

Abstract
The genomic integration of HIV into cells results in long-term persistence of virally infected cell populations. This integration event acts as a heritable mark that can be tracked to monitor infected cells that persist over time. Previous reports have documented clonal expansion in people and have linked them to proto-oncogenes; however, their significance or contribution to the latent reservoir has remained unclear. Here, we demonstrate that a directed pattern of clonal expansion occurs in vivo, specifically in gene pathways important for viral replication and persistence. These biological processes include cellular division, transcriptional regulation, RNA processing, and posttranslational modification pathways. This indicates preferential expansion when integration events occur within genes or biological pathways beneficial for HIV replication and persistence. Additionally, these expansions occur quickly during unsuppressed viral replication in vivo, reinforcing the importance of early intervention for individuals to limit reservoir seeding of clonally expanded HIV-infected cells.
Keywords: AIDS/HIV, Virology
Keywords: AIDS vaccine
During in vivo infections, clonal expansion of HIV-infected cells occurs in specific biological pathways, including cellular division, transcriptional regulation, RNA processing, and posttranslational modification.
Introduction
HIV is the causative agent of AIDS and was first characterized in humans over 3 decades ago (1, 2). It is estimated that over 35 million people have died from HIV infection, with an additional 36.7 million people worldwide currently infected with the virus (Joint United Nations Programme on HIV/AIDS [UNAIDS]; www.unaids.org). Potent drug regimens administered as combination antiretroviral therapy (cART) are able to suppress viral replication to undetectable levels in most individuals; however, viral rebound occurs rapidly following any disruption in treatment (3–5). These reactivations increase the risk of selecting for mutations in the viral population that are resistant to previously administered antiretroviral drugs (6, 7). Like all retroviruses, HIV reverse transcribes its genome and permanently inserts itself into selected chromosomal locations within the infected cell, leading to long-term viral persistence (8, 9). Viral rebound occurs when an infected cell containing an integrated provirus reactivates transcription and emerges from its latent state (10). The process behind site selection of integration is still not entirely understood, but HIV is known to preferentially integrate into regions of open chromatin and active gene transcription (11). Once HIV has successfully integrated within a chromosomal locus, all subsequent cells arising through cellular division will contain the identical viral integration site (IS), functioning as a unique marker for each independent viral infection event. This results in a unique and heritable viral integration signature, which can be sequenced and tracked over time using IS analysis (ISA).
Previous reports have characterized IS within HIV-infected individuals while they were suppressed on cART (12, 13). These studies established that clonal expansion of HIV-infected cells contribute, on some level, to viral persistence (14). In agreement with historical data, they observed that the majority of integrations occurred within gene transcripts (approximately 80% of IS), and about 12.5% of these genes have been associated with cancer development (13). It remains unclear what link, if any, there is between IS site selection of HIV and the potential for oncogenic development. These reports suggested that the persistence and clonal expansion of infected cells may be influenced by the specific gene harboring the viral IS. Despite providing important initial insights into the viral integration landscape of individuals infected with HIV, several important questions remain. First, the driving force behind these clonal expansions and their potential clinical relevance remains to be determined. Additionally, it has also been reported that the majority of infected and clonally expanded cells contain a defective provirus, implying that these expansions are not driven by viral integration (15). However, it is still possible that, despite having a defective provirus, intact long terminal repeat (LTR) regions could still alter local gene expression through recruitment of transcriptional factors (16). Therefore, even dead proviral elements might provide important insight into clonal expansion.
These observed clonal expansions raise some interesting possibilities. After HIV infection, the virus represses cellular division by using accessory genes to mediate several intracellular signaling pathways (17). As a result, clonal expansion of HIV-infected cells would only occur under certain circumstances: (a) if the integrated provirus became defective; (b) if the provirus silenced expression, resulting in latency; (c) if an infected cell underwent antigen-stimulated expansion, overwhelming viral antiexpansion signals; or (d) if the integration itself initiated cellular proliferation due to insertional transformation. The 2 circumstances of highest clinical interest are situations in which the virus transitioned to a latent infection and those occurring due to integration induced expansion. While HIV has not been shown to directly cause oncogenic transformation, the occurrence of lymphomas is higher in AIDS patients (18, 19). These AIDS-related lymphomas are thought to arise due to systemic immune dysregulation caused by HIV-mediated CD4+ T cell depletion (20). Another possibility for clonal expansion occurs when integrated proviral elements become dormant and transition into latently infected cells. It is thought that these cells contribute to viral rebound after cART withdrawal, since they are refractory to treatment — either due to low or no viral gene expression (21). It is still unclear exactly what causes an infected cell to become latent and, subsequently, where these cells reside in virally suppressed individuals (22). One hypothesis is that latency is a randomly occurring event in a rare subset of infected cells due to changes in cellular transcription or chromatin status (23). Another possibility is that the specific site of integration could drive cells to latency due to either local gene expression patterns or integration into a temporarily transcriptionally silent region of the genome.
In this manuscript, we use a preclinical HIV animal model to link, for the first time to our knowledge, IS locations within specific gene families to their subsequent clonal expansion and persistence over time. We can then compare these IS identified using our in vivo model system to IS observed during in vitro infection conditions. While they may appear comparable in terms of total chromosomal distribution, there are significant differences in specific enrichment and expansion locations of integrations. We identify significantly expanded clones primarily during in vivo infection and occasionally in known proto-oncogenes, and we also determine that cellular expansions are specifically driven by IS occurring within genes from biological pathways that the virus would benefit from manipulating. Using a data set of almost 240,000 IS, we demonstrate that HIV-infected cells preferentially expand when integrated within genes linked to cellular processes of transcription, protein modification, cellular replication, and other viral processes. Additionally, we also observed significant clonal expansion of IS within 2 known proto-oncogenes involved in cellular proliferation pathways, which has not been previously reported, to our knowledge. This data confirms that clonal expansion occurs early in infection and appears specific to processes important for viral replication or persistence. This reiterates the importance of IS tracking moving forward for all HIV treatment protocols and trials as an essential benchmark for measuring the effect on and elimination of the latent viral reservoir.
Results
Generating in vivo HIV-infected samples for ISA.
In order to generate in vivo HIV-infected samples for ISA, we adapted a previously characterized mouse model of infection (24). Humanized mice were generated by engrafting human fetal liver CD34+ hematopoietic stem and progenitor cells (HSPCs) into neonatal nonobese diabetic–SCID (NOD-SCID), common γ–/– (NSG) mice (25, 26). Cells from 2 unique donors were engrafted in 2 different litters of mice for a total of 13 animals. Beginning at week 8, blood samples were collected every other week to monitor engraftment and lineage development. Mice were challenged with HIV 16 weeks after engraftment once peripheral levels of human CD45+ and CD3+ T cells stabilized. At time of challenge, average peripheral human engraftment was 70.1%. This level of engraftment resulted in 23.3% of total blood cells being human CD3+ T cells, of which 15.4% were CD4+, resulting in a CD4/CD8 ratio of 1.96. Three animals were unchallenged controls, and 2 animals that were challenged with HIV did not successfully initiate an infection, as determined by quantitative viral load PCR and were excluded from the remainder of this study. After HIV challenge, all 8 mice that were successfully infected demonstrated a rapid loss of peripheral human CD45+ cells (Figure 1A), which corresponded to a loss of CD3+ (Figure 1B) and CD4+ cells (Figure 1C) when compared with uninfected mock controls. Viral load in mice that successfully initiated infection ranged between 1 × 105 and 1 × 107 viral copies/ml throughout the experiment (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.99127DS1). After 28–30 weeks after engraftment, 12–14 weeks after infection, mice were sacrificed and lymphoid tissues analyzed for human engraftment and lineage contribution. While all mice exhibited significant depletion in peripheral human CD45+ engraftment at 28 weeks (P < 0.0001, unpaired, 2-tailed t test), human cells were still detected in BM, spleen, and thymus (Figure 1D). Both BM and spleen samples were analyzed for proviral ISA. A summary of all samples used for ISA throughout the manuscript is provided (Supplemental Table 1).
Figure 1. Depletion of circulating human CD4+ cells in periphery of infected mice.

Neonate NSG mice were engrafted with human CD34+ cells at birth and challenged with HIV at 16 weeks of age after development of CD4+ T cells. (A) Peripheral human CD45+ engraftment in blood samples over time for 2 cohorts of mice. Each line represents a single mouse and is colored red after infection at 16 weeks, indicated by vertical dashed red line. (B) CD3+ and (C) CD4+ T cell development over time for individual mice, represented as percent of cells in total blood. Mock mice represented as black lines (n = 3) and HIV-infected mice represented as red lines (n = 8). (D) Human engraftment at time of necropsy in peripheral blood, BM, spleen, and thymus of mice as indicated on upper x axis. Bar length corresponds to total human CD45+ engraftment and is broken down into stacked boxes representing specific cell lineages. Individual animal numbers listed on y axis and horizontal dashed black line separates mock from HIV-infected mice. One mock mouse died early and was excluded from necropsy analysis.
IS preferentially cluster on specific chromosomes.
A total of 6,259 unique HIV IS were identified in BM and spleen samples analyzed from the 8 mice (Table 1). An average of 80.51% of all IS fell within known gene transcripts and 6.95% within known oncogenes. We first wanted to determine the distribution of these IS located on each individual chromosome of the human genome. Assuming an entirely random distribution, IS would occur more frequently on larger chromosomes than smaller chromosomes for any set number of unique sites. When the proportion of observed-to-expected IS was calculated based on the specific length of each individual chromosome, 3 chromosomes exhibited a 2.5- to 4-fold enrichment for HIV integrations (Figure 2A). It has previously been documented that HIV preferentially integrates into locations of active gene transcription at time of infection (11), and these 3 chromosomes (chromosomes 16, 17, and 19) also have a higher gene density content (27). As a positive control for our in vivo IS database, we also analyzed IS in several in vitro infections of either primary human CD4+ cultured cells, or a well-characterized Jurkat cell line (Supplemental Figure 2). These in vitro infections were propagated for up to 28 days after challenge. When the same analysis was performed for the proportion of integrations relative to expectation in these in vitro infections, the results matched the in vivo data set (Figure 2B). The same pattern was also consistent for in vitro infections when using either a CXCR4 or CCR5 tropic virus (Supplemental Figures 3 and 4, and Supplemental Table 2), although the CXCR4 infection data sets only represented an n = 1 in these studies.
Table 1. Integration site characteristics within individual mice.

Figure 2. Distribution of HIV integration sites across all human chromosomes.

Visual representation of the frequency for HIV viral integration events on each individual chromosome. (A) All HIV integrations from both in vivo or in vitro data sets were combined and plotted based on frequency of integrations relative to unbiased expectation of chromosome size, with larger chromosomes expected to contain more unique integrations when compared with smaller ones. The y axis is proportion of integrations observed relative to expectation based on chromosome size, with a horizontal dashed black line at a value of 1. Overlaid heatmap at top of histogram corresponds to height of each bar and is color coded, with higher frequencies than expected as darker red, lower frequency than expected as darker blue, and expected frequencies as white. (B) Heatmap representation of different HIV data sets broken down by infection system or cell type, and color code in the same fashion as previously described.
When analyzed either together as 1 group (all HIV) or broken down by individual condition, the pattern was the same. This suggests that, regardless of cell type or model system used, the IS distribution — at least on the chromosomal level — is comparable. This trend was consistent when we analyzed IS orientation frequency in reference to either the chromosome or transcriptional direction of IS within genes (Table 2). There was essentially a 50/50 split of IS in the forward and reverse orientations for each data group. However, there was a slight increase, though not statistically significant, in frequency for IS in the forward direction relative to transcript orientation for those IS found within genes. Nevertheless, the HIV in vivo data set exhibited the greatest increase of integrations in frame with gene transcripts (3.58%), indicating an increased preference for the same transcriptional orientation as transcribed genes.
Table 2. Number and orientation of IS in each data set.

Specific regions of IS enrichment present on several chromosomes.
We next wanted to transition from a bulk chromosomal view to a higher-resolution analysis to determine what differences existed between the in vivo and in vitro IS data sets. Each chromosome was broken down into a series of consecutive 25 kB bins stretching the entire length of the chromosome, and the number of unique IS identified within each bin was calculated (Figure 3A). Similar to the total chromosomal view, the in vivo IS from the mice exhibited an almost identical pattern to the in vitro IS, despite a large difference in the total number of sites between these data sets: 6,229 and 233,684, respectively. A handful of identified IS (30 for in vivo and 822 for in vitro) could not reliably be mapped to a unique chromosomal location and were excluded from this analysis. Interestingly, the distribution of viral IS across each individual chromosome varied greatly, with some individual bins containing almost 10 times more integrations than the immediately surrounding bins. These specific hotspot regions are discussed in more detail later in this manuscript. Despite several of these enriched bins occurring on either end of chromosomes, no distinct pattern of integrations was observed (Figure 3B).
Figure 3. HIV integration sites cluster in specific chromosomal regions.

Integration sites identified in either in vivo NSG mouse experiments or in vitro tissue culture infections were analyzed. (A) Circos plot depicting IS sites across the genome. Each chromosome is represented on the exterior of the ring and is broken down into sequential bins, each 25 kB in size. The total number of unique integrations occurring within each bin is represented by the height of histogram bars, with black bars radiating outward depicting integrations found in vitro (233,684 IS) and red bars radiating inward depicting those found in vivo (6,229 IS). Concentric rings function as a y axis and have incremental values of 35 for the black histogram and 2 for the red histogram. (B) Specific location of integration relative to chromosomal length is plotted for either in vitro infections (black line) or in vivo infections (red line). The x axis represents relative chromosomal position with 0.25, meaning 25% of the distance from beginning of chromosome, and y axis represents the frequency of observing an integration at each position throughout the chromosome.
In order to ensure that the bioinformatics pipeline our lab developed for identifying and aligning IS to the genome was not artificially skewing our results, we performed an identical analysis comparing HIV IS to 2 completely unrelated IS data sets: (a) human nonhematopoietic-based cell lines and (b) human CD34+ HSPCs and lineage progeny, both transduced using VSVG-pseudotyped lentiviral vectors (Supplemental Figure 5 and Supplemental Table 3). There was still a high degree of similarity between HIV IS and lentiviral IS in primary HSCPs; however, there was a large difference in chromosomal IS distribution when compared with the nonhematopoietic cell populations. This indicates that the high degree of similarity is not a result of data skewing by our bioinformatics analysis but, rather, may arise due to similar transcription profiles in cells of hematopoietic origin. We also analyzed the proportional frequency of integrations across each individual chromosome for each of these data sets (Supplemental Figure 6). While some chromosomes exhibited slightly higher integration frequencies at unique locations, most indicated a consistent distribution. One exception to this trend was found at the midpoint of chromosome 11, which indicated a highly enriched location for integrations in both HIV data sets and in lentivirus-transduced hematopoietic cells. This could represent a region of chromatin that is consistently open and expressed in hematopoietic cells, resulting in a hotspot of viral integrations.
Specific genomic regions are enriched for HIV integrations in vivo.
Since the distribution of IS across individual bins within each chromosome appeared nearly identical, we next wanted to determine if there were any enriched bins that contained a significantly higher proportion of IS occurring specifically in vivo. To achieve this, each bin across the genome that contained IS was assigned a frequency for the total number of unique IS occurring in that bin relative to the entire data set. The HIV in vivo data set was directly compared with a data set containing all IS identified in vitro. To determine which individual bins exhibited a differential frequency of IS falling outside the expected normal distribution, the upper and lower 99% CIs were estimated from simulated SDs using a bootstrap method. This resulted in a total of 685 bins exhibiting an enrichment of IS within the HIV in vivo data set and 144 bins within the in vitro data set (Figure 4A). Almost all of these enriched bins within both the in vivo and in vitro data sets contain a gene transcript (93.9% and 97.2%, respectively). The top 10 bins exhibiting the highest enrichment frequency all contained gene transcripts within their boundaries (Table 3). The highest enriched gene bin was found to contain the gene NEAT1, a long noncoding RNA transcript previously hypothesized to play a role in HIV infection (28). Other top genes identified, including MINK1, SMARCC1, SEPT9, and POLR2A, encode for genes important in cellular signaling cascades or mRNA transcription. When all significantly enriched bins in either in vivo or in vitro data sets were analyzed for their specific chromosomal locations, different patterns of enrichment were observed (Figure 4B). While there was a relatively equal chromosomal distribution of bins identified in the in vivo data set, the in vitro data set was concentrated on chromosomes 16, 17, 19, and 22. Other chromosomes exhibited only sporadic enrichment or none at all. Since this analysis is a measure of unique genomic regions enriched for integrations, the results indicate that there is a higher propensity for HIV integrations to occur in specific locations during in vivo infection — hence, more regions of enrichment — while in vitro IS are more equally distributed across the genome, resulting in fewer locations of enrichment.
Figure 4. Specific chromosomal regions are enriched for viral integration sites.

The HIV in vivo IS data set was directly compared with in vitro IS to determine if there were specific locations within the genome that were significantly enriched for IS during in vivo infection. (A) Dot plot representing significantly enriched 25 kB bin segments in 1 data set compared with the other. Bins in the positive direction on y axis were enriched for during in vivo infection (685 bins), while bins in the negative direction were enriched for during in vitro infections (144 bins). The x axis represents relative chromosomal position but is not normalized for chromosome length. Top 4 bins enriched in vivo are labeled with the genes contained at that locus. Horizontal red lines indicate the 99th percentile of simulated data, similar to P = 0.01. (B) Circos plot highlighting the bin locations for each enriched data set. Bins containing integrations enriched during in vivo infection are plotted in gray, and those enriched during in vitro infection are plotted in black. Chromosomes represented on exterior of ring.
Table 3. Top 10 chromosomal bins enriched in vivo.

Expanded clones occur more frequently during in vivo infection.
We next analyzed the breakdown of individual clones identified within each experimental infection group to determine both the frequency of expanded clones and the degree of expansion. Our IS protocol utilizes randomized acoustic shearing to fragment the DNA prior to linker ligation. This typically results in 2 different cells containing identical IS to shear at different locations, yielding various lengths of intervening genomic sequences. Counting these various fragment lengths provides a minimum estimate for the total number of cells containing the same IS, indicating an expanded clone. These clones were broken down into 3 groups: (a) unexpanded clones found in 1 cell, (b) low-level expanded clones found in 2–4 cells, or (c) high-level expanded clones found in 5 or more cells. A cell and its progeny must have undergone at least 4 cellular divisions to yield 5 or more identical clones. Since HIV typically shuts down cellular replication machinery during infection, we set a clonal expansion of 5 as our threshold to be considered an expanded clone. When each group was analyzed for clonal expansion, the in vivo infection data set exhibited the highest frequency of expansion at both low-level (23.90% of clones) and high-level (1.92% of clones) expansion (Figure 5 and Table 4). The total frequency of highly expanded clones in either of the in vitro primary or cell line infections was 0.01% and 0.04%, respectively, despite having a significantly higher number of total IS detected. The top expanded clones for the in vivo infection data set also exhibited higher total cell numbers (36–72 clones) compared with either in vitro primary cells (4–5 clones) or the cell line (5–6 clones).
Figure 5. Expanded clones occur more frequently during in vivo infection.

All IS identified were classified into 3 groups of expansions and plotted in pie charts for (A) HIV in vivo integrations, (B) HIV in vitro primary cell integration, and (C) HIV in vitro cell line integrations. Proportion of IS found in 1 cell for each group represented by white area, 2–4 cells represented with gray area, and 5 or more cells represented as black area. Actual percentages for each category are listed in each pie chart.
Table 4. Top 8 expanded clones within genes.

Two of the top expanded clones found within the in vivo infection data set occurred in known oncogenes JAK2 (Figure 6A) and SEPT9 (Figure 6B). SEPT9 was also the gene identified containing an enriched frequency of integrations during in vivo infections. While there were several other low-level expanded clones occurring in both of these genes, there was only 1 in JAK2 and 1 in SEPT9 (Figure 6, circled red arrows) that were classified as highly expanded. Additionally, these 2 clones that exhibited the highest expansion — 72 and 50, respectively — were both in the same transcriptional orientation as the genes. The expanded integration in SEPT9 occurred in what appears to be a hotspot of integrations in intron 2 of the full-length gene transcript. The remaining expanded clones identified over 50 times did not occur in known oncogenes. We also detected numerous integrations within 2 other genes that have previously been reported in the literature to contain expanded clones in HIV-infected individuals (MKL2 and BACH2) (12, 13); however, we did not detect clones expanded over 3 cells in any of our data sets (Supplemental Figure 7).
Figure 6. Clonal expansion observed in known oncogenes.

Individual gene plots indicating the location of all detected integrations within 2 oncogenes identified as containing substantially expanded clones (A) JAK2 and (B) SEPT9. Gene name is listed at top left for each group, and specific location on chromosome is highlighted by red box. The gene transcript is expanded below chromosome; each arrow indicates a unique IS, and arrow direction depicts orientation. Black arrows represent an IS found in 1 cell, and red arrows represent an IS found in 2 or more, with the number of clones detected noted next to each arrow. Circled red arrows indicate expanded clones over 5 cells. Blue lines at the bottom of each graph represent all known transcript variants of each gene.
Clonally expanded cells are enriched in specific biological pathways.
In order to determine if there is a correlation between the individual genes demonstrating the highest level of clonal expansion in each data set, we utilized the DAVID Bioinformatics Database through the National Institute of Allergy and Infectious Disease (NIAID) (29, 30). We first calculated the average clonal expansion for integrations occurring in each gene identified by ISA and then sorted for the top 1,000 genes in each data set. These genes were analyzed using the biological processes output tool in DAVID and graphed in a heatmap according to the significance of gene enrichment (Figure 7 and Supplemental Table 4). A total of 44 pathways were very significantly enriched (P < 0.005) within the HIV in vivo data set specifically, while only 1 or 3 pathways were identified in either the in vitro primary or cell line data sets, respectively. In addition to analyzing the HIV IS, we also included in the analysis data sets from both the lentiviral-transduced hematopoietic and nonhematopoietic cells and a randomly generated IS library. There was very little overlap of enriched pathways between the 6 data sets, with each one containing a unique cluster of several pathways. For example, the processes identified for lentiviral-transduced nonhematopoietic cells were primarily involved in metabolic pathways. Within the HIV in vivo data set, the most significantly enriched pathway contained genes involved in viral processes (P = 3.63 × 10–8), indicating a significant preferential expansion of clones containing IS within these genes. In addition to the viral process pathway, numerous other relevant pathways that encompass biological processes that HIV would benefit from manipulating — such as RNA splicing (P = 3.39 × 10–5), protein phosphorylation (P = 8.31 × 10–4), transcriptional regulation (P = 8.12 × 10–4), and cell division (P = 2.65 × 10–3) — were significantly enriched.
Figure 7. Clonally expanded cells are significantly enriched in specific gene pathways.

Heatmap of biological processes identified using publically available DAVID database. The top 1,000 genes containing expanded clones were analyzed for each group listed at top of each column. All biological processes identified are plotted as a row of individual boxes within each column and color coded based on significance. Red boxes indicate a biological pathway that was very significantly enriched in the data set (P < 0.005), blue boxes were significantly enriched (P < 0.01), gray boxes were observed but not significant (P ≥ 0.01), and white boxes indicate a pathway that was not found in the given data set. The top 30 pathways represented with red boxes in HIV in vivo data set are broken down at left with each biological process named and with associated P value listed. Statistics were performed by DAVID database using a modified Fisher’s exact test (EASE score).
Discussion
The use of viral IS as an important facet for understanding HIV pathogenesis and persistence in clinical studies is becoming more apparent (31–33). Gaining a better understanding of the processes behind IS selection, how the transition to latent viral infection occurs, and if clonal expansion of infected cells might contribute to the latently infected pool of cells is essential. Here, we document for the first time to our knowledge targeted clonal expansions of HIV-infected cells in genes significantly enriched for specific biological pathways, despite only representing a small fraction of total IS. The number of IS demonstrating clonal expansion in this study is most likely due to both the shorter timeline of infection (3 months) and the lack of ART suppression, which would lead to higher levels of viral induced cytotoxicity compared with humans with HIV on treatment. Analyzing samples in this manner lets us determine what the IS profile looks like early during infection when the latent reservoir is most likely seeded, as opposed to after long-term treatment before significant clonal outgrowths are expected. Furthermore, these IS frequently occur in pathways that the virus would benefit from mediating at the genetic level, including multiple pathways involved in transcription, translation, and posttranslational modification of gene products, as well as pathways involved in mediating cell cycle progression and cellular division. We also observed expanded clones containing an intact LTR sequence in several previously undocumented proto-oncogenes known to play a role in hematopoietic malignant development (34, 35). These findings only occurred during in vivo HIV infection of humanized mice and not during in vitro cell culture infections. This further emphasizes the need for validated preclinical models of HIV infection designed for treatment- and cure-related strategies in order to accurately recapitulate an infection setting.
Viral integrations were consistently detected in genes represented in numerous biological pathways, and these integrations exhibited specific clonal expansion only during in vivo infection. The accessory genes of HIV are known to broadly alter the internal composition of infected cells by hijacking normal phosphorylation (36) and ubiquitin processes (37), mediating viral gene transcription (38), and suppressing immune surveillance and detection (39). These data indicate that cells containing viral integrations within genes relevant to these pathways have a higher frequency of expansion, especially during in vivo infection, indicating preferential IS expansion. Thus, after a random integration event, preferential expansion occurs within the infected pool of cells through manipulation of specific signaling pathways. This ultimately might provide a selective advantage for the virus, such as altering transcription/translation or cell cycle regulation. While these studies are not able to determine whether the provirus is intact and capable of creating infectious viral particles, the presence of an LTR promoter sequence at these loci could be enough to alter transcriptional regulation of these genes (40). Further investigations will be required to determine if the transcriptional profile of these types of integrations actually alter gene expression, especially if they are observed in human clinical patient data.
Recent publications by several groups have documented that clonal expansion does occur in people during viral suppression on cART (12, 13, 41). Furthermore, these expansions often are associated with specific genes and frequently occur in the same transcriptional orientation and intronic location. Specific viral integration events within unique genes have also been linked to increasing cellular proliferation and survival of infected cells (42). While we did observe significantly expanded clonal populations in several genes, including numerous oncogenes, we did not observe expansion in 2 previously reported genes by other research groups (12, 13, 43). Both BACH2 and MKL2 genes were identified in our data set as containing viral integrations, but none were expanded beyond 2 cells. We also did not observe the phenomenon of a high-frequency identical IS within the same intronic region or same transcriptional orientation, implying that such expansions might only occur during long-term ART suppression. If such a restriction of IS persistence is observed during treatment through multiple-round sampling over time, then this would provide strong selective evidence for targeted clonal expansion. The pattern of equal orientation distribution held true in all data sets, with frequencies of IS occurring in either the same or opposite orientation as gene expression at approximately 50%. The only exception to this was for HIV integrations within genes occurring in vivo, where a slight preference was observed for IS in the same orientation as gene transcription.
Another publication also documented clonal expansion occurring early during acute HIV infection and demonstrated that expansion was more likely to occur when the provirus was integrated in proximity to genes linked to cell activation and chromatin regulation (44). However, their data set was limited to 18,104 IS, most of which were obtained from in vitro infections. In this manuscript, we document almost 240,000 IS, including over 6,000 from in vivo samples. Similarly, we observed a clonal expansion of HIV-infected cells that occurs within 12 weeks of infection. A handful of viral IS were greatly expanded (detected in over 50 individual cells) and were obtained from only a fraction of the total tissue or peripheral reservoir. One explanation for clonal expansion of infected cells is through T cell receptor antigen-stimulated cell division. However, these humanized mice are maintained in an enhanced pathogen-free barrier within the mouse facility, and all consumable nutrients are sterilized. Therefore, pathogen-mediated antigen stimulation should occur relatively infrequently, although it cannot be completely ruled out. Using a data set from 13 individuals, including 6,719 unique IS, another publication demonstrated that most clonally expanded HIV-infected cells contained a defective provirus (15). These cells most likely would be detected in assays to measure total proviral content, artificially inflating the frequency of latent cells capable of reinitiating an infection. However, while they might not be functionally able to reactivate, the presence of proviral elements, especially the transcriptionally active LTR sequences, could alter local gene expression through recruitment of positive or negative regulators of transcription.
These data demonstrate that unsuppressed replication during acute infection stages can seed a larger reservoir of clonally expanded cells, which may ultimately lead to persistence of these infected cells over time. Additionally, these early clonal expansions are significantly enriched in biological processes beneficial to viral replication and persistence. Several pathways involved in histone regulation, mitotic progression, or cell division were also enriched, supporting an intriguing possibility of IS selection being a determinant for latency. It would also be of great interest to analyze a time course of infection, including IS associated with acute, chronic, and treated stages of infection. Such an analysis could provide further details of how cART treatment or other interventions shift the IS landscape and enable direct comparisons between early clonal expansion and the latent reservoir population.
While the mouse model of HIV infection utilized here is frequently used for testing different treatment strategies, we cannot rule out that findings are limited by this fact. In this study, we present data generated from unsuppressed viral replication in humanized mice, while most IS data generated to date have occurred in cART suppressed, chronically infected humans. Additionally, human data have primarily been collected from peripheral blood samples, while we analyzed viral IS in the lymphoid tissue compartments of BM and spleen due to low volume of blood, which can reliably be collected from mice. This does limit the potential interpretations that can be made and could result in any discrepancies between these data and previously published work. Additional studies are certainly required to further investigate these questions and to determine how similar viral IS and clonal expansions observed in animal models are when compared with people infected with HIV. These studies will undoubtedly prove invaluable in better understanding viral persistence and inform treatment options aimed at reducing or eliminating the reservoir and associated clonal expansions of virally infected cells.
Together, our data demonstrate that, although clonal expansion can and does occur in all infection settings, clonal outgrowth is statistically correlated to relevant gene pathways during in vivo infections in an animal model of HIV infection, reinforcing the importance of analyzing such preclinical model systems for any experimental treatment protocol. Indeed, new methodologies for either treating HIV infection or attempting to reduce the latent reservoir need to perform ISA to quantify the effect these treatments have on this population of cells. These findings also open new possibilities for developing protocols of therapeutic interventions during HIV treatment to mediate identified pathways and aid in the elimination of latently infected cells that persist through standard patient care.
Methods
Human CD34 processing and isolation. Human CD34+ cells were isolated from fetal liver tissue purchased from Advanced Bioscience Resources. Tissues were first manually broken down with scalpels and incubated in RPMI medium (Thermo Fisher Scientific) under gentle agitation for 1 hour at 37°C in the presence of 25 μg/ml DNAse (MilliporeSigma) and 5 μg/ml Liberase TH (Roche Diagnostics). Cell suspension was then filtered through a 70-μm filter (BD Biosciences) and lysed using hemolytic solution. CD34+ cells were then labeled and isolated using the CD34 MicroBead UltraPure Kit (Miltenyi Biotec) according to manufacturer’s protocol.
Mice. NOD.Cg-PrkdcscidIl2rgtm1Wjl/Szj (NSG) mice were purchased from The Jackson Laboratory or bred in-house under approved protocols and in pathogen-free housing conditions. Neonatal mice between 1 and 3 days after birth received 150 cGy of radiation, followed 3–4 hours later by a single intrahepatic injection of 1 × 106 CD34+ cells resuspended in 30 μl of PBS (Thermo Fisher Scientific) containing 1% heparin (Abraxis BioScience). Since mice were injected as neonates, sex was not taken into account for this study.
NSG mouse sample collection and processing. Blood collection began 8 weeks after transplant and continued every other week through retro-orbital puncture using glass capillary pipettes and collected into EDTA Microtainers (BD Biosciences). A maximum of 200 μl of blood was collected at each time point and diluted 1:1 with PBS. Blood was then centrifuged and plasma collected for RNA isolation. Cellular portion was then used for antibody staining and flow cytometry to determine engraftment levels and lineage contribution. At time of necropsy, tissues harvested included BM, spleen, and thymus, in addition to peripheral blood. Tissue samples were passed through a 70-μm filter (BD Biosciences) and washed with PBS. Blood and tissue samples were stained with appropriate fluorescently conjugated antibodies for FACS for 15 minutes at room temperature. RBCs were removed by incubation in BD FACS Lysing Solution (BD Biosciences), which was diluted out using PBS prior to analysis. All cells were acquired on a FACS Canto II (BD Biosciences) and analyzed using FlowJo software v10.1 (BD Biosciences). Up to 20,000 viable cells from blood and 100,000 viable cells from tissues were acquired when possible. Gates were established using full minus 1–stained (FMO-stained) controls. Samples were stained at a 1:20 dilution using human CD45-PerCP (clone 2D1), mouse CD45.1/CD45.2-V500 (clone 30-F11), CD3-FITC or allophycocyanin (APC) (clone UCHT1), CD4-V450 (clone RPA-T4), CD8-APC-Cy7 (clone SK1), CD20-phycoerythrin (PE) (clone 2H7), CD14-APC or PE-Cy7 (clone M5E2), and CD34-APC (clone 581). All antibodies were acquired from BD Bioscience. Once mice were infected with HIV, all samples were fixed using 10% neutral-buffered formalin solution (MilliporeSigma) for 10 minutes after antibody staining and RBC lysis.
HIV virus preparation and mouse challenge. Mice were challenged by a single i.p. injection of 200 μl of HIV-1 virus containing 2.5 × 105 infections units. Viral strains used included HIV-1 BaL, NL4.3, and JRCSF. All 3 viral strains were obtained through the NIH AIDS Reagent Program (ARP) Division of AIDS, NIAID. The BaL strain (catalog 510) was deposited by Suzanne Garter, Mikula Popovic, and Robert Gallo (45). The NL4.3 strain (catalog 114) was deposited by Malcolm Martin (46). The JRCSF strain (catalog 2708) was deposited by Irvin S.Y. Chen and Yoshio Koyanagi (47). BaL virus was propagated in PM1 cells (catalog 3038) obtained through the NIH ARP, Division of AIDS, NIAID, and deposited by Marbin Reitz (48). NL4.3 and JRCSF were obtained as proviral clones and transfected into 293T cells using TurboFect Transfection Reagent (Thermo Fisher Scientific) according to manufacturer’s protocols. In all cases, supernatant was collected from cells and filtered through 0.22-mm filters (MilliporeSigma), tittered on GHOST cells (catalog 3942) obtained through the NIH ARP, and deposited by Vineet N. Kewal Ramani and Dan R. Littman (49) according to provided protocols.
In vitro HIV infections. Jurkat reporter cells (JLTRG) were obtained from the NIH ARP, Division of AIDS, NIAID (catalog 11587) and deposited by Olaf Kutsch (50, 51). Primary CD4+ T cells were isolated from adult human apheresis product purchased from the Hematopoietic Cell Processing and Repository facility at Fred Hutchinson Cancer Research Center. Cells were enriched using the CD4 MicroBead kit (Miltenyi Biotec) according to manufacturer’s protocols. Cells were infected with HIV at an MOI ranging between 0.1–0.001 diluted in RPMI (Thermo Fisher Scientific) containing 1% penicillin/streptomycin (pen/strep; Thermo Fisher Scientific) for 4 hours at 10 × 106 cells per ml. Cells were then washed with PBS to remove unbound viral particles and were resuspended at 1 × 106 cells per ml in culture media (RMPI, 1% pen/strep, 10% FBS; Atlas Biological). CD4+ T cells were cultured in the same media supplemented with 50 μg/ml human recombinant IL-2 and 5 μg/ml phytohaemagglutinin (PHA). Cells were propagated for up to 3 weeks and split 1:2 every 3–4 days with fresh culture media. Cells were pelleted and genomic DNA extracted for ISA. Infection in JLTRG cells was tracked by flow cytometry for GFP expression after collection and cellular fixation using 10% neutral buffered formalin solution (MilliporeSigma) for 10 minutes. Gates were set using uninfected cell controls to contain <1% GFP+ cells.
Quantitative viral load PCR. Viral RNA was extracted from mouse plasma or tissue culture supernatant using the QIAamp Viral RNA Mini Kit (QIAGEN) as previously described (52). Briefly, viral RNA was then analyzed using the TaqMan RNA-to-Ct 1-Step Kit (Thermo Fisher Scientific) using primer and probes specific to the LTR region (F: 5′-GCCTCAATAAAGCTTGCCTTGAG-3′, R: 5′-GGCGCCACTGCTAGAGATTTTC-3′; probe FAM 5′-AAGTAGTGTGTGCCCGTCTGTTRTKTGACT-3′ TAMARA). Plates were analyzed on an ABI TaqMan 7500 real-time PCR system (Thermo Fisher Scientific).
Lentivirus-transduced cell populations. Human hematopoietic cells containing integrated lentiviral vectors were compiled from historical data, some of which have previously been published (52, 53), using ISA from either clinical or preclinical samples. For a nonhematopoietic IS library, HeLa (obtained through ATCC) or GHOST (obtained through NIH ARP) cells were transduced at an MOI of 10 using a SIN LV (pRSC-hPGK.eGFP) produced with a third-generation split packaging system and pseudotyped by the vesicular stomatitis virus G protein (VSVG). These vectors were produced by our institutional Vector Production Core (director HPK) at the Fred Hutchinson Cancer Research Center. Infectious titer was determined by flow cytometry evaluating EGFP expression following titrated transduction of HT1080 human fibrosarcoma–derived cells.
IS processing. ISA was performed on spleen and BM samples from mice and cell culture samples as previously described (53, 54), with the following modifications: DNA was extracted from cells using the DNeasy Blood and Tissue Kit (QIAGEN), and up to 3 μg was randomly sheared using an M220 focused ultrasonicator (Covaris). Fragmented DNA was purified, polished (End-It DNA End Repair Kit, Epicenter), and ligated to modified linker cassettes containing known primer binding sites. This product was amplified using sequential nested exponential PCR. Product from first PCR was purified, and eluted DNA was diluted prior to a second nested PCR, which added both barcodes and sequences required for compatibility with the next-generation–sequencing MiSeq platform (Illumina). Sequencing was performed by the Genomics Core Facility at the Fred Hutchinson Cancer Research Center. Sequences for all primers and linker cassettes used are provided in the supplement (Supplemental Table 5). IS were identified using a bioinformatics platform as previously described in detail (52).
Transduction filter methods. Crossover IS appearing in distinct samples originating from unique transduction events were present in the data. Theoretically, this should never be observed and is likely the result of contamination, index swapping, or other errors in processing. In some cases, it is possible to determine which sample the IS originates from by comparing the number of genomically aligned sequence reads representing the IS in each sample. When examining collisions, the genomically aligned sequence counts were used instead of normalized frequencies to avoid biases introduced by low capture frequency in samples with few genomically aligned reads because the log-base 10-fold difference between the most (131,641 reads) and fewest (25 reads) genomically aligned reads across samples was large (3.72 reads).
Using a custom python script, a list of all collisions was generated. Each transduction event was parsed for observations of IS in the collision list. For each transduction event in which a collision IS was detected, the mean count of the IS for samples in which it was detected was recorded. For example, an IS at chr9:5,069,731 was observed in 4 transduction events. In the first transduction event, it was observed in 2 samples where it was represented by 174 and 77 genomically aligned sequence reads. It was observed in 1 sample from each of the other transduction events where it was represented by a single genomically aligned read. The mean count of the IS in the first transduction event was 125.5 and was 1 for all other transduction events.
The ratio of mean counts from each transduction event was compared with the maximum mean count from a single transduction event. If a transduction event had a mean count greater than or equal to one-half of the maximum mean count for the IS, the IS was discarded from the data set; otherwise, the IS was kept for the transduction event in which it had the highest count and removed from the other samples. In other words, if the ratio of the maximum mean count to the next highest mean count was greater than 1:1, the IS was discarded. If the ratio was less than 1:1, the IS was retained in the transduction event where it had the highest count and removed from all others. Returning to the previous example, 1:125.5 is 0.008. In this case, all nonmaximum genomically aligned read counts fall below 0.5, and the IS is retained in the first transduction event data set and removed from all other transduction event data sets. Overall, from an initial set of 377,643 unique IS, 6,072 collisions (1.6%) were detected and 2,166 (0.57%) were unresolvable (removed from all data sets).
Significant bin comparison. For each sample group, unique IS were converted to C-Start positions on a concatenation of the entire genome to ensure the maximum number of equally sized bins. An IS’s new position, L, is given by the equation:
(Equation 1)
where c is a chromosome number, m is an IS’s chromosome number, Sc is the size of chromosome c, and l is the C-Start of an IS. For this equation, the size of chromosome zero is assumed to be zero, and chromosomes X and Y are coded as chromosomes 23 and 24, respectively. Chromosome sizes were obtained from the Genome Reference Consortium (https://www.ncbi.nlm.nih.gov/grc/human/data). IS that mapped to unincorporated contigs were not included in this analysis. As an example, in the HIV in vitro data set, there is an IS at chr4: 55,428,089. To determine its new position, L, we summed the sizes of all preceding chromosomes (chr1, chr2, chr3) and added the C-Start, or:
(Equation 2)
The bin number, B, in which the new position falls is given by the equation:
(Equation 3)

where W is the window size (or number of bp in each bin). Using our previous example and a window size of 25 kb, the bin number, B, is the ceiling of the quotient obtained from dividing the new position, L, by the window size, W, or:
(Equation 4)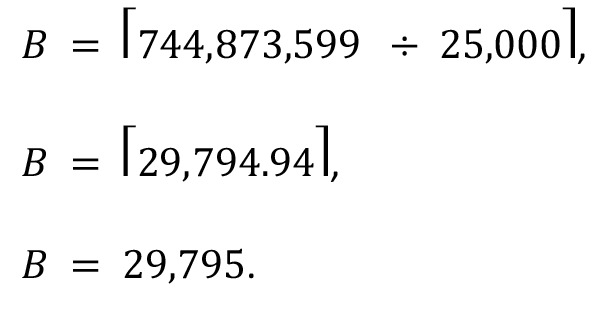
The number of IS falling within each bin of the concatenated genome was then counted for each sample group. The normalized frequency of integrations for a given bin B, f(B), was calculated using the equation:
(Equation 5)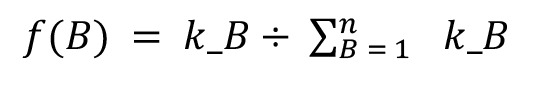
where kB is the number of IS falling within a given bin B, and n is the number of bins in the linearized genome. For example, in the bin identified previously, B = 29,795, there were a total of 3 unique IS, a total of 123,531 bins, and 7,295 unique IS fell within those bins; therefore:
(Equation 6)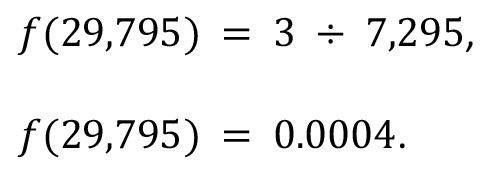
Once the normalized frequencies were calculated for the 2 samples of interest, a bootstrap analysis was conducted to estimate which bins have differential frequencies falling outside the expected distribution. First, any bins with zero IS in both samples were removed from the analysis because their inclusion can cause slight differences to appear significant if the number of bins with integrations is small relative to the total number of bins in the concatenated genome (data not shown). The difference in frequency, d, or distance between a given bin in the first sample, B1, and a given bin in the second sample, B2, is given by the equation:
(Equation 7)
The upper and lower limits, D, of the expected distribution were calculated as the points falling 3 SDs above or below the mean frequency difference (where 99% of frequencies should fall) and is given by the equation:
(Equation 8)
For the actual bootstrapping, the boot function in R was used (https://cran.r-project.org/web/packages/boot/boot.pdf). The boot function allowed for the construction of simulated comparisons. A simulated data set was constructed for each sample by randomly sampling from actual bin frequencies, f(B), for each sample until a full set of bin frequencies was obtained. The synthetic sets of bin frequencies were used to calculate new estimates of the upper and lower limits, D, of the expected distribution of differential frequencies. Using the boot function, 2,000 simulations were conducted for both the upper and lower limits. The limits reported in the figures represent the mean value of the 2,000 simulations for the upper and lower limits. For the figures, only differential bin values falling above or below those extremes are plotted.
Annotating IS. We compared the genomic locations of all HIV IS to the latest human genome (Genome Reference Consortium human build 38 [GRCh38]) RefSeq gene list available from UCSC genome browser. This was achieved using a custom python script. Several attributes of each gene in the RefSeq list were memorized: the NCBI gene name, chromosome, strand, transcription start site, transcription end site, exon count, exon and intron positions, and alternate gene name. For each sample group, the genomic location of each IS was compared with the data obtained from the RefSeq file. Integrations were annotated with distance to the nearest transcription start site, with NCBI and alternate gene names for the gene with the nearest transcription start site, and whether the IS falls within a gene or not. If the IS falls within a gene, some additional information is recorded: the NCBI and alternate gene names for the genes in which the IS falls within, from which strand (forward or reverse) the genes are transcribed, whether the IS is within an intron or exon, and which intron or exon the integration falls within.
DAVID analysis. For each sample group, the annotated integrations were combined and used as input in a custom Java script to identify and tally all IS that occurred within genes. For each gene, basic statistics were calculated from the integrations falling within the gene: the highest, average, and median number of genomically aligned reads and read fragment lengths; the percentage of integrations located on the forward and reverse strands; and the percentage of integrations falling within introns and exons. COSMIC database (https://cancer.sanger.ac.uk/cosmic) was used to identify oncogenic genes. Each output file was sorted on highest average read fragment length, and the top 1,000 genes were used as input gene lists in the DAVID Bioinformatics tool (https://david.ncifcrf.gov/summary.jsp) (29, 30). Gene ontology information concerning significant biological pathways was attained through the Functional Annotation Tool.
Circos plot generation. A custom python script was used to split each chromosome in the human genome into consecutive 25 kb regions. Additionally, for each sample group, the number of integration events that occurred in each bin was recorded. The output of this script was then used as input to Circos (http://circos.ca/) to create dual histogram plots. An additional Circos plot was created to visualize the presence or absence of enriched bins identified using the bin comparison method.
Statistics. Statistical analysis was performed as described in detail in previous methods sections. For the transduction filter applied to samples, statistics were performed in R; for significant bin comparisons, statistics were performed in python scripts; and for DAVID analysis, statistics were performed within the publically available analysis package and provided as P values, which were considered significance starting at P < 0.01. When t tests were used for statistical analysis, they were performed as unpaired, 2-tailed t tests.
Study approval. All animal studies were carried out in compliance with approved protocol number 1864 by the IACUC at the Fred Hutchinson Cancer Research Center.
Author contributions
HPK is the principal investigator of the study and coordinated the overall execution of the project. KGH conceived, designed, coordinated, and executed the experiments. LES and ZKN wrote all bioinformatics scripts used in data analysis, LES created visual representation of data including Circos plot outputs, and ZKN created the bin comparison method. CI processed mouse and culture samples for analysis and prepared samples for sequencing. JEA provided critical feedback for experimental design and data analysis, as well as provided lab space and reagents for sample processing. KGH wrote the manuscript, which was critically reviewed by JEA and HPK.
Supplementary Material
Acknowledgments
We thank Helen Crawford for help preparing and formatting this manuscript. We also thank Sarah Weitz, Melissa Comstock, and Don Parrilla for assistance with performing many of the mouse procedures and general colony maintenance, Biswajit Paul for assistance with the in vitro HIV infections, Cristina McAllister for sample processing and preparation, Daniel Humphrys for assistance with ISA processing, and Martin Wohlfahrt and Donald Gisch for producing the lentiviral vector controls used in this manuscript. We also thank all former Kiem lab members who contributed to IS processing and analysis from hematopoietic-derived cells that was used in this manuscript, as well as the Comparative Medicine staff at Fred Hutchinson Cancer Research Center for support with vivarium maintenance. HPK is a Markey Molecular Medicine Investigator and received support as the inaugural recipient of the Jose Carreras/E. Donnall Thomas Endowed Chair for Cancer Research and the Endowed Chair for Cell and Gene Therapy. This work was supported by HPK’s endowment fund.
Version 1. 07/12/2018
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2018;3(12):e99127. https://doi.org/10.1172/jci.insight.99127.
Contributor Information
Christina Ironside, Email: cironsid@fhcrc.org.
Hans-Peter Kiem, Email: hkiem@fhcrc.org.
References
- 1.Gallo RC, et al. Isolation of human T-cell leukemia virus in acquired immune deficiency syndrome (AIDS) Science. 1983;220(4599):865–867. doi: 10.1126/science.6601823. [DOI] [PubMed] [Google Scholar]
- 2.Barré-Sinoussi F, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS) Science. 1983;220(4599):868–871. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- 3.Fischer M, et al. HIV RNA in plasma rebounds within days during structured treatment interruptions. AIDS. 2003;17(2):195–199. doi: 10.1097/00002030-200301240-00009. [DOI] [PubMed] [Google Scholar]
- 4.Harrigan PR, Whaley M, Montaner JS. Rate of HIV-1 RNA rebound upon stopping antiretroviral therapy. AIDS. 1999;13(8):F59–F62. doi: 10.1097/00002030-199905280-00001. [DOI] [PubMed] [Google Scholar]
- 5.Taylor S, Boffito M, Khoo S, Smit E, Back D. Stopping antiretroviral therapy. AIDS. 2007;21(13):1673–1682. doi: 10.1097/QAD.0b013e3281c61394. [DOI] [PubMed] [Google Scholar]
- 6.Henrich TJ, et al. Antiretroviral-free HIV-1 remission and viral rebound after allogeneic stem cell transplantation: report of 2 cases. Ann Intern Med. 2014;161(5):319–327. doi: 10.7326/M14-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peterson CW, et al. Lack of viral control and development of combination antiretroviral therapy escape mutations in macaques after bone marrow transplantation. AIDS. 2015;29(13):1597–1606. doi: 10.1097/QAD.0000000000000702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Craigie R, Bushman FD. HIV DNA integration. Cold Spring Harb Perspect Med. 2012;2(7):a006890. doi: 10.1101/cshperspect.a006890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mullins JI, Frenkel LM. Clonal Expansion of Human Immunodeficiency Virus-Infected Cells and Human Immunodeficiency Virus Persistence During Antiretroviral Therapy. J Infect Dis. 2017;215(suppl_3):S119–S127. doi: 10.1093/infdis/jiw636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siliciano RF, Greene WC. HIV latency. Cold Spring Harb Perspect Med. 2011;1(1):a007096. doi: 10.1101/cshperspect.a007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schröder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110(4):521–529. doi: 10.1016/S0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 12.Maldarelli F, et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science. 2014;345(6193):179–183. doi: 10.1126/science.1254194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wagner TA, et al. HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science. 2014;345(6196):570–573. doi: 10.1126/science.1256304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hughes SH, Coffin JM. What Integration Sites Tell Us about HIV Persistence. Cell Host Microbe. 2016;19(5):588–598. doi: 10.1016/j.chom.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohn LB, et al. HIV-1 integration landscape during latent and active infection. Cell. 2015;160(3):420–432. doi: 10.1016/j.cell.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lusic M, Marcello A, Cereseto A, Giacca M. Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 2003;22(24):6550–6561. doi: 10.1093/emboj/cdg631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malim MH, Emerman M. HIV-1 accessory proteins--ensuring viral survival in a hostile environment. Cell Host Microbe. 2008;3(6):388–398. doi: 10.1016/j.chom.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 18.Goedert JJ. The epidemiology of acquired immunodeficiency syndrome malignancies. Semin Oncol. 2000;27(4):390–401. [PubMed] [Google Scholar]
- 19.Grogg KL, Miller RF, Dogan A. HIV infection and lymphoma. J Clin Pathol. 2007;60(12):1365–1372. doi: 10.1136/jcp.2007.051953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Epeldegui M, Vendrame E, Martínez-Maza O. HIV-associated immune dysfunction and viral infection: role in the pathogenesis of AIDS-related lymphoma. Immunol Res. 2010;48(1-3):72–83. doi: 10.1007/s12026-010-8168-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alexaki A, Liu Y, Wigdahl B. Cellular reservoirs of HIV-1 and their role in viral persistence. Curr HIV Res. 2008;6(5):388–400. doi: 10.2174/157016208785861195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dahabieh MS, Battivelli E, Verdin E. Understanding HIV latency: the road to an HIV cure. Annu Rev Med. 2015;66:407–421. doi: 10.1146/annurev-med-092112-152941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang GP, Ciuffi A, Leipzig J, Berry CC, Bushman FD. HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 2007;17(8):1186–1194. doi: 10.1101/gr.6286907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holt N, et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol. 2010;28(8):839–847. doi: 10.1038/nbt.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lepus CM, et al. Comparison of human fetal liver, umbilical cord blood, and adult blood hematopoietic stem cell engraftment in NOD-scid/gammac-/-, Balb/c-Rag1-/-gammac-/-, and C.B-17-scid/bg immunodeficient mice. Hum Immunol. 2009;70(10):790–802. doi: 10.1016/j.humimm.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patton J, Vuyyuru R, Siglin A, Root M, Manser T. Evaluation of the efficiency of human immune system reconstitution in NSG mice and NSG mice containing a human HLA.A2 transgene using hematopoietic stem cells purified from different sources. J Immunol Methods. 2015;422:13–21. doi: 10.1016/j.jim.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Q, Chen CY, Yedavalli VS, Jeang KT. NEAT1 long noncoding RNA and paraspeckle bodies modulate HIV-1 posttranscriptional expression. MBio. 2013;4(1):e00596–e00512. doi: 10.1128/mBio.00596-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 30.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kiselinova M, De Spiegelaere W, Vandekerckhove L. The use of HIV-1 integration site analysis information in clinical studies aiming at HIV cure. J Virus Erad. 2016;2(3):175–176. [PMC free article] [PubMed] [Google Scholar]
- 32.Maldarelli F. The role of HIV integration in viral persistence: no more whistling past the proviral graveyard. J Clin Invest. 2016;126(2):438–447. doi: 10.1172/JCI80564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maldarelli F. HIV-infected cells are frequently clonally expanded after prolonged antiretroviral therapy: implications for HIV persistence. J Virus Erad. 2015;1(4):237–244. [PMC free article] [PubMed] [Google Scholar]
- 34.Mahon FX. JAK the trigger. Oncogene. 2005;24(48):7125–7126. doi: 10.1038/sj.onc.1208885. [DOI] [PubMed] [Google Scholar]
- 35.Futreal PA, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4(3):177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Francis AC, Di Primio C, Allouch A, Cereseto A. Role of phosphorylation in the nuclear biology of HIV-1. Curr Med Chem. 2011;18(19):2904–2912. doi: 10.2174/092986711796150478. [DOI] [PubMed] [Google Scholar]
- 37.Arora S, Verma S, Banerjea AC. HIV-1 Vpr redirects host ubiquitination pathway. J Virol. 2014;88(16):9141–9152. doi: 10.1128/JVI.00619-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karn J, Stoltzfus CM. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb Perspect Med. 2012;2(2):a006916. doi: 10.1101/cshperspect.a006916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamp W, Berk MB, Visser CJ, Nottet HS. Mechanisms of HIV-1 to escape from the host immune surveillance. Eur J Clin Invest. 2000;30(8):740–746. doi: 10.1046/j.1365-2362.2000.00697.x. [DOI] [PubMed] [Google Scholar]
- 40.Geiss GK, et al. Large-scale monitoring of host cell gene expression during HIV-1 infection using cDNA microarrays. Virology. 2000;266(1):8–16. doi: 10.1006/viro.1999.0044. [DOI] [PubMed] [Google Scholar]
- 41.Kok YL, et al. Monocyte-derived macrophages exhibit distinct and more restricted HIV-1 integration site repertoire than CD4(+) T cells. Sci Rep. 2016;6:24157. doi: 10.1038/srep24157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ali ASM, et al. Targeting Deficiencies in the TLR5 Mediated Vaginal Response to Treat Female Recurrent Urinary Tract Infection. Sci Rep. 2017;7(1):11039. doi: 10.1038/s41598-017-10445-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ikeda T, Shibata J, Yoshimura K, Koito A, Matsushita S. Recurrent HIV-1 integration at the BACH2 locus in resting CD4+ T cell populations during effective highly active antiretroviral therapy. J Infect Dis. 2007;195(5):716–725. doi: 10.1086/510915. [DOI] [PubMed] [Google Scholar]
- 44.Satou Y, et al. Dynamics and mechanisms of clonal expansion of HIV-1-infected cells in a humanized mouse model. Sci Rep. 2017;7(1):6913. doi: 10.1038/s41598-017-07307-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gartner S, Markovits P, Markovitz DM, Kaplan MH, Gallo RC, Popovic M. The role of mononuclear phagocytes in HTLV-III/LAV infection. Science. 1986;233(4760):215–219. doi: 10.1126/science.3014648. [DOI] [PubMed] [Google Scholar]
- 46.Adachi A, et al. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59(2):284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koyanagi Y, Miles S, Mitsuyasu RT, Merrill JE, Vinters HV, Chen IS. Dual infection of the central nervous system by AIDS viruses with distinct cellular tropisms. Science. 1987;236(4803):819–822. doi: 10.1126/science.3646751. [DOI] [PubMed] [Google Scholar]
- 48.Lusso P, et al. Growth of macrophage-tropic and primary human immunodeficiency virus type 1 (HIV-1) isolates in a unique CD4+ T-cell clone (PM1): failure to downregulate CD4 and to interfere with cell-line-tropic HIV-1. J Virol. 1995;69(6):3712–3720. doi: 10.1128/jvi.69.6.3712-3720.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mörner A, et al. Primary human immunodeficiency virus type 2 (HIV-2) isolates, like HIV-1 isolates, frequently use CCR5 but show promiscuity in coreceptor usage. J Virol. 1999;73(3):2343–2349. doi: 10.1128/jvi.73.3.2343-2349.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ochsenbauer-Jambor C, Jones J, Heil M, Zammit KP, Kutsch O. T-cell line for HIV drug screening using EGFP as a quantitative marker of HIV-1 replication. BioTechniques. 2006;40(1):91–100. doi: 10.2144/000112072. [DOI] [PubMed] [Google Scholar]
- 51.Kutsch O, et al. Bis-anthracycline antibiotics inhibit human immunodeficiency virus type 1 transcription. Antimicrob Agents Chemother. 2004;48(5):1652–1663. doi: 10.1128/AAC.48.5.1652-1663.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haworth KG, Ironside C, Norgaard ZK, Obenza WM, Adair JE, Kiem HP. In Vivo Murine-Matured Human CD3+ Cells as a Preclinical Model for T Cell-Based Immunotherapies. Mol Ther Methods Clin Dev. 2017;6:17–30. doi: 10.1016/j.omtm.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adair JE, et al. Extended survival of glioblastoma patients after chemoprotective HSC gene therapy. Sci Transl Med. 2012;4(133):133ra57. doi: 10.1126/scitranslmed.3003425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adair JE, et al. Gene therapy enhances chemotherapy tolerance and efficacy in glioblastoma patients. J Clin Invest. 2014;124(9):4082–4092. doi: 10.1172/JCI76739. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.