 was considered significant.
Author contributions
BG, RS, YD, and RLW designed the research. RLW, DN, HZ, J. Moss, J. Magenheim, IF, OA, and SP performed the research. TD, MG, GL, BLL, AS, and KO contributed reagents. RLW, AS, KO, GL, MG, BG, RS, and YD wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Ibrahim Knani and Shoshi Shpitzen for excellent technical help, Lilach Gavish for coordination of sample collection, Gideon Zamir, Eithan Galun, Howard Cedar, Muli Ben-Sasson, and Aharon Razin for stimulating discussions, and the donors who participated in the study. This work was supported by grants from the Human Islet Research Network of the NIH (HIRN; RRID:SCR_014393 and UC4 DK104216-01 to Y. Dor) and the Kahn Foundation. This work was supported in part by a grant from the American Schools and Hospitals Abroad Program of the United States Agency for International Development for the upgrading of the Hebrew University Medical School Sequencing Facility.
Version 1. 06/21/2018
Electronic publication
Funding Statement
This research was performed using funding provided by the NIDDK-supported Human Islet Research Network (HIRN, RRID:SCR_014393; https://hirnetwork.org; UC4 DK104216-01).
Footnotes
Conflict of interest: RLW, HZ, J. Magenheim, J. Moss, DN, SP, RS, BG, and YD are inventors on a patent entitled “New set of probes for determining tissue of origin by methylation pattern” (patent number 62/531,988). J. Magenheim, RS, BG, and YD are inventors on a patent entitled “A dual-probe digital droplet PCR strategy for specific detection of tissue-specific circulating DNA molecules” (patent number 62/531,983).
Reference information: JCI Insight. 2018;3(12): e120687. https://doi.org/10.1172/jci.insight.120687.
Contributor Information
Roni Lehmann-Werman, Email: roni.werman@mail.huji.ac.il.
Judith Magenheim, Email: judithm@ekmd.huji.ac.il.
Joshua Moss, Email: joshua.moss@mail.huji.ac.il.
Daniel Neiman, Email: daniel.neiman@mail.huji.ac.il.
Ofri Abraham, Email: ofriky@gmail.com.
Sheina Piyanzin, Email: sheinadreamer@gmail.com.
Hai Zemmour, Email: hai.zemmour@mail.huji.ac.il.
Markus Grompe, Email: grompem@ohsu.edu.
Giora Landesberg, Email: giora.lan@mail.huji.ac.il.
Bao-Li Loza, Email: polly0806@gmail.com.
Abraham Shaked, Email: abraham.shaked@uphs.upenn.edu.
Kim Olthoff, Email: kim.olthoff@uphs.upenn.edu.
Benjamin Glaser, Email: ben.glaser@mail.huji.ac.il.
Ruth Shemer, Email: shemer.ru@mail.huji.ac.il.
Yuval Dor, Email: yuvald@ekmd.huji.ac.il.
References
- 1.Pratt DS, Kaplan MM. Evaluation of abnormal liver-enzyme results in asymptomatic patients. N Engl J Med. 2000;342(17):1266–1271. doi: 10.1056/NEJM200004273421707. [DOI] [PubMed] [Google Scholar]
- 2.Kim WR, Flamm SL, Di Bisceglie AM, Bodenheimer HC, Public Policy Committee of the American Association for the Study of Liver Disease Serum activity of alanine aminotransferase (ALT) as an indicator of health and disease. Hepatology. 2008;47(4):1363–1370. doi: 10.1002/hep.22109. [DOI] [PubMed] [Google Scholar]
- 3.Adams LA, Sanderson S, Lindor KD, Angulo P. The histological course of nonalcoholic fatty liver disease: a longitudinal study of 103 patients with sequential liver biopsies. J Hepatol. 2005;42(1):132–138. doi: 10.1016/j.jhep.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 4.Mofrad P, et al. Clinical and histologic spectrum of nonalcoholic fatty liver disease associated with normal ALT values. Hepatology. 2003;37(6):1286–1292. doi: 10.1053/jhep.2003.50229. [DOI] [PubMed] [Google Scholar]
- 5.Alvarez F, et al. International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol. 1999;31(5):929–938. doi: 10.1016/S0168-8278(99)80297-9. [DOI] [PubMed] [Google Scholar]
- 6.Machado MV, Cortez-Pinto H. Non-alcoholic fatty liver disease: what the clinician needs to know. World J Gastroenterol. 2014;20(36):12956–12980. doi: 10.3748/wjg.v20.i36.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wan JCM, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17(4):223–238. doi: 10.1038/nrc.2017.7. [DOI] [PubMed] [Google Scholar]
- 8.De Vlaminck I, et al. Circulating cell-free DNA enables noninvasive diagnosis of heart transplant rejection. Sci Transl Med. 2014;6(241):241ra77. doi: 10.1126/scitranslmed.3007803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bianchi DW, et al. DNA sequencing versus standard prenatal aneuploidy screening. N Engl J Med. 2014;370(9):799–808. doi: 10.1056/NEJMoa1311037. [DOI] [PubMed] [Google Scholar]
- 10.Fan HC, Gu W, Wang J, Blumenfeld YJ, El-Sayed YY, Quake SR. Non-invasive prenatal measurement of the fetal genome. Nature. 2012;487(7407):320–324. doi: 10.1038/nature11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lo YM, et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med. 2010;2(61):61ra91. doi: 10.1126/scitranslmed.3001720. [DOI] [PubMed] [Google Scholar]
- 12.Lehmann-Werman R, et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc Natl Acad Sci USA. 2016;113(13):E1826–E1834. doi: 10.1073/pnas.1519286113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun K, et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc Natl Acad Sci USA. 2015;112(40):E5503–E5512. doi: 10.1073/pnas.1508736112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akirav EM, et al. Detection of β cell death in diabetes using differentially methylated circulating DNA. Proc Natl Acad Sci USA. 2011;108(47):19018–19023. doi: 10.1073/pnas.1111008108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korones DN, Brown MR, Palis J. “Liver function tests” are not always tests of liver function. Am J Hematol. 2001;66(1):46–48. doi: 10.1002/1096-8652(200101)66:1<46::AID-AJH1007>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 16.Balaji P, Sankaranarayanan S, Venkataraman V, Sankaranarayanan VS. Incidentally detected elevated liver enzymes: from liver to muscle. Indian Pediatr. 2017;54(4):331–332. [PubMed] [Google Scholar]
- 17.Tay SK, Ong HT, Low PS. Transaminitis in Duchenne’s muscular dystrophy. Ann Acad Med Singap. 2000;29(6):719–722. [PubMed] [Google Scholar]
- 18.Guo S, Diep D, Plongthongkum N, Fung HL, Zhang K, Zhang K. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat Genet. 2017;49(4):635–642. doi: 10.1038/ng.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lo YM, Zhang J, Leung TN, Lau TK, Chang AM, Hjelm NM. Rapid clearance of fetal DNA from maternal plasma. Am J Hum Genet. 1999;64(1):218–224. doi: 10.1086/302205. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.