

Abstract
With the development of precision medicines based on small molecules, antibodies, RNAs and gene therapy, technological innovation is providing some exciting possibilities to treat the most severe genetic diseases. However, these treatments do not always lead to a cure for the disease, and there are several factors that may hinder their overall success. Patients living during a period of great medical change and innovation may benefit from these technological advances but may also just face failures, both in terms of frustrated hopes as well as suffering. In this article, we are telling the stories of three children with rare and severe disorders, who live in an age of significant medical changes, bearing the burden of difficult scientific and ethical choices. The first two cases that are suffering respectively from severe immunodeficiency and beta thalassemia have already been described in scientific journals, as well as in popular magazines. Although similar when considering the medical challenges, the two cases had opposite outcomes, which resulted in distinct ethical implications. The third case is a baby with spinal muscular atrophy, living at a time of continued innovation in the treatment of the disease. With these cases, we discuss the challenges of providing correct information and proper counseling to families and patients that are making the bumpy journey on the road of medical innovation.
Keywords: X-severe combined immunodeficiency, Primary immunodeficiency, Beta thalassemia, Spinal muscular atrophy, Bioethics, History of medicine, Precision medicine, Genetics
Core tip: Precision therapies are heralded to change the prognosis of rare and severe genetic disorders. However, the new treatments are not always curative and what can be viewed on one hand as a partial improvement, on the other may result, at least for some patients, in prolonged suffering. During this period of change, great hopes but unpredictable outcomes can mark the lives of patients. Recalling and considering the ethical debates on historical cases can help doctors in giving the right advice to the families of patients with rare and severe disorders.
INTRODUCTION
The history of medicine is riddled with successes and failures with exciting innovations and untold suffering. The contrast between the hopes pinned on innovation and the prolonged suffering in real life is particularly reflected by the experience of suffering from complex and rare disorders. It has been argued that the timetable of research sadly does not fit the life expectancy of patients. Even when progress seems to make headway, we can identify at least three groups of patients: those who are too late to benefit from novel treatments, those who will witness the success of innovative therapies, and those who will journey on the bumpy ride of innovation too slowly, facing new medical problems and ethical concerns. Subjects in the third group call for attention, as their lives will be challenged by difficult choices, whether to accept the risks brought on by changes, often ignoring whether innovation will have results of improvement that are clinically relevant. Moreover, they must cope with the difficulties of obtaining novel medications, which may be offered to them following a label prescription or as part of clinical trials that can require extraordinary willingness to cooperate and sometimes are never granted marketing authorization[1-3]. Doctors will inform patients and/or their caregivers about the costs/benefits of the new treatments, based on the best expertise at any given moment. However, the partial medical knowledge and highly heterogeneous progression of the disease in addition to the response to treatments may hinder a reliable prediction of the disease outcome. This is particularly true for those patients with severe rare diseases. Even if regulatory agencies established robust rules to protect patients, their journey across major changes in medical knowledge and practice can be full of unforeseen difficulties.
In the first part of this article we mention two stories from the past, which both raised bioethical concerns and received media attention. For both cases, significant advances in medical care were paralleled by the personal stories of patients, who lived during remarkable scientific innovation. During this period, these patients went through moments of great hope and painful adversity. In retrospect, our judgment concerning their cases cannot be impartial to the respective clinical outcomes and the social achievements during that time, in particular concerning birth control, prenatal genetic testing, and facing the prospect of death. However, it is not our intention to judge, but just to remember how difficult it can be to make the right choice. Even if each disease is considered in its own right, in the second part of the article we discuss how these lessons learnt from history can help us to cope with new challenges, such as the medical innovation that is on its way regarding spinal muscular atrophy (SMA).
DISCUSSION CASE #1
The case
These are the days of miracle and wonder, Simon P: The bubble boy: Immunodeficiencies at the time of molecular immunology. The story of David Vetter, also known as “the bubble boy”, has been described in detail elsewhere, both in scientific manuscripts and across some of the world’s magazines, as well as in a movie (The Boy in the Plastic Bubble, 1976, by Douglas Day Stewart) and in a documentary (PBS American Experience, 2006)[4-6]. The life of the boy inside a plastic bubble was at the same time a theme for scientific and ethical debate and also a popular real show, probably offering a much more cheerful portrait than in reality.
David’s parents had their first son in 1970. Unfortunately, he died of severe combined immunodeficiency (SCID). Since there was no family history for immunodeficiency and the molecular defects underlying SCID were still unknown, it was impossible to predict the exact risk of familial recurrence of the disease. However, doctors informed the Vetters that, if they would have another child with SCID, the newborn could be delivered by cesarean section and immediately placed in a sterile environment by means of a plastic “isolator” that would protect him from infections until a cure was found. Being animated by a catholic sense of procreation and a fair trust in the doctors, the Vetters soon had a second boy, David, who was also diagnosed with SCID. The isolation of David in a sterile environment was made possible due to a dedicated research project at the Children Hospital in Houston. Despite great efforts to create a domestic environment in the bubble, the life inside could not have been so comfortable, not only because of the difficulty to interact with others, but also for the constant noise of the bubble’s blower motors, turning the room into a scientific laboratory. As a research project, it led to several scientific publications, but also to ethical debates[4,5,7-12].
It is difficult to try and interpret David’s story only from indirect data. However, much of his life was dominated by the conflict between his natural curiosity about the world and the fear of microbes, which was taught to him to decrease his desire to come out of the bubble. When he was six years old, National Aeronautics and Space Administration (NASA) made him a special spacesuit coat to allow for safe walking outside of the bubble (the so called “Mobile Biologistical Isolation System”). His reluctance to use this device only after several walks reflects the contrast between his fears and his dreams. As he grew older, he would live through even more conflicts. He soon became aware of how his life was different from his peers and how senseless it was to study things that he could not experience in reality. It does not come as a surprise that he started developing unbearable sadness and depression despite the support from Mary Murphy, a psychologist dedicated to the prevention and treatment of emotional crises. Murphy wrote a concerning tale about her experience with the boy, which was never published due to the contrariety of the parents and the medical staff. She also wrote a scientific manuscript about David’s psychological development[13].
At the age of 12 years, due to the untenable life conditions and recent medical advances, David underwent histocompatibility leukocyte antigen (HLA)-mismatched bone marrow transplantation from his older sister. Unfortunately, he developed Burkitt’s lymphoma and died a few months later in February 1984. His death was followed by a scientific and ethical debate with several contributions in JAMA[14-16]. Great attention was given to a letter from the Reverend Raymond J Lawrence, the chaplain of the Houston Children Hospital, who manifested his disappointment in the case, criticizing the excessive trust in science that had led a boy to be conceived “for research”, not considering the potential damages that a possible medical failure could cause to him[14].
It will remain a matter for debate whether the case of David represents more of a triumph for technology or an error in estimating the power of medicine. Even if ethical issues were discussed at a conference in 1975, years before the death of the boy, a different outcome after transplantation could probably have influenced everyone’s judgment. What remains true, apart from any ethical considerations on the case, is the fact that David lived through a period of great changes, both in medical science and in bioethics.
The context
This story is well placed at the beginning of a scientific revolution that witnessed memorable breakthroughs in the field of immunology. Some of his doctors even envisaged the possibility that, once the causative gene would be identified (and it would only be identified in 1993)[17] the disease could be cured with gene therapy. In Figure 1, the life of David is paralleled by a brief history of knowledge on X-SCID. Only a few babies, before David, survived SCID due to bone marrow transplantation from matched related or unrelated donors[18-21]. More recently, several reports showed that the outcome of hematopoietic stem cell transplantation has greatly improved in the last two decades. The early diagnosis, thanks to neonatal screening or based on family history, is a major determinant of this success, allowing the procedure to be performed before the development of chronic infections, which are the greatest threat to the patients’ survival[22,23]. Hematopoietic stem cell protocols have been optimized in such a way that matched unrelated donors are now considered a suitable source of stem cells, with a rate of success not much lower than for HLA-matched siblings. Gene therapy was accomplished at the turn of the third millennium, and X-SCID was one of the first applications for this technology[24]. Indeed, the observation of attenuated X-SCID phenotypes in subjects who developed T cell clones with spontaneous reversion of the genetic mutation highlighted a favorable proliferative fitness of corrected cells and predicted the success of gene therapy[25]. However, initial attempts of gene therapy were burdened by the development of leukemia due to insertional activation of the LMO2 oncogene[26]. The simultaneous improvements in the outcome of bone marrow transplantation hindered further applications of gene therapy for X-SCID until recent advances of the development of safer vectors and of the preclinical development of gene editing approaches[27,28].
Figure 1.
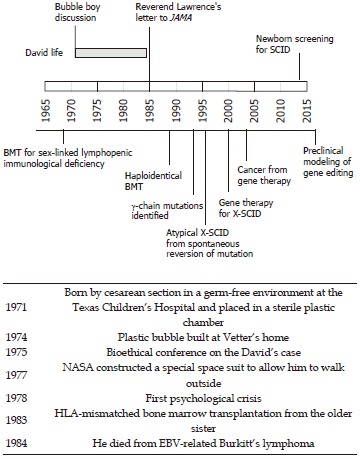
The timeline of David’s life paralleled by the most significant advances in understanding and treating X-severe combined immunodeficiency. SCID: Severe combined immunodeficiency; BMT: Bone marrow transplantation; HLA: Histocompatibility leukocyte antigen; EBV: Epstein-Barr virus; NASA: National Aeronautics and Space Administration.
CASE DISCUSSION #2
The case
You are soul’s unbreakable, Muse: The history of Khaled, an “immortal” boy. This is the history of a boy born in 1962 who was affected by severe anemia due to beta thalassemia[29,30]. His parents were Jewish and of Syrian and Iranian lineage living in Portugal. At that time, thalassemia major was invariably fatal in infancy. Subjects with the most severe form of thalassemia had only transient benefits from transfusions, which were quickly outweighed by the consequences of iron overload. Indeed, when Khaled’s parents sought the opinion of the foremost pediatric hematologist in Europe, Guido Fanconi at the Kinderspital in Zurich, he explained to them that there was no recommendation on treating the boy with transfusions. Blood transfusions could allow him to survive several years longer but there would be complications and suffering from enriched iron overload. There was also the possibility that he could have had a milder form of the disease than was first thought, and therefore he could survive without transfusions. Dr. Fanconi gave a well thought out opinion, following the best of the available expertise, based both on science and humanity. Four years later, the boy had survived, albeit with severe anemia, hepatosplenomegaly, gross bone deformities and recurrent fractures. Thus, the parents decided to seek the attention of David G Nathan, a hematologist in Boston, who was making significant progress with the treatment of thalassemia. As shown in Figure 2, patients’ survival was slowly increasing in the cohort of patients born in the late 1960s. However, there was no medication available to efficiently reduce iron overload, and thus it was hard to predict possible changes in the natural history of the disease in subsequent years. When Dr. Nathan decided to start regular transfusions, the increasing iron overload contributed to the possibility of heart failure making the treatment even more challenging. Fortunately, iron chelation was entering clinical protocols for thalassemia major, even if intramuscular administration of the drug allowed insufficient urinary excretion of the metal[31,32]. Great advances were achieved with continuous intravenous administration, and in 1977 subcutaneous delivery of the medication was implemented. Patients were required to stay at the hospital for long periods until portable infusion pumps were designed and became available. As shown in Figure 2, Khaled’s life paralleled the most significant advances in the treatment of the disease, witnessed by a continuous improvement in life expectancy in subsequent generations. Dr. Nathan[29] characterized this story in a beautiful book, showing how the life of his patient paralleled the history of medical successes and defeats (like viral infections) in thalassemia. An update of this story can be read in the Harvard magazine[30].
Figure 2.
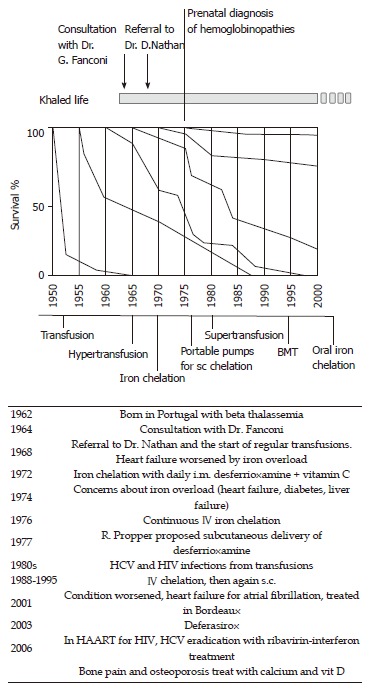
The timeline of Khaled’s life paralleled by the most significant advances in understanding and treating Beta thalassemia major. BMT: Bone marrow transplantation; HCV: Hepatitis C virus; HIV: Human immunodeficiency virus; HAART: Highly active antiretroviral therapy.
The context
The graph in Figure 2 summarizes the experience of Vullo and his collaborators, in Ferrara Italy, but it can also fit in the experience of other groups dealing with this disease[33]. Indeed, most pediatricians caring for patients with thalassemia experienced these exciting changes. Only an effective super-transfusion iron chelation regimen can completely suppress the patient’s hemopoiesis and prevent the complex cascade typical of the disease: inhibition of hemopoiesis prevented both anemia and bone deformities; iron chelation prevented organ damage to the heart, liver and endocrine glands[34-38].
Today the burden of beta thalassemia has been greatly reduced because of several factors including careful identification of the carriers, birth prevention of newborns with beta thalassemia major, early treatment of patients with blood transfusion and iron chelators, in some cases with bone marrow transplantation, and gene therapy[39]. Fortunately, the incidence of thalassemia major has decreased in countries with a higher prevalence of heterozygous individuals due to public information campaigns, population screenings, genetic counseling, and prenatal diagnosis, either voluntary or through institutional basis[40,41].
CASE DISCUSSION #3
The case
“How many roads must a man walk down before you call him a man?” Dylan B: A case of SMA at the time of a major medical progress. The story of Alessandro begins on a night in December 2015 in Italy. Following reduced fetal movements and the mother’s hypertension, he was born prematurely at 32 wk of gestation. After birth, severe hypotonia was noted. Alessandro spent his first two months of life in a neonatal intensive care unit with a breathing machine and a feeding tube to sustain his vital functions. After a hard and complicated time, he was discharged with persisting hypotonia. He never achieved enough strength to sustain his head and his parents became aware that something was wrong. At 6 mo old, he was diagnosed with SMA. At that time, no cure for SMA was available and the treatment was based on respiratory and feeding assistance (standard care, see Figure 3). At 7 mo old, the parents took part in a special program on how to assist children with SMA and learned how to manage the ordinary and extraordinary practice of their child’s daily life. At 10 mo old, Alessandro had his first breathing failure and a non-invasive ventilation (NIV) was started. This experience led his parents to understand the meaning of palliative care. They refused aggressive treatment and tracheotomy. However, in the same month Nusinersen, a new and promising drug for SMA became available, and the parents entered into the expanded access program (EAP) for this drug. At 12 mo old, Alessandro received the first intrathecal drug infusion. After the fourth infusion, positive results became evident: small limb movements reappeared, NIV was reduced from 18 h/d to 10 h/d, and the ability to swallow was effectively increased. Nusinersen infusion, a lifelong treatment, was continued for another year and albeit further small improvements were noticed. The child also took part in an advertising campaign, which aimed to gather funds for research in SMA. In January 2018, at 25 mo old, Alessandro had aspiration pneumonia. He was hospitalized for three weeks, and he lost the strength and confidence gained in the previous year. NIV was necessarily increased to 16 h/d, and a percutaneous gastrostomy was proposed to reduce the stress of eating. After 15 d, the child was hospitalized again because of a new respiratory failure. Alessandro and his parents’ state of mind completely changed in the last two months: they faced the prospect of death as they realized that SMA was getting worse despite Nusinersen. The drug was able to improve the quality of life, but it was not a cure. The last respiratory failure was crucial in the parents’ choice. With great pain and fear, but also with the peacefulness of having accepted the fatal course of the disease, the parents chose palliative care. A few days later Alessandro died without pain. His mother said, “I never allowed a cold to tear away my son, but I also never asked him to win the Olympic games just for the fear of living with torment. Love also means letting it go”.
Figure 3.
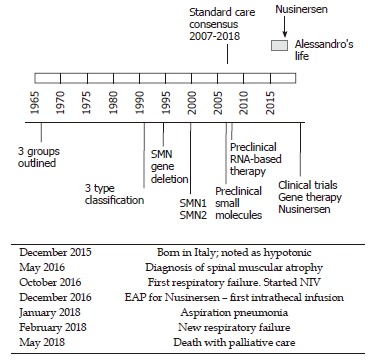
The timeline of Alessandro’s life paralleled by the most significant advances in understanding and treating spinal muscular atrophy 1. NIV: Non-invasive ventilation; EAP: Expanded access program.
The context
SMA is the most common genetic cause of infant mortality in developed countries. The first International Consensus Statement for Standard of Care in SMA in 2007 has represented a fundamental milestone in the history of SMA[42]. In fact, the large variation in the disease phenotype of SMA hinders a reliable prediction of the natural course of the disease in each case. Moreover, the existence of relevant differences in clinical care at different hospitals is a serious challenge for families in making a balanced decision. Until recent developments, almost all subjects with SMA1 died in their first year of life due to respiratory failure and infections. SMA does not affect cognitive development, making it even more painful when considering the conflict in children between their growing interests and their limited abilities. Death usually occurs after prolonged agonies in children receiving quite aggressive care with artificial nutrition and ventilation. Thus, in SMA1, this point has been and still is critical to determine the threshold for choosing between palliative care and intervention, considering caregivers beliefs and opinions, as well as their capacity to shoulder the burden of the disease[43]. Parents/caregivers must receive complete information on the possible treatment options. Facing these ethical problems is part of the experience of most pediatricians as described in the example by Gray and his collaborators[44]. To make matters even more difficult, we are now living in a period of significant medical advances for the treatment of SMA, thus the threshold for decision-making may change accordingly within the lifespan of patients[45]. For example, pharmacologic treatment with Nusinersen may improve motor skills and prolong life expectancy, but in the most severe cases it is not clear if these effects can represent a desirable benefit or therapeutic persecution. Sometimes it may just be impossible to know. Moreover, the launch of Nusinersen for SMA raised several medical concerns[46]. First, the primary outcome of the ENDEAR trial, which led to the authorization of the drug, was changed while the study was ongoing[47,48]. Even though the overall survival, free of permanent mechanical ventilation, was the original primary outcome, this was shifted during the trial to “motor milestone response”, which is defined as “more milestones improved than worsened” (considering the following milestones from the Hammersmith Infant Neurological Examination: kicking, head control, rolling, sitting, crawling, standing, and walking). The result of this change was the approval of the drug based on marginal albeit significant improvements in motor function. Based on the result of the ENDEAR trial and on other uncontrolled trials, Nusinersen was granted marketing authority for all types of SMA. However, the achievements shown in ENDEAR were not as clearly proven for all types of the disease and at any age[46]. Secondly, whilst the proof of efficacy of the drug was obtained in subjects with SMA1 who were younger than 6 mo old at the time Nusinersen was first administered, the drug had been approved for all types of the disease. Thirdly, the extremely high costs of the treatment risked making it not equitably distributable. Moreover, it is even debatable if the same amount of money could not have been used in better ways to improve the care for patients. Finally, the most serious concern is that Nusinersen does not represent a cure and in some cases may be too risky. Paradoxically, the availability of Nusinersen may have an unpredictable impact on decisions about prenatal screening. It is of note that the availability of iron chelators, increasing chronicity, and healthcare costs in beta thalassemia had a role in driving prevention programs in Cyprus, where the disease is prevalent[49]. Similarly, it could be expected that the availability of Nusinersen may increase screening programs for SMA. Some would argue that Nusinersen may increase patients’ survival to allow for better care (perhaps gene therapy) in the near future. However there is no reliable prediction about this. Thus, we must recognize that children with SMA are living today during major periods of change, like in other stories described in this article[50].
CONCLUSION
Making the best choice during periods of mounting medical advances among prenatal screening, palliative care, and aggressive treatment can be difficult when dealing with severe diseases. On the one hand, medical progress is increasing the probability of survival for children with severe genetic disorders; on the other, genetic screenings are offering a way to reduce the birth of newborns with relatively common severe disorders like cystic fibrosis, SMA, and thalassemia[51,52]. It may be difficult to find the correct direction between full fruition of life and the maintenance of some vital function with unbearable sufferance. Parents’ ethical, cultural, and religious feelings should be considered together with the best medical evidence to elaborate shared bioethical opinions in each case.
ACKNOWLEDGEMENTS
We acknowledge all the patients and families who have gone through so much, facing the challenges of coping with a rare disease, and whose entire life have been paralleled by major medical changes with the opportunities and the challenges.
Footnotes
Conflict-of-interest statement: The authors declare that they have no conflict of interest concerning the present article.
Manuscript source: Invited manuscript
Peer-review started: June 30, 2018
First decision: July 9, 2018
Article in press: August 13, 2018
Specialty type: Pediatrics
Country of origin: Italy
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B, B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
P- Reviewer: Pandey A, Teng RJ, Watanabe T S- Editor: Ji FF L- Editor: Filipodia E- Editor: Song H
Contributor Information
Alberto Tommasini, Department of Pediatrics, Institute of Maternal and Child Health, IRCCS Burlo Garofolo, Trieste 34137, Italy. alberto.tommasini@burlo.trieste.it.
Andrea Magnolato, Department of Pediatrics, Institute of Maternal and Child Health, IRCCS Burlo Garofolo, Trieste 34137, Italy.
Irene Bruno, Department of Pediatrics, Institute of Maternal and Child Health, IRCCS Burlo Garofolo, Trieste 34137, Italy.
References
- 1.Dooms M, Carvalho M. Compounded medication for patients with rare diseases. Orphanet J Rare Dis. 2018;13:1. doi: 10.1186/s13023-017-0741-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kempf L, Goldsmith JC, Temple R. Challenges of developing and conducting clinical trials in rare disorders. Am J Med Genet A. 2018;176:773–783. doi: 10.1002/ajmg.a.38413. [DOI] [PubMed] [Google Scholar]
- 3.Giannuzzi V, Landi A, Bosone E, Giannuzzi F, Nicotri S, Torrent-Farnell J, Bonifazi F, Felisi M, Bonifazi D, Ceci A. Failures to further developing orphan medicinal products after designation granted in Europe: an analysis of marketing authorisation failures and abandoned drugs. BMJ Open. 2017;7:e017358. doi: 10.1136/bmjopen-2017-017358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malinak LR, Wilson R, South MA, Montgomery JR, Mumford DM, Flowers CE Jr. Germ-free delivery. The initiation of management of infants with a high probability of congenital immune deficiency states. Am J Obstet Gynecol. 1973;116:201–204. doi: 10.1016/0002-9378(73)91051-x. [DOI] [PubMed] [Google Scholar]
- 5.Bealmear PM, South MA, Wilson R. David’s story: the gift of 12 years, 5 months, and 1 day. Prog Clin Biol Res. 1985;181:475–489. [PubMed] [Google Scholar]
- 6.Kirk RG. “Life in a germ-free world”: isolating life from the laboratory animal to the bubble boy. Bull Hist Med. 2012;86:237–275. doi: 10.1353/bhm.2012.0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mukhopadhyay N, Richie E, Mackler BF, Montgomery JR, Wilson R, Fernbach DJ, South MA. A longitudinal study of T and B lymphocytes from a three-year-old patient with severe combined immunodeficiency (SCID) in ‘gnotobiotic protection’. Exp Hematol. 1978;6:129–134. [PubMed] [Google Scholar]
- 8.Mukhopadhyay N, Richie E, Montgomery JR, Wilson R, Fernbach DJ. Letter: T and B cell characteristics in combined immunodeficiency. N Engl J Med. 1974;291:678. doi: 10.1056/NEJM197409262911313. [DOI] [PubMed] [Google Scholar]
- 9.Mukhopadhyay N, Richie E, Montgomery J, Wilson R, Fernbach DJ, South MA. Peripheral blood T and B cell characteristics in a patient with severe combined immune deficiency (SCID) maintained in a gnotobiotic environment. Exp Hematol. 1976;4:1–9. [PubMed] [Google Scholar]
- 10.Freedman DA, Montgomery JR, Wilson R, Bealmear PM, South MA. Further observations on the effect of reverse isolation from birth on cognitive and affective development. J Am Acad Child Psychiatry. 1976;15:593–603. doi: 10.1097/00004583-197601540-00001. [DOI] [PubMed] [Google Scholar]
- 11.Simmons K. ‘Bubble boy’ reacts well to marrow transplant. JAMA. 1983;250:2751. [PubMed] [Google Scholar]
- 12.South MA. David the bubble boy: some lessons he has taught us. Cutis. 1977;19:568, 572, 574. [PubMed] [Google Scholar]
- 13.Murphy MA, Vogel JB. Looking out from the isolator: David’s perception of the world. J Dev Behav Pediatr. 1985;6:118–121. [PubMed] [Google Scholar]
- 14.Lawrence RJ. David the “Bubble Boy’ and the boundaries of the human. JAMA. 1985;253:74–76. [PubMed] [Google Scholar]
- 15.Rennie D. “Bubble Boy’. JAMA. 1985;253:78–80. [PubMed] [Google Scholar]
- 16.Bubble Boy. JAMA. 1985;254:1036–1037. [PubMed] [Google Scholar]
- 17.Puck JM, Deschênes SM, Porter JC, Dutra AS, Brown CJ, Willard HF, Henthorn PS. The interleukin-2 receptor gamma chain maps to Xq13.1 and is mutated in X-linked severe combined immunodeficiency, SCIDX1. Hum Mol Genet. 1993;2:1099–1104. doi: 10.1093/hmg/2.8.1099. [DOI] [PubMed] [Google Scholar]
- 18.Gatti RA, Meuwissen HJ, Allen HD, Hong R, Good RA. Immunological reconstitution of sex-linked lymphopenic immunological deficiency. Lancet. 1968;2:1366–1369. doi: 10.1016/s0140-6736(68)92673-1. [DOI] [PubMed] [Google Scholar]
- 19.Meuwissen HJ, Gatti RA, Terasaki PI, Hong R, Good RA. Treatment of lymphopenic hypogammaglobulinemia and bone-marrow aplasia by transplantation of allogeneic marrow. Crucial role of histocompatiility matching. N Engl J Med. 1969;281:691–697. doi: 10.1056/NEJM196909252811302. [DOI] [PubMed] [Google Scholar]
- 20.Rubinstein A, Speck B, Jeannet M. Successful bone-marrow transplantation in a lymphopenic immunologic deficiency syndrome. N Engl J Med. 1971;285:1399–1402. doi: 10.1056/NEJM197112162852503. [DOI] [PubMed] [Google Scholar]
- 21.Buckley RH, Amos DB, Kremer WB, Stickel DL. Incompatible bone-marrow transplantation in lymphopenic immunologic deficiency. Circumvention of fatal graft-versus-host disease by immunologic enhancement. N Engl J Med. 1971;285:1035–1042. doi: 10.1056/NEJM197111042851901. [DOI] [PubMed] [Google Scholar]
- 22.Gennery AR, Slatter MA, Grandin L, Taupin P, Cant AJ, Veys P, Amrolia PJ, Gaspar HB, Davies EG, Friedrich W, et al. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: entering a new century, do we do better? J Allergy Clin Immunol. 2010;126:602–10.e1-11. doi: 10.1016/j.jaci.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 23.Heimall J, Logan BR, Cowan MJ, Notarangelo LD, Griffith LM, Puck JM, Kohn DB, Pulsipher MA, Parikh S, Martinez C, et al. Immune reconstitution and survival of 100 SCID patients post-hematopoietic cell transplant: a PIDTC natural history study. Blood. 2017;130:2718–2727. doi: 10.1182/blood-2017-05-781849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, Gross F, Yvon E, Nusbaum P, Selz F, Hue C, Certain S, Casanova JL, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288:669–672. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- 25.Stephan V, Wahn V, Le Deist F, Dirksen U, Broker B, Müller-Fleckenstein I, Horneff G, Schroten H, Fischer A, de Saint Basile G. Atypical X-linked severe combined immunodeficiency due to possible spontaneous reversion of the genetic defect in T cells. N Engl J Med. 1996;335:1563–1567. doi: 10.1056/NEJM199611213352104. [DOI] [PubMed] [Google Scholar]
- 26.Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, Radford I, Villeval JL, Fraser CC, Cavazzana-Calvo M, et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2003;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- 27.De Ravin SS, Wu X, Moir S, Anaya-O’Brien S, Kwatemaa N, Littel P, Theobald N, Choi U, Su L, Marquesen M, et al. Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immunodeficiency. Sci Transl Med. 2016;8:335ra57. doi: 10.1126/scitranslmed.aad8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alzubi J, Pallant C, Mussolino C, Howe SJ, Thrasher AJ, Cathomen T. Targeted genome editing restores T cell differentiation in a humanized X-SCID pluripotent stem cell disease model. Sci Rep. 2017;7:12475. doi: 10.1038/s41598-017-12750-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nathan DG. Genes, blood, and courage : a boy called Immortal Sword. Cambridge, Mass. Harvard University Press; 1995. [Google Scholar]
- 30.Nathan DG. Lessons from an Unexpected Life. Harvard Magazine. Boston: Harvard University; 2009. pp. 36–41. [Google Scholar]
- 31.McDonald R. Deferoxamine and diethylenetriaminepentaacetic acid (DTPA) in thalassemia. J Pediatr. 1966;69:563–571. doi: 10.1016/s0022-3476(66)80041-0. [DOI] [PubMed] [Google Scholar]
- 32.Diwany M, Gabr M, el Hefni A, Mokhtar N. Desferrioxamine in thalassaemia. Arch Dis Child. 1968;43:340–343. doi: 10.1136/adc.43.229.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borgna-Pignatti C, Rugolotto S, De Stefano P, Piga A, Di Gregorio F, Gamberini MR, Sabato V, Melevendi C, Cappellini MD, Verlato G. Survival and disease complications in thalassemia major. Ann N Y Acad Sci. 1998;850:227–231. doi: 10.1111/j.1749-6632.1998.tb10479.x. [DOI] [PubMed] [Google Scholar]
- 34.Propper RD, Button LN, Nathan DG. New approaches to the transfusion management of thalassemia. Blood. 1980;55:55–60. [PubMed] [Google Scholar]
- 35.SMITH CH, SCHULMAN I, ANDO RE, STERN G. Studies in Mediterranean (Cooley’s) anemia. II. The suppression of hematopoiesis by transfusions. Blood. 1955;10:707–717. [PubMed] [Google Scholar]
- 36.Cazzola M, De Stefano P, Ponchio L, Locatelli F, Beguin Y, Dessi C, Barella S, Cao A, Galanello R. Relationship between transfusion regimen and suppression of erythropoiesis in beta-thalassaemia major. Br J Haematol. 1995;89:473–478. doi: 10.1111/j.1365-2141.1995.tb08351.x. [DOI] [PubMed] [Google Scholar]
- 37.De Sanctis V, Roos M, Gasser T, Fortini M, Raiola G, Galati MC; Italian Working Group on Endocrine Complications in Non-Endocrine Diseases. Impact of long-term iron chelation therapy on growth and endocrine functions in thalassaemia. J Pediatr Endocrinol Metab. 2006;19:471–480. [PubMed] [Google Scholar]
- 38.Olivieri NF, Brittenham GM, McLaren CE, Templeton DM, Cameron RG, McClelland RA, Burt AD, Fleming KA. Long-term safety and effectiveness of iron-chelation therapy with deferiprone for thalassemia major. N Engl J Med. 1998;339:417–423. doi: 10.1056/NEJM199808133390701. [DOI] [PubMed] [Google Scholar]
- 39.Thompson AA, Walters MC, Kwiatkowski J, Rasko JEJ, Ribeil JA, Hongeng S, Magrin E, Schiller GJ, Payen E, Semeraro M, et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N Engl J Med. 2018;378:1479–1493. doi: 10.1056/NEJMoa1705342. [DOI] [PubMed] [Google Scholar]
- 40.Cao A, Furbetta M, Galanello R, Melis MA, Angius A, Ximenes A, Rosatelli C, Ruggeri R, Addis M, Tuveri T, et al. Prevention of homozygous beta-thalassemia by carrier screening and prenatal diagnosis in Sardinia. Am J Hum Genet. 1981;33:592–605. [PMC free article] [PubMed] [Google Scholar]
- 41.Angastiniotis M, Kyriakidou S, Hadjiminas M. The Cyprus Thalassemia Control Program. Birth Defects Orig Artic Ser. 1988;23:417–432. [PubMed] [Google Scholar]
- 42.Wang CH, Finkel RS, Bertini ES, Schroth M, Simonds A, Wong B, Aloysius A, Morrison L, Main M, Crawford TO, et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22:1027–1049. doi: 10.1177/0883073807305788. [DOI] [PubMed] [Google Scholar]
- 43.Mercuri E, Bertini E, Iannaccone ST. Childhood spinal muscular atrophy: controversies and challenges. Lancet Neurol. 2012;11:443–452. doi: 10.1016/S1474-4422(12)70061-3. [DOI] [PubMed] [Google Scholar]
- 44.Gray K, Isaacs D, Kilham HA, Tobin B. Spinal muscular atrophy type I: do the benefits of ventilation compensate for its burdens? J Paediatr Child Health. 2013;49:807–812. doi: 10.1111/jpc.12386. [DOI] [PubMed] [Google Scholar]
- 45.van der Ploeg AT. The Dilemma of Two Innovative Therapies for Spinal Muscular Atrophy. N Engl J Med. 2017;377:1786–1787. doi: 10.1056/NEJMe1712106. [DOI] [PubMed] [Google Scholar]
- 46.Burgart AM, Magnus D, Tabor HK, Paquette ED, Frader J, Glover JJ, Jackson BM, Harrison CH, Urion DK, Graham RJ, et al. Ethical Challenges Confronted When Providing Nusinersen Treatment for Spinal Muscular Atrophy. JAMA Pediatr. 2018;172:188–192. doi: 10.1001/jamapediatrics.2017.4409. [DOI] [PubMed] [Google Scholar]
- 47.Gerrity MS, Prasad V, Obley AJ. Concerns About the Approval of Nusinersen Sodium by the US Food and Drug Administration. JAMA Intern Med. 2018;178:743–744. doi: 10.1001/jamainternmed.2018.0869. [DOI] [PubMed] [Google Scholar]
- 48.inkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, Chiriboga CA, Saito K, Servais L, Tizzano E, et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med. 2017;377:1723–1732. doi: 10.1056/NEJMoa1702752. [DOI] [PubMed] [Google Scholar]
- 49.Ashiotis T, Zachariadis Z, Sofroniadou K, Loukopoulos D, Stamatoyannopoulos G. Thalassaemia in Cyprus. Br Med J. 1973;2:38–42. doi: 10.1136/bmj.2.5857.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Groen EJN, Talbot K, Gillingwater TH. Advances in therapy for spinal muscular atrophy: promises and challenges. Nat Rev Neurol. 2018;14:214–224. doi: 10.1038/nrneurol.2018.4. [DOI] [PubMed] [Google Scholar]
- 51.Boardman FK, Sadler C, Young PJ. Newborn genetic screening for spinal muscular atrophy in the UK: The views of the general population. Mol Genet Genomic Med. 2018;6:99–108. doi: 10.1002/mgg3.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Archibald AD, Smith MJ, Burgess T, Scarff KL, Elliott J, Hunt CE, Barns-Jenkins C, Holt C, Sandoval K, Siva Kumar V, et al. Reproductive genetic carrier screening for cystic fibrosis, fragile X syndrome, and spinal muscular atrophy in Australia: outcomes of 12,000 tests. Genet Med. 2018;20:513–523. doi: 10.1038/gim.2017.134. [DOI] [PubMed] [Google Scholar]