

Abstract
Purpose
This study aimed to characterize the pharmacokinetic (PK) properties, safety, and tolerability of asenapine, and to develop a population PK model in pediatric patients with schizophrenia, bipolar disorder, or other psychiatric disorders.
Methods
Two Phase I multiple ascending-dose studies were conducted to evaluate the PK, safety, and tolerability of sublingual asenapine in pediatric patients (age 10–17 years) with schizophrenia or bipolar I disorder. Patients received asenapine 1–10 mg twice daily for up to 12 days. PK parameters (maximum concentration [Cmax], area under the curve from 0 to 12 hours [AUC0–12], time to Cmax [Tmax], and half-life) were summarized for asenapine with descriptive statistics, and safety parameters were collected. A population PK model, which included the two Phase I studies and two additional Phase III efficacy studies (asenapine 2.5–10 mg twice daily for up to 8 weeks, age 10–17 years), was developed using nonlinear mixed-effect modeling based on a previously developed adult PK model. The final model was used in simulations to obtain asenapine-exposure estimates across pediatric subgroups and to determine if intrinsic covariates warrant dose adjustments.
Results
The PK of asenapine showed rapid absorption (Tmax ~1 hour) with an apparent terminal half-life between 16 and 32 hours. Increases in mean Cmax and AUC0–12 appeared to be dose-proportional in one study and near dose-proportional in the second study. Steady state was attained within 8 days. The most frequently occurring treatment-emergent adverse events were dysgeusia, sedation, and oral hypoesthesia. Simulation-based estimates of Cmax and AUC0–12 were similar for pediatric and adult patients; age, body-mass index, race, and sex were not associated with changes in asenapine exposure.
Conclusion
Asenapine was generally safe and well tolerated in pediatric patients aged 10–17 years. PK and safety data were similar to that observed in the adult population. Intrinsic factors had no significant impact on asenapine exposure, indicating there is no need for dose adjustments in the pediatric population.
Keywords: asenapine, pharmacokinetics, schizophrenia, bipolar disorder, child and adolescent, atypical antipsychotic
Plain-language summary
The time course of asenapine blood concentrations after a dose range of 1–10 mg twice daily in patients aged 10–17 years is overall similar to that observed in adults. Age, body-mass index, race, and sex have no significant impact on asenapine concentrations, indicating that it is not necessary to adjust asenapine dose for these factors in pediatric patients. Children aged 10–11 years appear to be more sensitive to side effects during initial asenapine treatment, and this can be prevented by starting with a low dose and increasing the dose over a few days. This simplifies treatment of patients with early-onset schizophrenia or bipolar I disorder.
Introduction
Atypical antipsychotics differ across a wide range of pharmacokinetic (PK) and pharmacodynamic properties. As a class, atypical antipsychotics are nonselective for the dopamine type 2 receptor, but they also display a wide range of variation in their antagonism against dopamine, serotonin (5-hydroxytryptamine [5HT]), norepinephrine, and histamine receptors.1–3 Asenapine is an atypical antipsychotic characterized by higher binding affinity for the 5HT2A receptor compared to the D2 receptor. Additionally, asenapine has a broad range of effects on other neurotransmitter systems, including antagonism against dopamine, 5HT, histamine, and α-adrenergic receptors.4 Antagonism of 5HT7 and 5HT2C receptors is of particular clinical interest, based on preclinical findings of possible beneficial effects on cognition and mood.2 Additionally, low affinity for muscarinic receptors may result in fewer anticholinergic side effects.3
In adults, asenapine is indicated in the US for the treatment of schizophrenia and acute treatment of manic or mixed episodes associated with bipolar I disorder (monotherapy or adjunct to lithium or valproate), as well as maintenance monotherapy for bipolar I disorder.5 Efficacy and tolerability have been demonstrated in adults in multiple clinical trials.6–13 Asenapine is also approved in the US as monotherapy for bipolar I disorder in pediatric patients aged 10–17 years, based on a Phase III acute study and long-term extension in bipolar I disorder in which asenapine demonstrated significant superiority to placebo in improving Young Mania Rating Scale (YMRS) total score.14,15 In pediatric schizophrenia patients, improvement in Positive and Negative Syndrome Scale (PANSS) total score was numerically greater for asenapine than for placebo, although differences were not statistically significant.16
The PK profile of asenapine in adults is well characterized; however, data have been limited in the pediatric population. In adults, asenapine is rapidly absorbed after sublingual administration and reaches peak plasma levels (Tmax) within 30–90 minutes following a single 5 mg dose.2 Its absolute bioavailability is approximately 35%, which is reduced if drinking or eating occurs within 10 minutes after administration.2,3 Steady-state concentration is reached within 3 days of twice daily (BID [bis in die]) dosing. Increasing the dose from 5 mg to 10 mg BID results in less than linear (1.7 times) increases in both the extent of exposure and maximum concentration.17,18 Asenapine is metabolized in the liver primarily through glucuronidation by UGT1A4 and through oxidative metabolism by CYP1A2 and CYP3A4.2,3 Although there are numerous metabolites, none is considered clinically relevant, as they have low affinity for the relevant receptors or are unable to cross the blood–brain barrier.2 The estimated terminal half-life (t1/2) in adults is approximately 24 hours.2,3,5,17
The goal of this publication is to characterize the PK profile, safety, and tolerability of multiple asenapine doses from two Phase I studies of pediatric patients aged 10–17 years. A population PK-modeling analysis, including additional PK data from two Phase III studies in pediatric patients, was also performed to determine whether differences in age or weight warrant asenapine dose adjustments.
Methods
Overview
Two Phase I multiple ascending-dose studies evaluated the PK, safety, and tolerability of sublingual asenapine (up to 10 mg BID) in pediatric patients with schizophrenia, bipolar I disorder, or other conditions (eg, autism, conduct disorder, oppositional defiant disorder) in which the chronic use of antipsychotic medication may be warranted. All patients received rapidly dissolving sublingual asenapine tablets. A population PK-modeling analysis was performed using data from these two Phase I studies and two Phase III fixed-dose studies.
Ethical conduct
Study 1 and study 2 were each conducted at two study centers in the US. For both Phase I studies, independent review boards at each study site (study 1, Aspire, San Diego, CA; study 2, Copernicus Group, Research Triangle Park, NC) reviewed and approved protocols (with amendment for study 2) and informed consent forms, which included all required elements in accordance with International Conference on Harmonisation E6 guidelines and local laws. Studies were conducted in accordance with good clinical practice standards and applicable country and/or local statutes and regulations. Protocol requirements were provided and discussed with each potential subject and their legal representative, and written informed consent was obtained from all subjects and their authorized legal representative(s) prior to any study-related procedures.
Study 1: design, participants, and dosing
Design
Study 1 was a placebo-controlled, double-blind, randomized, parallel-group, multiple-dose study of asenapine (up to 10 mg BID) in pediatric patients 12–17 years of age (Figure 1A). Briefly, patients were enrolled into four treatment cohorts (n=10 each) and then randomized to receive either asenapine (n=8) or matching placebo (n=2), with at least four subjects in each cohort between 12 and 15 years of age. The trial consisted of a ~3-week screening period, background medication tapering/discontinuation period up to 3 days, placebo run-in on day 0 for familiarization with dosing procedures, asenapine/placebo treatment period, posttreatment restabilization period, and a follow-up visit.
Figure 1.

Design and dosing schedule for (A) study 1 and (B) study 2.
Notes: In study 1, screening was performed within 3 weeks of baseline. Dosing was BID, except for the last day of dosing (only morning dose administered). In study 2, screening was performed within 28 days prior to baseline. Sublingual asenapine was given BID. Cohort 3a was originally scheduled similarly to cohorts 3b–3d (5 mg BID on day 1 followed by 10 mg BID thereafter); however, after safety results of cohort two became available, a protocol amendment modified the original treatment regimen (slower titration to 10 mg asenapine) to improve tolerability. a≥4 subjects per group must have been between 12 and 15 years of age.
Abbreviation: BID, bis in die (twice daily).
Participants
All patients had a current Diagnostic and Statistical Manual of Mental Disorders (DSM)-IV-TR diagnosis of schizophrenia, bipolar disorder, autism, conduct disorder, oppositional defiant disorder, or any condition for which the chronic use of antipsychotic medication was clinically warranted and/or administered. Patients were in good physical health without conditions that could confound the safety and tolerability results and had body weight ≥37 kg and body-mass index (BMI) between 18 and 30 kg/m2 (inclusive). Patients may have continued to receive concomitant treatment for extrapyramidal symptoms (EPSs; propranolol and anticholinergics) or for agitation or insomnia (benzodiazepine or partial benzodiazepine agonist, respectively). Concomitant use of medications that induce or inhibit CYP1A2 and/or CYP3A4 (primary metabolizing enzymes) was avoided, such as fluvoxamine, omeprazole, rifampin, ketoconazole, and erythromycin.
Study 2: design, participants, and dosing
Design
Study 2 (NCT01206517) was an open-label, multiple- ascending-dose study of asenapine (up to 10 mg BID) in pediatric patients 10–17 years of age (Figure 1B). The trial consisted of a ~3-week screening period, background antipsychotic-medication tapering/discontinuation period up to 5 days, placebo run-in on day 0, asenapine-treatment period, posttreatment restabilization period, and a follow-up visit. Patients were hospitalized for the entire duration of the study.
Participants
All patients had a current DSM-IV-TR diagnosis of schizophrenia of paranoid, disorganized, catatonic, or undifferentiated subtype; or bipolar I disorder, with a current manic or mixed episode determined by a structured clinical interview at screening. In addition to a documented history of schizophrenia or bipolar disorder, patients in cohort 3a could also have had a diagnosis of autism, conduct disorder, oppositional defiant disorder, or any condition for which the chronic use of antipsychotic medication was warranted and/or administered. Patients discontinued all psychotropic medication during the treatment period, except for those specified in the protocol. They also discontinued the use of β-blockers and strong inhibitors or inducers of CYP1A2 and/or CYP2D6 (primary metabolizing enzymes). Concomitant medications to treat EPSs (anticholinergics, short-acting benzodiazepines) were allowed. Potential patients were excluded if they had an uncontrolled clinically significant general medical condition, had history of psychiatric or personality disorders that may impact participation, or had a primary diagnosis other than schizophrenia or bipolar I disorder (with the exception of cohort 3a). Patients were not excluded based on body weight or BMI for this study. Smoking was not allowed during the study.
Pharmacokinetic assessments and analyses (studies 1 and 2)
Blood samples for PK evaluation of asenapine and its primary metabolite, N-desmethylasenapine, were collected on the final day of dosing for both studies (days 10 or 11 for study 1, days 7, 8, or 12 for study 2). Samples were drawn at predose and multiple time points between 15 minutes and 72 hours or between 30 minutes and 48 hours postdose for study 1 and study 2, respectively. Additional predose samples were collected for both studies on varying days. Samples were assayed for asenapine and N-desmethylasenapine concentrations using a validated liquid chromatography tandem mass-spectrometry method. The lower limit of quantification was 0.025 ng/mL for asenapine and 0.050 for N-desmethylasenapine. Samples below the lower limit were included as zero. The upper limit of quantification was 20 ng/mL for both asenapine and N-desmethylasenapine.
PK parameters for asenapine and N-desmethylasenapine were calculated using noncompartmental analysis of plasma concentration–time data using WinNonlin (version 4.0.1 for study 1 and version 5.2.1 for study 2). Maximum plasma concentration (Cmax), Tmax, area under the plasma concentration–time curve from 0 to 12 hours (AUC0–12), and terminal t½ were determined from each individual plasma concentration–time curve. Additional PK parameters evaluated for asenapine included apparent total body clearance (Cl/F) and apparent volume of distribution during the terminal phase (Vz/F).
Safety and efficacy assessments (studies 1 and 2)
Safety in both studies was evaluated by adverse event (AE) reporting, physical examination, vital signs (blood pressure, heart rate, and body temperature), electrocardiography (ECG), and laboratory values (hematology, blood chemistry, and urinalysis). Patients in study 2 were also administered psychological assessment tools at baseline and on the last day of treatment and on day five for cohorts 1, 2, and 3b–3d and day eight for cohort 3a. Assessments included the Columbia Suicide Severity Rating Scale, Extrapyramidal Symptom Rating Scale, Children’s Depression Rating Scale – revised, Clinical Global Impression, PANSS (only to patients with schizophrenia), and YMRS (only to patients with bipolar disorder).
Determination of sample size (studies 1 and 2)
For study 1, the number of subjects enrolled represented a sample size sufficient to meet the objectives of the study, based on recommendations of internal clinical pharmacology guidelines for multiple-dose safety and tolerance studies. For study 2, the power calculation assumed the variability of asenapine PK in pediatric patients was similar to that in adults. Between-group subject variability (percentage coefficient of variation [CV]) in Cmax and AUC across a range of PK studies was found to be on average 45% and 37%, respectively. Assuming this %CV, a group size of six for the age-group 10–11 years will result in a relative standard error of geometric mean estimates of 18% for Cmax and 15% for AUC. The larger group for cohort three was needed to characterize the PK at 10 mg BID across the full age range of 10–17 years. A group size of six was planned for patients 10–11 years old and a group size of four was planned for the other three age-groups (12–13, 14–15, and 16–17 years). Assuming %CV and in the absence of an age effect on asenapine PK, a total group size of 18 will result in a relative standard error of the geometric mean estimates of 11% for Cmax and 9% for AUC.
Statistical analyses (studies 1 and 2)
The safety population was defined as all subjects that received at least one dose of the study drug. The PK population was defined as all subjects who had at least one PK parameter of interest. PK parameters were summarized with descriptive statistics by dose and/or age-group. In study 2, attainment of a steady state was assessed using a repeated-measures ANOVA model on log-transformed trough concentrations with day and day-by-cohort interaction as fixed effects. Steady-state attainment was determined with Helmert contrasts.
Population PK-modeling analysis
A population PK model was constructed to determine whether differences in age or weight would warrant dose adjustment. The model was fitted utilizing data from the two Phase I PK studies (A7501022 and P06522) and two fixed-dose Phase III studies evaluating the efficacy and safety of asenapine in pediatric patients. One was a 3-week study in patients aged 10–17 years with manic or mixed episodes associated with bipolar I disorder receiving asenapine 2.5, 5, or 10 mg BID (P06107; NCT01244815),14 and the other was an 8-week study in patients aged 12–17 years with schizophrenia receiving asenapine 2.5 or 5 mg BID (P05896; NCT01190254).16 The model development, refinement, and evaluation process is shown in Figure S1. NONMEM version 7 with PsN version 3.2.4 was used in the population PK analysis and R version 3.0.2 was used for the exploratory analysis.19
Population PK analyses were performed using nonlinear mixed-effect modeling. Briefly, the existing population PK model in adults20 was used as the base structural model to develop an initial population PK model in pediatric patients with data from the two Phase I studies. The initial model was further refined to include covariate effects and updated with data from the Phase III studies to obtain the final model.
Model selection was based on log-likelihood criterion, goodness-of-fit plots, and scientific plausibility, with model reliability assessed through diagnostic plots, residual plots, visual predictive checks, and bootstrap analysis. Interindividual variability for PK parameters was evaluated using an exponential error model, and residual variability was modeled using a log-additive error model that included two separate random-effect parameters for the Phase I and Phase III efficacy studies. Statistics of interest (eg, fifth, 50th, and 95th percentiles) were also calculated from the simulated and observed data for comparison. Prespecified covariates (specifically, age, body weight, BMI, sex, race, and dose) were investigated for their impact on asenapine PK using a stepwise covariate-selection method. This procedure involved stepwise testing of linear and nonlinear relationships in a forward-inclusion (at a significance level of P<0.01) and backward-exclusion (P<0.001) step. Selected covariates were retained in the final model based on parameter precision and biological plausibility. Simulations were used to determine the effect of covariates on asenapine PK and compare steady-state exposure (AUC0–12 and Cmax) between pediatric and adult patients. Note that N-desmethylasenapine was not included in the population PK model or simulations, because it is not considered to contribute to the effects of asenapine.
Finally, an exploratory exposure–response analysis was performed to examine AUC0–12 as a potential predictor of response on the YMRS at day 21 end point from the Phase III bipolar I study described (n=233).14 Individual estimates of AUC0–12 were derived from the final PK model and summarized as equally sized bins (approximately 40 values/bin).
Results
Patient characteristics and disposition
Study 1
A total of 40 patients were enrolled in study 1: 23 males, 17 females, 27 black/African-American, and 13 white. The mean age was 14.8 years and mean BMI 24.9 kg/m2. A total of 38 patients completed the study. One patient who received matching placebo was withdrawn on day 2 because of an AE (exacerbation of schizophrenia), while one patient who received 3 mg asenapine withdrew consent on day 11 (reason unknown). All patients were included in the PK and safety analyses.
Study 2
A total of 30 patients were enrolled in study 2: 17 males, 13 females, 26 black/African-American, and four white. The mean age for cohorts 1, 2, and 3a was 10.8, 10.5, and 10.3 years, respectively, and that for cohorts 3b, 3c, and 3d 12.5, 14.5, and 16.5 years, respectively. Mean BMI across all cohorts was 21.9 kg/m2. A total of 27 patients completed the study. Three withdrew consent and discontinued the study prematurely (n=2 from Cohort 3a, n=1 from Cohort 3c). These three patients were excluded from PK analyses; all 30 patients were included in safety evaluations.
Asenapine pharmacokinetics
Mean asenapine concentration–time profiles in pediatric patients showed rapid absorption and an initial rapid decline in plasma concentrations, followed by a slower elimination phase (Figure 2). Time to maximum plasma-asenapine concentration was ~1 hour, with median values of 0.71–1.5 hours (Table 1). Mean t1/2 fluctuated among dose groups, with a range of 16 hours (10 mg) to 32 hours (5 mg) across all cohorts. No consistent trend in t1/2 was observed with increasing dose (Table 1).
Figure 2.

Mean asenapine plasma concentration–time profiles.
Notes: Following administration of sublingual asenapine 1–10 mg BID (final dose) for study 1 (top) and study 2 (bottom). Inset represents concentrations on a semilogarithmic scale.
Abbreviation: BID, bis in die (twice daily).
Table 1.
PK parameters for asenapine following multiple BID doses by dose and age
| Dose | Cohort | Age, years | n | Cmax, ng/mL | Tmax, hours | AUC0–12, ng⋅h/mL | t1/2, hours |
|---|---|---|---|---|---|---|---|
| Study 1 | |||||||
| 1 mg | 1 | 12–17 | 8a | 1.0 (50) | 0.7 (0.3–1.5) | 6.6 (61) | 29 (41) |
| 3 mg | 2 | 12–17 | 8b | 2.6 (56) | 0.9 (0–1.5) | 16 (50) | 26 (25) |
| 5 mg | 3 | 12–17 | 8 | 3.5 (48) | 1.0 (0–2.8) | 23 (48) | 32 (38) |
| 10 mg | 4 | 12–17 | 8 | 2.8 (82) | 1.3 (0–3.0) | 20 (54) | 23 (22) |
| Study 2 | |||||||
| 2.5 mg | 1 | 10–11 | 6 | 1.8 (64) | 1.0 (0.5–2.0) | 11 (54) | 22 (28) |
| 5 mg | 2 | 10–11 | 6 | 3.5 (18) | 1.8 (1.5–3.0) | 24 (17) | 19 (17) |
| 10 mg | 3a–d | 10–17 | 15 | 7.8 (49) | 1.0 (0.5–3.0) | 44 (45) | 20 (45) |
| 10 mg dose by age cohort | |||||||
| 3a | 10–11 | 4 | 9.2 (56) | 1.5 (0.5–1.5) | 55 (38) | 16 (15) | |
| 3b | 12–13 | 4 | 6.8 (10) | 1.0 (1.0–1.0) | 41 (23) | 17 (20) | |
| 3c | 14–15 | 3 | 7.0 (93) | 0.5 (0.5–3.0) | 37 (101) | 24 (61) | |
| 3d | 16–17 | 4 | 7.9 (34) | 1.0 (0.5–1.5) | 42 (30) | 25 (45) | |
Notes: For both studies, Tmax shown as medians (range); all other PK parameters shown as arithmetic mean (%CV).
n=7 for t1/2;
n=5 for t1/2.
Abbreviations: AUC0–12, area under the plasma concentration–time curve from 0 to 12 hours; BID, bis in die (twice daily); Cmax, maximum plasma concentration; CV, coefficient of variation; PK, pharmacokinetic; t1/2, half-life; Tmax, time to Cmax.
Individual mean Cmax and AUC0–12 generally increased with increasing doses. However, in study 1, values were lower with 10 mg vs 5 mg asenapine (Table 1). The increase in exposure with increasing doses appeared to be dose-proportional in study 2 and slightly less than dose-proportional in study 1 (excluding the 10 mg dose). Upon comparison of the different age cohorts in study 2, who all received 10 mg asenapine, exposure was generally similar across all ages, though AUC and Cmax were somewhat higher (~30%) in the 10- to 11-year age-group compared with the other groups (Table 1).
Steady state in study 1 was attained within 8 days of BID dosing based on visual inspection of morning predose plasma-asenapine concentrations. In study 2, Helmert contrast results indicated that steady state was attained after 6 days of BID dosing in cohorts 1 and 2, after 7 days of BID dosing in cohorts 3b, 3c, and 3d, and after 11 days of BID dosing in cohort 3a.
Trends in PK parameters for the primary metabolite, N-desmethylasenapine, were generally similar to those observed with asenapine (Table S1). Notably, the lower Cmax and AUC0–12 values observed for asenapine in the 10 mg-dose cohort from study 1 were not observed for N-desmethylasenapine. The potential significance of this is discussed in the following.
Safety
Adverse events
In study 1, a total of 122 treatment-emergent AEs (TEAEs) were reported, 94 of which were considered by the investigators to be associated with study treatment. All TEAEs were mild (110 of 122) or moderate (12 of 122) in intensity. The most common TEAE of moderate intensity occurring in asenapine-treated patients was headache (one each in cohorts 2 and 4 and two in cohort 3). No other moderate TEAEs were reported in more than one patient. The incidence of TEAEs was higher among patients treated with ≥3 mg asenapine than those who received placebo or 1 mg asenapine (Table 2). The most frequent TEAEs, occurring in three or more patients in any group, were somnolence, dysgeusia, sedation, oral paresthesia, glossalgia, and oral hypoesthesia (Table 2). One patient withdrew because of an exacerbation of schizophrenia that was reported as an AE following treatment with placebo on day 1.
Table 2.
Treatment-emergent adverse events (TEAEs) by dose
| TEAE | Study 1 (n =40)
|
Study 2 (n= 30)
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Placebo (n=8) |
Asenapine
|
Asenapine
|
||||||||
| Cohort 1, 1 mg (n=8) |
Cohort 2, 3 mg (n=8) |
Cohort 3, 5 mg (n=16)a |
Cohort 4, 10 mg (n=8) |
Cohort 1, 2.5 mg (n=6) |
Cohort 2, 5 mg (n=6) |
Cohort 3, 10 mg (n=6)a |
Cohort 3, 10 mg (n=12)b–d |
Cohort 3, 10 mg (n=18)b |
||
| Any TEAE, n (%) | 6 (75) | 5 (62.5) | 8 (100) | 14 (87.5) | 8 (100) | 3 (50) | 4 (67) | 1 (16.7) | 10 (83.3) | 11 (61) |
| TEAEs by preferred term in ≥2 patients in any cohort, n (%) | ||||||||||
| Dysgeusia | 1 (12.5) | 2 (25) | 4 (50) | 3 (18.8) | 2 (25) | 1 (17) | 0 | 0 | 7 (58.3) | 7 (39) |
| Sedation | 0 | 2 (25) | 4 (50) | 7 (43.8) | 0 | 2 (33) | 2 (33) | 0 | 1 (8.3) | 1 (6) |
| Hypoesthesia, oral | 1 (12.5) | 1 (12.5) | 4 (50) | 1 (6.3) | 0 | 1 (17) | 0 | 0 | 9 (75) | 9 (50) |
| Somnolence | 1 (12.5) | 0 | 0 | 1 (6.3) | 4 (50) | 1 (17) | 0 | 0 | 5 (41.7) | 7 (39) |
| Dizziness | 1 (12.5) | 0 | 1 (12.5) | 1 (6.3) | 1 (12.5) | 0 | 2 (33) | 0 | 7 (58.3) | 7 (39) |
| Headache | 1 (12.5) | 0 | 2 (25) | 2 (12.5) | 1 (12.5) | 0 | 1 (17) | 0 | 2 (16.7) | 2 (11) |
| Glossalgia | 1 (12.5) | 0 | 0 | 6 (37.5) | 1 (12.5) | 0 | 0 | 0 | 0 | 0 |
| Nausea | 1 (12.5) | 1 (12.5) | 0 | 1 (6.3) | 0 | 0 | 2 (33) | 1 (16.7) | 1 (8.3) | 2 (11) |
| Dystonia | 0 | 0 | 0 | 0 | 0 | 0 | 4 (67) | 0 | 3 (25) | 3 (17) |
| Paresthesia, oral | 0 | 0 | 0 | 2 (12.5) | 4 (50) | 0 | 0 | 0 | 0 | 0 |
| Fatigue | 1 (12.5) | 0 | 1 (12.5) | 0 | 2 (25) | 0 | 0 | 0 | 0 | 0 |
| Contusion | 0 | 0 | 2 (25) | 2 (12.5) | 0 | 0 | 0 | 0 | 0 | 0 |
| Vomiting | 0 | 0 | 0 | 1 (6.3) | 0 | 0 | 2 (33) | 0 | 0 | 0 |
| Extrapyramidal disorder | 1 (12.5) | 0 | 0 | 0 | 2 (25) | 0 | 0 | 0 | 0 | 0 |
| Salivary hypersecretion | 0 | 0 | 0 | 0 | 0 | 0 | 2 (33) | 0 | 0 | 0 |
| Hostility | 0 | 0 | 0 | 0 | 2 (25) | 0 | 0 | 0 | 0 | 0 |
Notes: TEAEs in descending order across both studies.
Eight patients received 5 mg for 10 days (study 1, cohort 3) and another eight received 5 mg for 1 day as the titration step to subsequent 10 mg for 10 days (cohort 4);
cohort three includes cohort 3a, which consisted of patients aged 10–11 years who received a slower uptitration regimen, as well as cohorts 3b–3d (see Figure 1).
Of the 30 patients enrolled in study 2, 18 (60%) experienced at least 1 TEAE (Table 2). Most AEs were mild or moderate in intensity, though five asenapine-treated patients experienced AEs considered severe (n=1 sedation [cohort 2], n=1 dysgeusia and somnolence [cohort 3c], n=2 oral hypoesthesia and dysgeusia [cohort 3d], n=1 dystonia, headache, and somnolence [cohort 3d]). The most commonly reported TEAEs of moderate intensity were somnolence (n=6 [one each in cohorts 1 and 3b, two each in cohorts 3c and 3d]), dystonia (n=6 [one each in cohorts 3c and 3d, four in cohort 2]), dizziness (n=6 [three in cohort 3c, three in cohort 3d]), dysgeusia (n=5 [one each in cohorts 1, 3b, and 3d, two in cohort 3c]), and oral hypoesthesia (n=4 [one each in cohorts 3b and 3d, two in cohort 3c]). When comparing the 10 mg asenapine groups, incidence was lowest in the 10- to 11-year age cohort receiving the slower-uptitration regimen (cohort 3a, 17%) vs the older patients (cohort 3b, 50%; cohorts 3c and 3d, 100%). The most frequent TEAEs overall were oral hypoesthesia (33% of patients), dizziness (30%), dysgeusia (27%), somnolence (27%), dystonia (23%), sedation (17%), and nausea (13%; Table 2). A total of seven (23%) patients experienced dystonia during study 2, which was considered an event of clinical interest (Table 2). These events led to modification of dosing in cohort 3a by utilizing a slower regimen for uptitration. All episodes of dystonia resolved within 20 minutes to 4 hours of dosing with benztropine. No patients discontinued due to AEs in study 2. There were no deaths or serious AEs in either study.
Vital signs, ECG, and laboratory values
Values for systolic and diastolic blood pressure and heart rate tended to remain stable in study 1. Two patients had bradycardia reported as mild AEs that were deemed probably related to study treatment (one that began following 1 mg asenapine on day one and continued through day 10 and one during dosing with 5 mg asenapine on days 1–18). One patient had ECG recordings consistent with mild pericarditis that were reported as a mild AE possibly related to study treatment on day 11 (last dose) and was no longer present on day 18 (follow-up). This patient had possible ectopic atrial rhythm and anterolateral T-wave changes that were noted on ECG findings on day −5, and the ECG on day −1 was normal. Sinus arrhythmia (days 5 and 18) and extensive ST elevation suggesting pericarditis (days 5 and 11) were noted as significant changes from baseline. However, since the patient was asymptomatic and there was no associated clinical correlation with the ECG findings, the investigator concluded the patient did not have pericarditis and the ECG changes were not clinically significant, and only day 11 was reported as an AE. All three patients with cardiac AEs recovered. Other changes from pre- to postdose were minimal and not considered clinically significant.
In study 2, there were no dose-, age-, or time-related trends in mean vital-sign measurements. There were no reported AEs associated with ECG parameters nor dose- or age-related trends in mean ECG measurements. In study 1, one patient who received 10 mg asenapine had a possibly clinically significant increase in alkaline phosphatase. None of the clinical laboratory values for other patients met the criteria for a possibly clinically significant change. In study 2, none of the abnormal laboratory values (blood chemistry, hematology, urinalysis) were considered clinically relevant or reported as AEs.
Psychological assessment tools (study 2 only)
No patient reported suicidality or suicidal ideation assessed by the Columbia Suicide Severity Rating Scale. All Extrapyramidal Symptom Rating Scale scores were 0, indicating no EPSs were present at the time of assessment (day 4 for cohorts 1, 2, 3b, 3c, and 3d, day 7 for cohort 3a, and follow-up). Although this study was not powered to draw efficacy conclusions, efficacy-scale scores – Children’s Depression Rating Scale – revised, Clinical Global Impression, PANSS, and YMRS – trended toward a decrease overall.
Population PK-modeling analysis
Population PK-modeling analysis included PK data from the two Phase I PK studies and two additional Phase III efficacy studies, which included data from an additional 500 patients. A total of 2,451 concentration observations from 561 pediatric patients treated with asenapine were pooled. Asenapine PK in adult and pediatric populations was found to have good concordance when observed plasma concentrations from the pediatric population were overlaid on the prediction intervals derived from the adult-population PK model (Figure 3). This result justified the selection of the base structural model. The final pediatric PK model was best described as a two-compartment model with first-order absorption and first-order elimination. Parameter estimates and standard errors are summarized in Table 3. As the Phase III studies predominantly provided sparsely sampled concentration data and contained limited information on the exact time course of individual plasma concentrations, central volume of distribution (V2), peripheral volume of distribution (V3), and first-order absorption-rate constant (KA) were fixed to the respective parameter estimates obtained when fitting the concentration data from the Phase I studies only. Visual predictive checks showed that the model generally tracked the central tendency and distribution of observed data, indicating that the model adequately described observed asenapine data from the pediatric population (Figure 4 and Figure 5). For the final pediatric PK model, covariate analyses found no association of age, BMI, race, or sex with changes in asenapine exposure (Figure 6). As body weight and BMI were found to be highly correlated, care was taken to prevent both covariates from being included in relationships with the same PK parameter.
Figure 3.

Pediatric steady-state asenapine plasma concentrations (dots; studies 1 and 2) and adult model-based predictions (shaded areas) following clinically relevant doses.
Abbreviation: BID, bis in die (twice daily).
Table 3.
Population PK-model parameter estimates
| Final model parameter estimates
|
Estimates from bootstrap analysisa
|
|||
|---|---|---|---|---|
| Parameter | Population mean | RSE, % | Shrinkage, % | Median (90% CI) |
| Cl/F (L/h) | 296 | 3.09 | – | 296 (283–312) |
| V2/F (L)b | 2,740 | – | – | – |
| Q/F (L/h) | 120 | 14.3 | – | 120 (91.8–173) |
| V3/F (L)b | 2,490 | – | – | – |
| KA (hours)b | 2.98 | – | – | – |
| Interindividual variability, %CV | ||||
| IIV (Cl/F) | 66.2 | 19.5 | 27.3 | 66.4 (50.2–78.1) |
| Correlation (Cl/F–V2/F) | 0.921 | 20.5 | – | 0.918 (0.881–0.931) |
| IIV (V2/F) | 113 | 21.3 | 30.5 | 113 (87.8–137) |
| IIV (KA) | 68.9 | – | 73.5 | – |
| IIV (F) | 54.0 | 22.7 | 38.0 | 53.1 (42.0–66.8) |
| IIV (RV) | 19.2 | 29.8 | 45.3 | 18.5 (13.4–23.4) |
| Residual variability, % | ||||
| PK studies | 27.8 | 5.29 | – | 27.9 (25.6–30.3) |
| Efficacy studies | 56.0 | 5.05 | – | 56.0 (51.5–60.9) |
Notes:
Nineteen runs with minimization terminated were excluded when calculating bootstrap results;
V2/F, V3/F, and KA parameters were fixed to the obtained by using plasma-concentration data from PK and safety studies. Correlation (Cl/F–V2/F) calculated as covariance (Cl/F–V2/F)/square root (variance [Cl/F] × variance [V2/F]).
Abbreviations: Cl/F, apparent clearance; CV, coefficient of variation; F, bioavailability; IIV, interindividual variability; KA, first-order absorption-rate constant; PK, pharmacokinetic; Q/F, apparent intercompartmental clearance; RSE, relative standard error; RV, residual variability; V2/F, apparent central volume of distribution; V3/F, apparent peripheral volume of distribution.
Figure 4.

Goodness-of-fit plots for the final pediatric pharmacokinetic model.
Notes: (A, B) Closeness of observed plasma concentrations to population and individual predictions, respectively; (C, D) trends in residuals vs time and population predictions, respectively.
Figure 5.

Visual predictive check plots for each asenapine dose.
Notes: Adequacy of model is displayed by overlaying observations (o for Phase I studies and ♦ for Phase III studies) on model-predicted 95% prediction intervals (shaded area). Dots represent plasma-concentration data since last dose. Solid and dashed lines represent medians for binned time intervals of observations and model predictions, respectively.
Figure 6.
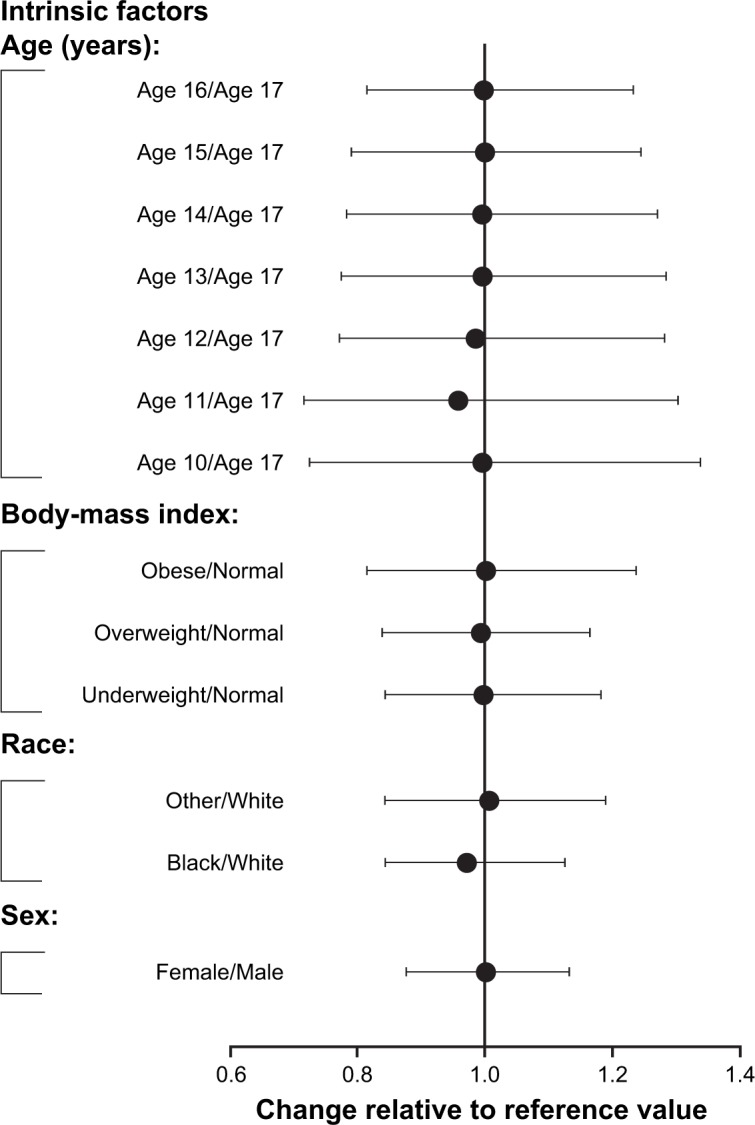
Impact of intrinsic factors on dose normalized AUC0–12 of asenapine.
Notes: Forest plot illustrates relative differences in asenapine AUC0–12 as a function of age, body-mass index, race, and sex. Black squares represent geometric mean ratios. Whiskers represent 90% confidence intervals. Body-mass index stratification: underweight (<18.5 kg/m2), normal (>18.5 and <25 kg/m2), overweight (≥25 and <30 kg/m2), obese (>30 kg/m2).
Abbreviation: AUC0–12, area under the plasma concentration–time curve from 0 to 12 hours.
Simulation of the population PK model showed that steady-state exposure metrics (eg, AUC0–12 and Cmax) for pediatric patients were similar to values in adult patients at the same dose, indicating similar exposure at the same asenapine dose, regardless of age (Table 4). In an exploratory exposure–response analysis of the Phase III study in patients with bipolar I disorder, differences in asenapine exposure were not associated with differences in observed YMRS scores (Figure S2). At most, a limited trend toward a decrease in YMRS score with increasing asenapine exposure was observed.
Table 4.
Steady-state asenapine pharmacokinetics in adult and pediatric populations
| Asenapine dose | AUC0–12, ng⋅h/mL, mean (%CV)
|
Cmax, ng/mL, mean (%CV)
|
||
|---|---|---|---|---|
| Adulta | Pediatricb | Adulta | Pediatricb | |
| 5 mg BID | 26.6 (38.4) | 19.3 (4.48–82.6) | 4.23 (45.3) | 4.56 (0.87–26.4) |
| 10 mg BID | 43.4 (53.1) | 37.8 (8.92–162) | 6.56 (50.9) | 8.64 (1.64–50.1) |
Notes:
Adult data derived from a double-blind, parallel, multicenter study to assess the effect of asenapine, quetiapine, and placebo on the corrected QT interval in patients with schizophrenia.36
Pediatric data derived from simulations. Values presented as medians (90% CI).
Abbreviations: AUC0–12, area under the plasma concentration–time curve from 0 to 12 hours; BID, bis in die (twice daily); Cmax, maximum plasma concentration; CV, coefficient of variation; PK, pharmacokinetic; t½, half-life; Tmax, time to Cmax.
Discussion
This publication aimed to evaluate the PK profile and tolerability of sublingual asenapine in pediatric patients with a psychotic disorder or bipolar I disorder across different age cohorts and dosing regimens. In pediatric patients aged 10–17 years, asenapine was rapidly absorbed (Tmax ~1 hour) and had an apparent terminal t½ of 16–32 hours, similar to the observed PK in adults. Across both studies, Cmax and AUC0–12 had near-dose-dependent increases, except for patients receiving 10 mg BID in study 1. The actual cause is unknown, but may have been due to the adolescent patients in the 10 mg group swallowing a greater portion of the administered dose. This assumption is based on the exposure results of the inactive metabolite N-desmethylasenapine and the ratio of N-desmethylasenapine to asenapine AUC0–12 values. The higher ratio of N-desmethylasenapine to asenapine in the 10-mg group relative to the other groups (1.75 vs 0.64–0.93, respectively) supports this hypothesis, since one would expect significant first-pass metabolism with oral administration. The PK parameters Cmax, AUC0–12, and t½ were similar for N-desmethylasenapine and asenapine, though Tmax was delayed relatively to asenapine, as expected since the formation of N-desmethylasenapine requires metabolism of asenapine.
Population PK modeling was similar between adults and pediatric patients. Age, BMI, race, and sex did not appear to be associated with clinically meaningful changes in asenapine exposure, suggesting that dose adjustments are not required based on these factors. Simulated Cmax and AUC0–12 values for asenapine in the pediatric population are broadly similar to what has been reported in adults,2,21 indicating similar exposure at the same asenapine dose regardless of age.
When age-groups that received the same dose were compared, the exposure observed was relatively similar; however, Cmax and AUC0–12 were ~30% higher in the youngest (10–11 years) age-group. Exploratory exposure–response analysis of the Phase III study in patients with bipolar I disorder showed that differences in asenapine exposure were not associated with measures of efficacy. Additionally, no differences in safety and tolerability were observed across either Phase I study. These results suggest that the increased exposure in the 10- to 11-year age-group was not clinically relevant. Increased exposure in the 10- to 11-year age-group relative to older cohorts was similar to results from a lurasidone study in which a 6- to 9-year-old cohort also showed increased exposure relative to older cohorts.22
Asenapine was generally safe and well tolerated in pediatric patients aged 10–17 years, and most AEs were mild or moderate in intensity. No patients experienced any serious AEs, and only one patient receiving placebo discontinued due to AEs. Though the higher doses had higher AE incidence, this may have been due to the titration schedule, as TEAE incidence was lower in the 10- to 11-year age-group receiving slower uptitration. Children aged 10–11 years may be more sensitive to initial asenapine treatment, and these results suggest that a slower titration schedule might have the potential to mitigate AEs. This approach to initiation of administration has been shown to decrease the frequency of AEs with other centrally acting drugs.23–26 The overall safety profile of asenapine is generally similar to what has been reported in the adult population and in older patients over the age of 65 years.13,27,28 The most frequently occurring AEs in the adult population (somnolence, sedation, dizziness, oral hypoesthesia, and dysgeusia)7,9,13,27,29 were also the most commonly occurring in the pediatric population. These AEs also occur commonly in other atypical antipsychotics used in the pediatric population.30,31
Children and adolescents may be more at risk for developing EPSs relative to adults, due to the increased number of striatal D2 receptors, which normally declines after childhood.32 Although EPSs and akathisia have been shown to be present in the adult population,7,27,29 there were only two reports of extrapyramidal disorder in the asenapine-treated pediatric population in study 1. In study 2, seven patients (four in the 10- to 11-age group receiving 5 mg BID) experienced dystonia; however, all episodes resolved following treatment with benztropine, suggesting that these side effects are manageable. Dystonia was not present in cohort 3a, which received the slower uptitration, and is infrequent in adults treated with asenapine.7,9
In the present pediatric population, asenapine was tolerated well with regard to vital signs, safety laboratory results, safety ECG, and/or other routine safety parameters. In the adult population, weight gain is a common side effect with atypical antipsychotics, although asenapine is generally associated with less weight gain relative to other treatments.33 In this pediatric population, although clinically relevant occurrences of weight gain were minimal, the studies were generally too short and underpowered to assess weight change adequately.
Overall, these results suggest that doses used in the adult population are also applicable to the pediatric population. These findings differ slightly from those of other atypical antipsychotics, which often have lower recommended starting and target doses for children and adolescents relative to the adult population.34,35 Additionally, this study found that variation in BMI did not result in clinically meaningful differences in exposure, which suggests that weight-based dosing adjustments are not needed in this population.
This study is the first to examine the PK profile of asenapine in children and adolescents using data from two Phase I studies; however, some limitations exist. A small number of patients were included in the dedicated PK and safety studies, which may prevent a reliable interpretation of the differences in PK profile, safety, and tolerability among the different age-groups. However, population-modeling analysis, which included PK data from two subsequently conducted Phase III studies, largely circumvented this limitation by including more patient samples. Although subjects of any race were recruited, the demographics among patients who met inclusion criteria only included white and black/African-American racial groups. A PK analysis on a more diverse patient population could be informative. The strengths of this study include the inclusion of various subtypes of psychiatric disorders, which may aid in overall generalizability.
Conclusion
Asenapine was generally safe and well tolerated in the pediatric population tested. Safety and PK data were similar to what is observed in the adult population. Younger children (10–11 years) did show higher exposure to asenapine than older children (>11 years); however, these results did not appear to be clinically meaningful. Taken together, the results from these analyses indicate that there is no need for dose adjustment in the pediatric population based on intrinsic factors.
Supplementary materials
Overview of population PK analysis of asenapine in a pediatric population. These studies consisted of 2 phase 1 PK studies (A7501022 and P06522), one phase 111 efficacy and safety study in pediatric patients aged 12 to 17 years with schizophrenia (P05896), and one phase 111 efficacy and safety study in pediatric patients aged 10 to 17 years with bipolar I disorder (P06107).
Abbreviation: PK, pharmacokinetic.
Exposure-response relationship of AUC0–12 and YMRS. Exposure-response relationship was examined through a visual exploration of the relationship between AUC0–12 and individual YMRS scores at endpoint in the 3-week, phase 111 study of asenapine in pediatric patients with bipolar I disorder. The boxes reflect the interquartile range (25–75th percentiles), the median denoted as solid line in each box, with the whiskers extending to the 5th–95th percentiles, and individual data outside these percentiles presented as symbols. For each bin, n is approximately 40.
Abbreviation: YMRS, Young Mania Rating Scale.
Table S1.
PK Parameters for N-desmethylasenapine following multiple BID doses by dose and age
| Dose | Cohort | Age, y | N | Cmax, ng/mL | % Cmax relative to parent | Tmax, hours | AUC0–12, ng⋅h/mL | % AUC0–12 relative to parent | t1/2, hours |
|---|---|---|---|---|---|---|---|---|---|
| Study 1 | |||||||||
| 1 mg | 1 | 12–17 | 8a | 0.4 (68) | 41.7 | 3.0 (0.5–12) | 4.0 (60) | 61.4 | 23 (28) |
| 3 mg | 2 | 12–17 | 8a | 1.0 (63) | 39.4 | 1.8 (0.3–6.0) | 10 (73) | 63.9 | 31 (101) |
| 5 mg | 3 | 12–17 | 8 | 1.4 (37) | 39.5 | 4.0 (0–11) | 13 (38) | 58.1 | 21 (36) |
| 10 mg | 4 | 12–17 | 8b | 3.0 (75) | 106.9 | 3.6 (0.8–4.0) | 26 (63) | 131.0 | 15 (23) |
| Study 2 | |||||||||
| 2.5 mg | 1 | 10–11 | 6 | 0.6 (34) | 35.2 | 4.0 (4.0–6.0) | 6.5 (33) | 56.9 | 14.6 (19) |
| 5 mg | 2 | 10–11 | 6 | 1.6 (43) | 44.5 | 5.0 (3.0–6.0) | 15 (44) | 64.4 | 13.9 (21) |
| 10 mg | 3a–d | 10–17 | 15 | 3.9 (67) | 49.9 | 3.0 (0.0–6.0) | 37 (65) | 82.6 | 14.7 (13) |
| 10 mg dose by age cohort | |||||||||
| 3a | 10–11 | 4 | 6.8 (38) | 73.5 | 3.0 (3.0–6.0) | 63 (38) | 113.9 | 12.6 (7) | |
| 3b | 12–13 | 4 | 2.7 (34) | 39.3 | 3.5 (2.0–4.0) | 26 (35) | 63.7 | 16.3 (10) | |
| 3c | 14–15 | 3 | 3.5 (92) | 49.4 | 2.0 (0.0–4.0) | 33 (90) | 90.7 | 14.2 (8) | |
| 3d | 16–17 | 4 | 2.5 (46) | 31.8 | 4.0 (0.0–6.0) | 23 (31) | 54.8 | 15.5 (11) | |
Notes: For both studies Tmax is shown as median (range); all other PK parameters are shown as arithmetic mean (%CV).
n=5 for t1/2;
n=6 for t1/2.
Abbreviations: AUC0–12, area under the plasma concentration-time curve from 0 to 12 hours; BID, twice daily; Cmax, maximum plasma concentration; %CV, percent coefficient of variation; SD, standard deviation; t1/2, elimination half-life; Tmax, time to Cmax.
Acknowledgments
Writing and editorial assistance was provided by Jacqueline Benjamin, PhD and Krystina Neuman, PhD of Prescott Medical Communications Group (Chicago, IL), a contractor of Allergan. The study was sponsored by Merck & Co Inc. This manuscript was supported by funding from Allergan (Irvine, CA).
Footnotes
Author contributions
Data analysis was conducted by the authors, and all authors met International Committee of Medical Journal Editors authorship criteria. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
Neither honoraria nor payments were made for authorship. PD acknowledges a potential conflict as a former employee of Merck Sharp and Dohme. RR has no conflicts of interest. RG acknowledges a potential conflict as a former employee of Merck Sharp and Dohme and a current employee of Certara. JD acknowledges a potential conflict as a former employee of Merck and Daiichi Sankyo. MJ acknowledges a potential conflict as a former employee of Merck and current employee of AstraZeneca UK. VPR acknowledges a potential conflict as a former employee of Merck and a current employee of AstraZeneca UK. AMMM acknowledges a potential conflict as an employee of Merck Sharp and Dohme. RLF receives or has received research support, acted as a consultant, and/or served on a speaker’s bureau for Aevi, Akili, Alcobra, Amerex, American Academy of Child and Adolescent Psychiatry, American Psychiatric Press, Bracket, ePharma Solutions, Forest, Genentech, Guilford Press, Ironshore, Johns Hopkins University Press, KemPharm, Lundbeck, Merck, NIH, Neurim, Nuvelution, Otsuka, PCORI, Pfizer, Physicians Postgraduate Press, Purdue, Roche, Sage, Shire, Sunovion, Supernus Pharmaceuticals, SyneuRx, Teva, Tris, TouchPoint, Validus, and WebMD. AJ acknowledges a potential conflict as a current employee of Allergan. TJC acknowledges a potential conflict as a current employee of Allergan. MDT acknowledges a potential conflict as a former employee of Merck and holds stock in Merck. Select data from this paper were presented at the 59th and 63rd American Academy of Child and Adolescent Psychiatry (AACAP) annual meetings as poster presentations with interim findings. The abstract from the poster presented at the 59th AACAP annual meeting was not published. The abstract from the 63rd AACAP annual meeting was published in the Journal of the American Academy of Child and Adolescent Psychiatry (2016;55[10 Suppl]). The authors report no other conflicts of interest in this work.
References
- 1.Preskorn SH. Clinically important differences in the pharmacokinetics of the ten newer “atypical” antipsychotics: part 1. J Psychiatr Pract. 2012;18(3):199–204. doi: 10.1097/01.pra.0000415076.28497.8e. [DOI] [PubMed] [Google Scholar]
- 2.Citrome L. Asenapine review, part I: chemistry, receptor affinity profile, pharmacokinetics and metabolism. Expert Opin Drug Metab Toxicol. 2014;10(6):893–903. doi: 10.1517/17425255.2014.908185. [DOI] [PubMed] [Google Scholar]
- 3.Mauri MC, Paletta S, Maffini M, et al. Clinical pharmacology of atypical antipsychotics: an update. EXCLI J. 2014;13:1163–1191. [PMC free article] [PubMed] [Google Scholar]
- 4.Minassian A, Young JW. Evaluation of the clinical efficacy of asenapine in schizophrenia. Expert Opin Pharmacother. 2010;11(12):2107–2115. doi: 10.1517/14656566.2010.506188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saphris [prescribing information] Dublin: Allergan; 2017. [Google Scholar]
- 6.Mcintyre RS, Cohen M, Zhao J, Alphs L, Macek TA, Panagides J. A 3-week, randomized, placebo-controlled trial of asenapine in the treatment of acute mania in bipolar mania and mixed states. Bipolar Disord. 2009;11(7):673–686. doi: 10.1111/j.1399-5618.2009.00748.x. [DOI] [PubMed] [Google Scholar]
- 7.Mcintyre RS, Cohen M, Zhao J, Alphs L, Macek TA, Panagides J. Asenapine in the treatment of acute mania in bipolar I disorder: a randomized, double-blind, placebo-controlled trial. J Affect Disord. 2010;122(1–2):27–38. doi: 10.1016/j.jad.2009.12.028. [DOI] [PubMed] [Google Scholar]
- 8.Mcintyre RS, Cohen M, Zhao J, Alphs L, Macek TA, Panagides J. Asenapine for long-term treatment of bipolar disorder: a double-blind 40-week extension study. J Affect Disord. 2010;126(3):358–365. doi: 10.1016/j.jad.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 9.Kane JM, Cohen M, Zhao J, Alphs L, Panagides J. Efficacy and safety of asenapine in a placebo- and haloperidol-controlled trial in patients with acute exacerbation of schizophrenia. J Clin Psychopharmacol. 2010;30(2):106–115. doi: 10.1097/JCP.0b013e3181d35d6b. [DOI] [PubMed] [Google Scholar]
- 10.Szegedi A, Calabrese JR, Stet L, et al. Asenapine as adjunctive treatment for acute mania associated with bipolar disorder: results of a 12-week core study and 40-week extension. J Clin Psychopharmacol. 2012;32(1):46–55. doi: 10.1097/JCP.0b013e31823f872f. [DOI] [PubMed] [Google Scholar]
- 11.Szegedi A, Zhao J, van Willigenburg A, Nations KR, Mackle M, Panagides J. Effects of asenapine on depressive symptoms in patients with bipolar I disorder experiencing acute manic or mixed episodes: a post hoc analysis of two 3-week clinical trials. BMC Psychiatry. 2011;11:101. doi: 10.1186/1471-244X-11-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schoemaker J, Stet L, Vrijland P, Naber D, Panagides J, Emsley R. Long-term efficacy and safety of asenapine or olanzapine in patients with schizophrenia or schizoaffective disorder: an extension study. Pharmacopsychiatry. 2012;45(5):196–203. doi: 10.1055/s-0031-1301310. [DOI] [PubMed] [Google Scholar]
- 13.Landbloom RL, Mackle M, Wu X, et al. Asenapine: efficacy and safety of 5 and 10 mg bid in a 3-week, randomized, double-blind, placebo-controlled trial in adults with a manic or mixed episode associated with bipolar I disorder. J Affect Disord. 2016;190:103–110. doi: 10.1016/j.jad.2015.06.059. [DOI] [PubMed] [Google Scholar]
- 14.Findling RL, Landbloom RL, Szegedi A, et al. Asenapine for the acute treatment of pediatric manic or mixed episode of bipolar I disorder. J Am Acad Child Adolesc Psychiatry. 2015;54(12):1032–1041. doi: 10.1016/j.jaac.2015.09.007. [DOI] [PubMed] [Google Scholar]
- 15.Findling RL, Landbloom RL, Mackle M, et al. Long-term safety of asenapine in pediatric patients diagnosed with bipolar I disorder: a 50-week open-label, flexible-dose trial. Paediatr Drugs. 2016;18(5):367–378. doi: 10.1007/s40272-016-0184-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Findling RL, Landbloom RP, Mackle M, et al. Safety and efficacy from an 8 week double-blind trial and a 26 week open-label extension of asenapine in adolescents with schizophrenia. J Child Adolesc Psychopharmacol. 2015;25(5):384–396. doi: 10.1089/cap.2015.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Citrome L. Role of sublingual asenapine in treatment of schizophrenia. Neuropsychiatr Dis Treat. 2011;7:325–339. doi: 10.2147/NDT.S16077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dogterom P, Timmer C, de Greef R, et al. Asenapine safety, tolerability, and pharmacokinetics after single and multiple doses in healthy volunteers. Clin Pharmacol Drug Dev. 2012;1(4):131–143. doi: 10.1177/2160763X12455328. [DOI] [PubMed] [Google Scholar]
- 19.Lindbom L, Ribbing J, Jonsson EN. Perl-Speaks-NONMEM (PsN): a Perl module for NONMEM related programming. Comput Methods Programs Biomed. 2004;75(2):85–94. doi: 10.1016/j.cmpb.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 20.Prohn M, de Greef R, Chapel S, Kerbusch T. Population pharmacokinetics of asenapine in patients with schizophrenia or bipolar disorder. Eur Neuropsychopharmacol. 2009;9(Suppl 3):S542–S543. [Google Scholar]
- 21.Stoner SC, Pace HA. Asenapine: a clinical review of a second-generation antipsychotic. Clin Ther. 2012;34(5):1023–1040. doi: 10.1016/j.clinthera.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 22.Findling RL, Goldman R, Chiu YY, et al. Pharmacokinetics and tolerability of lurasidone in children and adolescents with psychiatric disorders. Clin Ther. 2015;37(12):2788–2797. doi: 10.1016/j.clinthera.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Biton V, Edwards KR, Montouris GD, et al. Topiramate titration and tolerability. Ann Pharmacother. 2001;35(2):173–179. doi: 10.1345/aph.10093. [DOI] [PubMed] [Google Scholar]
- 24.Lyseng-Williamson KA, Perry C. Ziconotide. CNS Drugs. 2006;20(4):331–338. doi: 10.2165/00023210-200620040-00007. [DOI] [PubMed] [Google Scholar]
- 25.Mattson RH. Cognitive, affective, and behavioral side events in adults secondary to antiepileptic drug use. Rev Neurol Dis. 2004;1(Suppl 1):S10–S17. [PubMed] [Google Scholar]
- 26.Rauck R, Ma T, Kerwin R, Ahdieh H. Titration with oxymorphone extended release to achieve effective long-term pain relief and improve tolerability in opioid-naive patients with moderate to severe pain. Pain Med. 2008;9(7):777–785. doi: 10.1111/j.1526-4637.2007.00390.x. [DOI] [PubMed] [Google Scholar]
- 27.Gonzalez JM, Thompson PM, Moore TA. Review of the safety, efficacy, and side effect profile of asenapine in the treatment of bipolar 1 disorder. Patient Prefer Adherence. 2011;5:333–341. doi: 10.2147/PPA.S10968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dubovsky SL, Frobose C, Phiri P, de Greef R, Panagides J. Short-term safety and pharmacokinetic profile of asenapine in older patients with psychosis. Int J Geriatr Psychiatry. 2012;27(5):472–482. doi: 10.1002/gps.2737. [DOI] [PubMed] [Google Scholar]
- 29.Schoemaker J, Naber D, Vrijland P, Panagides J, Emsley R. Long-term assessment of asenapine vs. olanzapine in patients with schizophrenia or schizoaffective disorder. Pharmacopsychiatry. 2010;43(4):138–146. doi: 10.1055/s-0030-1248313. [DOI] [PubMed] [Google Scholar]
- 30.Findling RL, Pathak S, Earley WR, Liu S, Delbello MP. Efficacy and safety of extended-release quetiapine fumarate in youth with bipolar depression: an 8 week, double-blind, placebo-controlled trial. J Child Adolesc Psychopharmacol. 2014;24(6):325–335. doi: 10.1089/cap.2013.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sikich L, Hamer RM, Bashford RA, Sheitman BB, Lieberman JA. A pilot study of risperidone, olanzapine, and haloperidol in psychotic youth: a double-blind, randomized, 8-week trial. Neuropsychopharmacology. 2004;29(1):133–145. doi: 10.1038/sj.npp.1300327. [DOI] [PubMed] [Google Scholar]
- 32.Findling RL, Mcnamara NK. Atypical antipsychotics in the treatment of children and adolescents: clinical applications. J Clin Psychiatry. 2004;65(Suppl 6):30–44. [PubMed] [Google Scholar]
- 33.Kemp DE, Zhao J, Cazorla P, et al. Weight change and metabolic effects of asenapine in patients with schizophrenia and bipolar disorder. J Clin Psychiatry. 2014;75(3):238–245. doi: 10.4088/JCP.12m08271. [DOI] [PubMed] [Google Scholar]
- 34.Zyprexa [prescribing information] Indianapolis: Eli Lilly; 2009. [Google Scholar]
- 35.Seroquel [prescribing information] Wilmington (DE): AstraZeneca; 2009. [Google Scholar]
- 36.Chapel S, Hutmacher MM, Haig G, et al. Exposure-response analysis in patients with schizophrenia to assess the effect of asenapine on QTc prolongation. J Clin Pharmacol. 2009;49(11):1297–1308. doi: 10.1177/0091270009344855. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Overview of population PK analysis of asenapine in a pediatric population. These studies consisted of 2 phase 1 PK studies (A7501022 and P06522), one phase 111 efficacy and safety study in pediatric patients aged 12 to 17 years with schizophrenia (P05896), and one phase 111 efficacy and safety study in pediatric patients aged 10 to 17 years with bipolar I disorder (P06107).
Abbreviation: PK, pharmacokinetic.
Exposure-response relationship of AUC0–12 and YMRS. Exposure-response relationship was examined through a visual exploration of the relationship between AUC0–12 and individual YMRS scores at endpoint in the 3-week, phase 111 study of asenapine in pediatric patients with bipolar I disorder. The boxes reflect the interquartile range (25–75th percentiles), the median denoted as solid line in each box, with the whiskers extending to the 5th–95th percentiles, and individual data outside these percentiles presented as symbols. For each bin, n is approximately 40.
Abbreviation: YMRS, Young Mania Rating Scale.
Table S1.
PK Parameters for N-desmethylasenapine following multiple BID doses by dose and age
| Dose | Cohort | Age, y | N | Cmax, ng/mL | % Cmax relative to parent | Tmax, hours | AUC0–12, ng⋅h/mL | % AUC0–12 relative to parent | t1/2, hours |
|---|---|---|---|---|---|---|---|---|---|
| Study 1 | |||||||||
| 1 mg | 1 | 12–17 | 8a | 0.4 (68) | 41.7 | 3.0 (0.5–12) | 4.0 (60) | 61.4 | 23 (28) |
| 3 mg | 2 | 12–17 | 8a | 1.0 (63) | 39.4 | 1.8 (0.3–6.0) | 10 (73) | 63.9 | 31 (101) |
| 5 mg | 3 | 12–17 | 8 | 1.4 (37) | 39.5 | 4.0 (0–11) | 13 (38) | 58.1 | 21 (36) |
| 10 mg | 4 | 12–17 | 8b | 3.0 (75) | 106.9 | 3.6 (0.8–4.0) | 26 (63) | 131.0 | 15 (23) |
| Study 2 | |||||||||
| 2.5 mg | 1 | 10–11 | 6 | 0.6 (34) | 35.2 | 4.0 (4.0–6.0) | 6.5 (33) | 56.9 | 14.6 (19) |
| 5 mg | 2 | 10–11 | 6 | 1.6 (43) | 44.5 | 5.0 (3.0–6.0) | 15 (44) | 64.4 | 13.9 (21) |
| 10 mg | 3a–d | 10–17 | 15 | 3.9 (67) | 49.9 | 3.0 (0.0–6.0) | 37 (65) | 82.6 | 14.7 (13) |
| 10 mg dose by age cohort | |||||||||
| 3a | 10–11 | 4 | 6.8 (38) | 73.5 | 3.0 (3.0–6.0) | 63 (38) | 113.9 | 12.6 (7) | |
| 3b | 12–13 | 4 | 2.7 (34) | 39.3 | 3.5 (2.0–4.0) | 26 (35) | 63.7 | 16.3 (10) | |
| 3c | 14–15 | 3 | 3.5 (92) | 49.4 | 2.0 (0.0–4.0) | 33 (90) | 90.7 | 14.2 (8) | |
| 3d | 16–17 | 4 | 2.5 (46) | 31.8 | 4.0 (0.0–6.0) | 23 (31) | 54.8 | 15.5 (11) | |
Notes: For both studies Tmax is shown as median (range); all other PK parameters are shown as arithmetic mean (%CV).
n=5 for t1/2;
n=6 for t1/2.
Abbreviations: AUC0–12, area under the plasma concentration-time curve from 0 to 12 hours; BID, twice daily; Cmax, maximum plasma concentration; %CV, percent coefficient of variation; SD, standard deviation; t1/2, elimination half-life; Tmax, time to Cmax.