

Abstract
Children with ependymoma (EPN) are cured in less than 50% of cases, with little improvement in outcome over the last several decades. Chemotherapy has not impacted survival in EPN, due in part to a lack of preclinical models that has precluded comprehensive drug testing. We recently developed two human EPN cell lines harboring high-risk phenotypes which provided us with an opportunity to execute translational studies. EPN and other pediatric brain tumor cell lines were subject to a large-scale comparative drug screen of FDA-approved oncology drugs for rapid clinical application. The results of this in vitro study were combined with in silico prediction of drug sensitivity to identify EPN-selective compounds, which were validated by dose curve and time course modelling. Mechanisms of EPN-selective antitumor effect were further investigated using transcriptome and proteome analyses. We identified three classes of oncology drugs that showed EPN-selective anti-tumor effect, namely (i) fluorinated pyrimidines (5-fluorouracil, carmofur and floxuridine), (ii) retinoids (bexarotene, tretinoin and isotretinoin), and (iii) a subset of small-molecule multi-receptor tyrosine kinase inhibitors (axitinib, imatinib and pazopanib). Axitinib’s anti-tumor mechanism in EPN cell lines involved inhibition of PDGFRα and PDGFRβ, and was associated with reduced mitosis-related gene expression and cellular senescence. The clinically available, EPN-selective oncology drugs identified by our study have the potential to critically inform design of upcoming clinical studies in EPN, in particular for those children with recurrent EPN who are in the greatest need of novel therapeutic approaches.
Keywords: Ependymoma, drug screen, floxuridine, retinoids, axitinib
Introduction
Ependymoma (EPN) is the third most common pediatric brain tumor and remains incurable in over 50% of cases (1,2). Pediatric EPNs are comprised of a number of molecular subtypes, the most common being posterior fossa groups A (PFA) and B (PFB), and C11orf95-RELA fusion supratentorial, each with a distinct prognosis(1,3–5). Chemotherapy has not impacted survival in any EPN subtype, and lack of preclinical models has precluded comprehensive drug testing (6,7). Treatment of EPN has changed little over the last 30 years and standard treatment at presentation is limited to maximal surgery and radiation. In those patients who recur, outcome is dismal particularly for those high-risk patients whose tumors harbor gains of chromosome 1q and/or PFA molecular characteristics(5,8,9). Identification of novel therapeutic approaches for children with recurrent EPN is therefore critically needed.
A major challenge in advancing therapy for EPN is to make links between subtypes and drugs that can be used to stratify patient treatment, leading to improved outcomes and decreased toxicity. Atkinson et al. advanced this effort by establishing an EphB2-driven genetic mouse model of supratentorial EPN that was then subjected to high-throughput screening using a panel of 5303 unique compounds(10). Their preclinical study identified fluorinated pyrimidines (5-flurouracil (5-FU), floxuridine and carmofur) as EPN-selective compounds, and these results were subsequently used to drive a clinical trial of 5-FU in children with recurrent EPN(11).
In the present study, we extend pre-clinical testing into high-risk human posterior fossa EPN. Our laboratory recently succeeded in establishing 2 human EPN cell lines, derived from recurrent intracranial metastases of posterior fossa EPN(12). These cell lines each harbor two high-risk phenotypes, namely chromosome 1q gain and PFA subgroup characteristics. By subjecting these cell lines to screening of Food and Drug Administration (FDA)-approved oncology drugs, we intended to identify immediately actionable EPN-selective treatments that are urgently needed for upcoming clinical trials, in particular for those children whose high-risk posterior fossa EPN have recurred.
Materials and methods
Cell lines
This study utilized 2 EPN cell lines that were recently established and subjected to extensive characterization by our laboratory(12). Both cell lines (MAF811 and MAF928, hereafter 811 and 928 respectively) were established from metastatic posterior fossa EPN recurrences. Other pediatric brain tumor cell lines included in this study for comparative purposes were atypical teratoid/rhabdoid tumor (ATRT) cell lines: 794, 737, BT12 and BT16; medulloblastoma (MED) cell lines: DAOY, D458, ONS76 and 1097; and high-grade glioma (HGG) cell lines: (diffuse intrinsic pontine gliomas DIPG-IV, DIPG-VI and glioblastoma SF188). Cell lines were obtained as described previously(12–15) and 1097 was established from a primary patient sample (tumor type confirmed by molecular testing). All cell lines were subjected to routine authentication by DNA fingerprinting. For this study cell lines were propagated under standard tissue culture conditions and grown in fetal bovine serum-supplemented media as monolayers, apart from semi-adherent suspension cell line D458.
In vitro comparative drug screen
We screened a drug library that included 97 FDA approved anticancer active drugs (Selleck Chemicals, Houston, TX) (supplementary data). The panel included 45 cytotoxics, 25 kinase inhibitors, and 15 anti-hormonal/steroidal modulators. Cell lines were plated at ~50% confluence in 96-well plates, and treated with each drug at 1μM for 72 hours alongside untreated controls. Potency of each drug was measured using a tritiated thymidine incorporation proliferation assay as described previously(13). Cells were pulsed with 0.5mCi/ml tritiated thymidine for the final 24 hours of drug treatment. Treatment effect for each drug, measured as tritium scintillation counts per minute (cpm), were converted to “percent effect” ((Treatment cpm-background cpm)/(untreated cpm-background cpm)x100). The screen was repeated in EPN cell lines 811 and 928 to give 6 replicates for each cell line, and comparator cell lines treatments were repeated at least once to obtain a minimum of 5 replicates for each tumor type (ATRT, MED and HGG). The difference between average percent effect and significance of this difference (Student’s t-test) for both EPN cell line replicates combined (n=12) versus the average percent effect of all other pediatric brain tumor cell line replicates combined (n=23) was measured in order to identify the most EPN-selective and non-selective treatments.
In silico comparative drug screen
Known protein targets of FDA approved oncology drugs, as published in the literature and compiled in Ingenuity’s KnowledgeBase (Ingenuity Systems, Qiagen, Redwood City, CA), were identified. Expression levels of RNA transcripts corresponding to these protein targets (Supplementary data) were extracted from transcriptomic profiles of the EPN, ATRT, MED and HGG cell lines panel, previously generated (GEO dataset GSE86574) using HG-U133plus2 gene expression microarray analysis (Affymetrix, ThermoFisher Scientific, Waltham, MA) (12). Using a similar approach to the in vitro screen, we compared drug target gene expression in EPN to other tumor types in order to identify EPN-selective and EPN non-selective targets. For each target gene, the difference between average gene expression and significance of this difference (Student’s t-test) was measured for both EPN cell line replicates combined (n=6) versus the average gene expression of all other pediatric brain tumor cell line replicates combined (n=17). To compare expression of select drug targets in PFA patient samples and human pediatric autopsy cerebellum, we extracted specific transcripts (RARB, Affymetrix probeset 205080_at; PDGFRB, 202273_at; KDR(VEGFR2), 203934_at; VEGFC, 209946_at) from previously published data (GEO dataset GSE86574; Affymetrix HG-U133plus2 microarray) (16)
Dose effect curve modelling
To further validate the results of in vitro and in silico comparative drug sensitivity analyses, we generated dose curves for representative drugs from each of the 3-classes of agents that were shown to have EPN-selective potency. We treated EPN and other pediatric brain tumor cell lines with a range of doses of floxuridine, bexarotene and axitinib for exposure times that were normalized according to cell line doubling times (EPN 811 and 928 = 72 hours; BT12, DAOY, SF188, DIPG-IV and ONS = 24 hours). Cell proliferation was measured as above using tritiated thymidine incorporation. Treatment dose curves were modelled using Prism statistical software (GraphPad Software, Inc., La Jolla, CA).
IncuCyte growth monitoring
EPN 811 and 928 cells were seeded at ~25% confluence in 96-well plates (Costar, Corning, NY). CellEvent Caspase-3/7 Green Detection Reagent (Life Technologies) was added to track apoptosis. Cells were cultured at 37° and 5% CO2 and monitored using an IncuCyte Zoom live cell imaging system (Essen BioScience, Ann Arbor, MI). After 24 hrs cells were treated with the same drug dose ranges used for dose effect curve modelling described above. Images were captured at 4 hour intervals from four separate regions per well using a 10x objective over 4 to 10 days dependent on cell type and drug response kinetics. Each experiment was done in triplicate and growth curves were created from percent confluence and green object count (caspase 3/7 activity) measurements normalized to confluence at time of treatment (t=0).
Transcriptomic effect of axitinib treatment
EPN cell lines 811 and 928 were treated for 24 hours with axitinib at an approximate IC-50 dose as determined by dose curve analysis (500nM and 150nM respectively) after which RNA was extracted using an Allprep kit (Qiagen) per manufacturer’s instructions. Transcriptomic profiles from these and untreated controls were generated using HG-U133plus gene expression microarrays (Affymetrix) as described previously(12). Gene set enrichment analysis (GSEA) was used to examine enrichment of genes in predefined reference genesets available from the Broad Institute Molecular Signatures Database (MSigDB) (http://www.broadinstitute.org/gsea/msigdb)(17).
Cellular Senescence Analysis
EPN cell lines, at ~30% confluence, were treated with 1μM axitinib for 72 hours at which point we performed β-galactosidase staining in order to measure cellular senescence (14). Briefly, cells were fixed and stained using a senescence-associated-β-galactosidase kit (SA-β-gal, Cell Signaling Technology, Danvers, MA) per manufacturer’s instructions. Senescent cells (dark blue staining) and non-senescent cells (no staining) were enumerated microscopically. Twelve high-power fields per sample were counted in 3 replicates to quantify the percentage of senescent cells in treated and untreated samples.
Proteome profiler arrays
To investigate the activation/phosphorylation of receptor tyrosine kinases (RTKs), we used the Human Phospho-RTK antibody array (R&D Systems, Minneapolis, USA). The Human Phospho-RTK Antibody Array is a nitrocellulose membrane where forty-nine different anti-RTK antibodies have been spotted in duplicate, including four positive controls and five negative controls. To generate protein lysates for the proteome profiler array experiment, EPN and other cell lines were harvested per manufacturer’s instructions. Briefly, cells were washed in PBS then homogenized in lysis buffer using a rotostator. Cell lysates were gently rocked for 30 min at 4°C and then centrifuged at 14,000×g for 5 min (4°C), and the supernatants removed and stored at −80°C until use. A total of 250 μg of protein, as determined by the BCA assay, was used for each array and developed using chemiluminescent reagent provided. Positive signals on developed film were analyzed using a transmission mode scanner and collected as pixel density using Image J software (https://imagej.nih.gov/ij).
Results
In vitro comparative drug screen
Cancer cell line panels are important tools to characterize the in vitro activity of available oncology drugs. Here, we present a comparative drug sensitivity analysis to identify EPN-selective oncology drugs. We generated inhibition profiles of 97 FDA-approved anticancer agents using proliferation assays in 2 novel genetically-characterized EPN cell lines and a panel of comparator cell lines established from other pediatric brain tumor types.
Treatment effect for each drug at a 1μM dose for 72 hours on EPN cell lines 811 and 928 was compared to the effect in comparator cell lines established from ATRT (794, 737, BT12), MED (DAOY, D458) and high grade glioma (DIPG-IV, SF188). Drugs were ranked according to the difference in treatment effect and between these two groups to identify EPN-selective, i.e. those drugs that are more effective in EPN, and EPN non-selective drugs, i.e. those drugs that show less effect in EPN than others (Fig. 1A). Comparative drug effects were further analyzed to identify those drugs that were significantly different (p<0.05). Using this approach we identified 9 of the 97 drugs that were significantly more effective in EPN than others corresponding to 3 drug classes – fluorinated pyrimidines, retinoids and a subset of receptor tyrosine kinase inhibitors (Table 1A).
Figure 1. Identification of EPN-selective FDA-approved oncology drug candidates using in vitro and in silico screening.
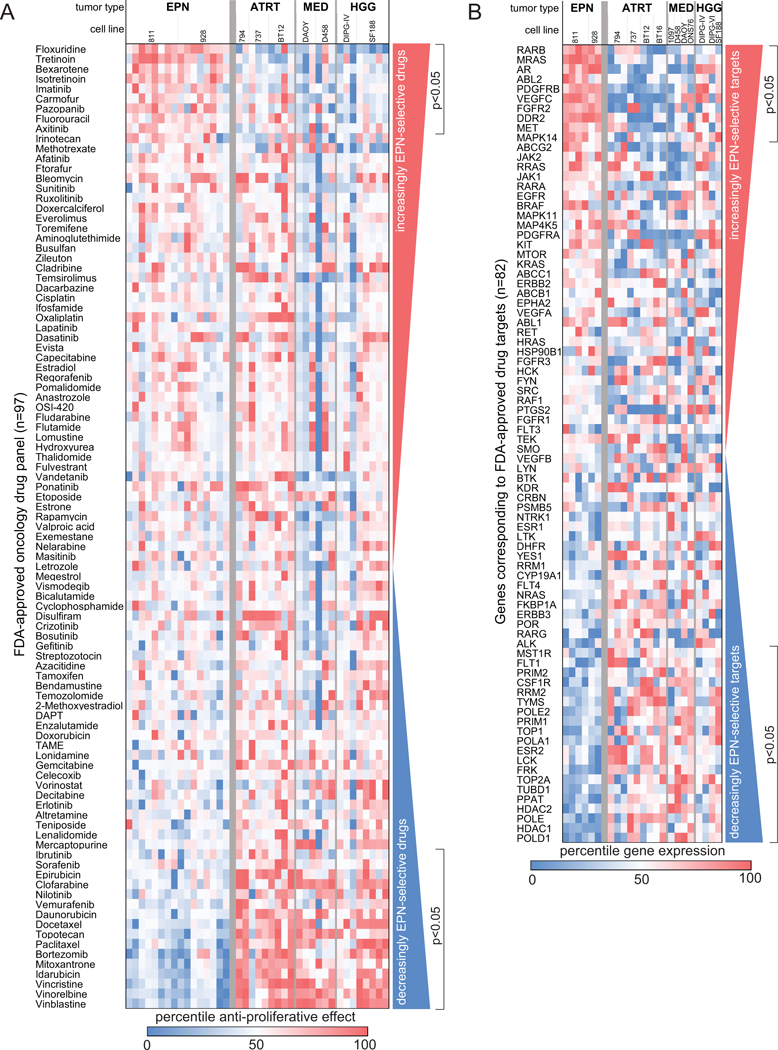
(A) Comparative drug sensitivity analysis of 97 FDA approved oncology drugs ranked from most EPN-selective (top) to most EPN non-selective (bottom). Treatment effect was measured using proliferation assays (tritiated thymidine incorporation) in a panel of pediatric brain tumor cell lines listed as follows with number of replicates in parenthesis (EPNs 811 (8) and 928 (8); ATRTs 794(2), 737 (3) and BT12 (4); MEDs DAOY (3) and D458 (3); HGGs DIPG-IV (3) and SF199 (5)). (B) In silico drug screening for putative targets of FDA-approved oncology drugs. RNA expression of 82 target genes was compared between EPN cell lines and other pediatric brain tumor cells lines to identify EPN-selective (top) and non-selective (bottom) drug targets. Cell lines included in this analysis, with replicates in parenthesis, were EPNs 811 (4) and 928 (2), ATRTs 794 (3), 737, BT12 and BT16 (2 each), MEDs 1097, D458, DAOY and ONS76 (1 each) and HGGs DIPG-IV (2), DIPG-VI and SF188 (1 each).
Table 1.
EPN-selective FDA-approved oncology drugs and target genes.
| A. FDA-approved oncology drugs* | |||||
|---|---|---|---|---|---|
| % effect at 1μM | Effect Δ | p-value | |||
| Drug | class | EPN | other | ||
| Floxuridine | Fluorinated pyrimidine | 27.3 | -124.5 | 151.8 | 4.4×10−7 |
| Carmofur | Fluorinated pyrimidine | 16.5 | -40.2 | 56.8 | 0.017 |
| 5-fluorouracil | Fluorinated pyrimidine | 26.5 | -17.6 | 44.1 | 0.027 |
| Tretinoin | Retinoid | 48.0 | 5.0 | 43.0 | 2.2×10−5 |
| Isotretinoin | Retinoid | 37.1 | -3.3 | 40.4 | 0.0015 |
| Bexarotene | Retinoid | 39.9 | 8.6 | 31.4 | 1.6×10−4 |
| Axitinib | Receptor tyrosine kinase inhibitor | 50.3 | 19.4 | 30.9 | 0.029 |
| Imatinib | Receptor tyrosine kinase inhibitor | 35.4 | 14.3 | 21.1 | 0.010 |
| Pazopanib | Receptor tyrosine kinase inhibitor | 34.8 | 15.6 | 19.3 | 0.026 |
| B. FDA-approved oncology drug target genes@ | |||||
|---|---|---|---|---|---|
| Gene expression (log2)# |
expression Δ | p-value | |||
| Gene | FDA-approved oncology drug | EPN | other | ||
| FGFR2 | regorafenib | 9.9 | 5.5 | 20.7 | 0.0088 |
| DDR2 | regorafenib, sorafenib | 11.6 | 7.5 | 17.2 | 0.023 |
| VEGFC | axitinib, imatinib, pazopanib, sunitinib, sorafenib, | 9.4 | 5.5 | 15.0 | 0.0054 |
| MET | crizotinib | 11.9 | 8.2 | 12.7 | 0.033 |
| PDGFRB | axitinib, dasatinib, imatinib, pazopanib, regorafenib, sorafenib, sunitinib | 8.2 | 4.6 | 12.3 | 0.0032 |
| RARB | bexarotene, isotretinoin, tretinoin | 8.4 | 5.1 | 9.5 | 0.00057 |
| MRAS | sorafenib | 9.5 | 6.4 | 8.7 | 0.00059 |
| AR | enzalutamide | 6.2 | 3.2 | 7.8 | 0.00088 |
| ABL2 | imatinib | 10.2 | 9.0 | 2.3 | 0.0026 |
| MAPK14 | regorafinib | 9.9 | 9.3 | 1.5 | 0.049 |
Ranked in order of difference in percent effect
Ranked in order of difference in difference in gene expression
normalized hybridization intensity (from Affymetrix gene chip HG-U133plus2)
All 3 fluorinated pyrimidines included in our FDA approved oncology drug panel (floxuridine, carmofur and 5-fluorouracil (5-FU)) were EPN-selective (Table 1A). In a prior high-throughput oncology drug screen in EphB2-driven murine supratentorial EPN, Atkinson et al. identified the same 3 fluorinated pyrimidines as having selective toxicity against EPN cells compared to neural stem cells(10). That discovery led to a Phase I study of 5-FU in patients with recurrent EPN, where responses, which were partial, were only observed in posterior fossa EPN (5 of 16, 4 of which were at sites of metastasis) and none in supratentorial EPN (0 of 6)(11). The EPN cell lines utilized in our study were established from posterior fossa EPN metastases, and their sensitivity to fluorinated pyrimidines correspond to the partial responses seen in the phase I clinical trial. Collectively, the results our pre-clinical drug screen and those of Atkinson et al. support further exploration of fluorinated pyrimidine-based treatment of EPN, and support the validity of our comparative drug screening approach as a means to identify EPN-selective oncology drugs.
The second EPN-selective drugs class identified was retinoids, where all 3 retinoids tested (tretinoin, isotretinoin and bexarotene) were significantly more potent in EPN than non-EPN cell lines (Table 1A). Research on the effect of retinoids in EPN is limited, with a single case report of an anaplastic EPN regressing in response to isotretinoin in an adult(18). Further investigation of retinoid treatment in EPN is therefore warranted.
The third class of EPN-selective drugs identified was a subset of receptor tyrosine kinase inhibitors (RTKI), specifically axitinib, imatinib and pazopanib. These small molecule multi-targeted compounds are each known to inhibit vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR) and stem cell factor receptor (c-kit). Atkinson et al. screened kinase inhibitors as part of their high-throughput screen, although of the 3 EPN-selective RTKI identified in our study, only imatinib was tested and did not demonstrate EPN-selectivity(10). A small number of other studies have explored the effects of select RTKIs in EPN, including some clinical trials(19,20), but none of these studies tested the EPN-selective RTKIs identified in our study.
A number of drug classes that were significantly less effective in EPN compared to other pediatric tumor type cell lines were identified (Fig. 1A, Table 2A). These included such cytotoxic drug classes as microtubule inhibitors (all 3 vinca alkaloids included in the panel (vinblastine, vincristine, vinorelabine) and both taxanes (paclitaxel and derivative docetaxel)). DNA synthesis inhibitors, such as topotecan, mitoxantrone, clofarabine and anthracyclines daunorubicin, idarubicin and epirubicin, showed a relatively diminished effect in EPN
Table 2.
EPN non-selective FDA-approved oncology drugs.*
| FDA-approved oncology drugs: EPN non-selective | |||||
|---|---|---|---|---|---|
| Average % effect at 1μM | Effect Δ | p-value | |||
| Drug | class | EPN | other | ||
| Sorafenib | Receptor tyrosine kinase inhibitor | -19.8 | 17.7 | 37.5 | 0.024 |
| Vinblastine | Microtubule inhibitor | 63.2 | 96.7 | 33.5 | 6.5×10−8 |
| Vincristine | Microtubule inhibitor | 62.7 | 95.7 | 33.0 | 5.7×10−7 |
| Vinorelbine | Microtubule inhibitor | 62.9 | 95.5 | 32.5 | 1.5×10−7 |
| Daunorubicin | DNA synthesis inhibitor | 68.6 | 96.8 | 28.2 | 0.0032 |
| Vemurafenib | BRAF inhibitor | -20.0 | 7.3 | 27.3 | 0.010 |
| Topotecan | Topoisomerase inhibitor | 69.1 | 95.7 | 26.6 | 1.0×10−4 |
| Nilotinib | Receptor tyrosine kinase inhibitor | 26.4 | 50.3 | 23.9 | 0.011 |
| Mitoxantrone | DNA synthesis inhibitor | 73.8 | 96.3 | 22.5 | 1.7×10−5 |
| Clofarabine | DNA synthesis inhibitor | 69.7 | 91.2 | 21.5 | 0.011 |
| Bortezomib | Proteosome inhibitor | 78.6 | 95.9 | 17.3 | 2.5×10−5 |
| Paclitaxel | Microtubule inhibitor | 78.8 | 96.0 | 17.2 | 3.4×10−5 |
| Mifepristone | hormone receptor antagonist | -8.1 | 8.9 | 17.0 | 0.029 |
| Docetaxel | Microtubule inhibitor | 77.3 | 92.8 | 15.4 | 0.0021 |
| Ibrutinib | Receptor tyrosine kinase inhibitor | -1.5 | 13.6 | 15.1 | 0.031 |
| Idarubicin | DNA synthesis inhibitor | 84.4 | 97.6 | 13.2 | 3.7×10−6 |
| Epirubicin | DNA synthesis inhibitor | 86.9 | 94.2 | 7.3 | 0.019 |
| B. FDA-approved oncology drug target genes@ | |||||
|---|---|---|---|---|---|
| Gene expression log2# | expression Δ | p-value | |||
| Gene | FDA-approved oncology drug | EPN | other | ||
| POLE2 | gemcitabine | 6.3 | 7.9 | 2.9 | 0.024 |
| POLD1 | clofarabine, gemcitabine, nelarabine | 6.9 | 8.4 | 2.7 | 0.0014 |
| PRIM1 | fludarabine | 8.4 | 9.7 | 2.5 | 0.022 |
| POLE | clofarabine, nelarabine | 5.9 | 7.2 | 2.5 | 0.0017 |
| PPAT | mercaptopurine | 8.7 | 9.9 | 2.4 | 0.0027 |
| PRIM2 | fludarabine | 7.4 | 8.6 | 2.3 | 0.048 |
| TOP2A | daunorubicin, doxorubicin, epirubicin, etoposide, idarubicin, mitoxantrone, teniposide | 11.0 | 12.1 | 2.2 | 0.014 |
| POLA1 | clofarabine, fludarabine, nelarabine | 7.8 | 9.0 | 2.2 | 0.019 |
| TUBD1 | docetaxel, vinblastine, vincristine, vinorelbine | 7.8 | 8.9 | 2.2 | 0.014 |
| CSF1R | sunitinib, imatinib | 3.1 | 4.2 | 2.1 | 0.046 |
| HDAC1 | vorinostat | 11.0 | 12.1 | 2.1 | 0.0016 |
| HDAC2 | vorinostat | 11.2 | 12.2 | 2.0 | 0.0017 |
| RRM2 | fludarabine, gemcitibine | 11.6 | 12.5 | 1.9 | 0.039 |
| TYMS | 5-fluorouracil, capecitabine, carmofur, floxuridine, | 12.2 | 13.1 | 1.9 | 0.027 |
| TOP1 | irinotecan, toptecan | 10.9 | 11.4 | 1.4 | 0.021 |
| FRK | regnorafenib | 2.7 | 2.9 | 1.2 | 0.017 |
| ESR2 | fulvestrant | 2.7 | 2.9 | 1.1 | 0.018 |
| LCK | dasatinib, pazopanib | 2.3 | 2.4 | 1.0 | 0.018 |
Ranked in order of difference in percent effect
Ranked in order of difference in difference in gene expression
normalized hybridization intensity (from Affymetrix gene chip HG-U133plus2)
In silico comparative drug screen
Molecular biomarkers of drug sensitivity/resistance are becoming widely used in therapeutic decision making, in particular those drugs that target specific gene mutations such as BRAF V600E in melanoma. Gene expression has also been shown to be associated with treatment response. A recent study compared the ability of different molecular biomarker types to predict drug responses and RNA expression patterns were shown to be the most predictive data type when compared to cancer gene mutations, copy number alterations, and DNA methylation (21). This finding supports the use of gene expression as a valid approach for drug selection. We therefore performed an in silico drug screen as an orthogonal approach to identify EPN-selective oncology drugs. Expression of RNAs corresponding to putative targets of FDA-approved oncology drugs included in the in vitro screen were compared between EPN and non-EPN cell lines. Target gene (n=82) RNA levels were ranked according to the difference between EPN cell lines 811 and 928 and comparator pediatric brain tumor cell lines (ATRT (794, 737, BT12, BT16), MED (1097, D458, DAOY, ONS76) and high-grade glioma (DIPG-IV, DIPG-VI, SF188)), identifying genes that were both relatively over- and under-expressed in EPN (Fig. 1B). RNA expression levels were further analyzed to identify those targets that were significantly higher or lower in EPN (p<0.05) (Table 1B, Table 2B).
Using this approach, we found that 10 of the 82 genes were significantly higher in EPN than other non-EPN pediatric brain tumors (Table 1B). We compared these EPN-specific genes to EPN-selective drugs to identify drugs and their target receptors which are consistent across both in vitro and in silico analysis. We identified consistent drug sensitivity and target receptor overexpression for 2 of the 3 classes of drugs identified in the in vitro screen. RARB (retinoic acid receptor-B) was the most significantly overexpressed target gene in EPN. This result is consistent with those of our in vitro screen showing all 3 drugs targeting RARB (bexarotene, isotretinoin, tretinoin) are EPN-selective (Table 1A). We examined RARB gene expression in primary PFA patient samples (n=12) and found that it was expressed 12.9-fold higher (p<0.001) than normal cerebellum (n=4) (Supplementary Fig. 1A), supporting further investigation of this class of agents in the treatment of EPN.
The majority of targets identified as EPN-selective were RTKs or related ligands, such as fibroblast growth factor receptor-2 (FGFR2), discoidin domain-containing receptor (2DDR2), vascular endothelial growth factor-C (VEGFC), met proto-oncogene (hepatocyte growth factor receptor; MET), platelet derived growth factor receptor-beta (PDGFRB) and v-abl Abelson murine leukemia viral oncogene homolog 2 (ABL2). These genes correspond to protein targets of a number of multi-target small-molecule RTKIs, of which axitinib, imatinib and pazopanib were also identified as EPN-selective drugs in our in vitro screen (Table 1A). Together, these data suggest that the sensitivity of EPN to these drugs is related to the relatively high expression of their common targets VEGFC and PDGFRB. We further showed overexpression of these potential shared targets of EPN-selective RTKIs in primary PFA patient samples versus normal cerebellum, specifically PDGFRB (2.5-fold; p<0.0001), VEGFR2 (KDR)(7.9-fold; p<0.0001) and VEGFC (11.5-fold, p<0.005)(Supplementary Fig. 1B-D). These data support further exploration of RTKIs axitinib, imatinib and pazopanib for the treatment of EPN.
We identified inconsistencies between EPN-selective drugs and their putative targets. TYMS (thymidylate synthase) and DHFR (dihydrofolate reductase), putative targets of EPN-selective fluorinated pyrimidines floxuridine, 5-fluorouracil and carmofur, were underexpressed in EPN versus comparator cell lines, TYMS significantly so (p<0.05). It is possible that in EPN reduced levels of TYMS, which provides sole de novo source of thymidylate necessary for DNA replication and repair, are further depleted by fluorinated pyrimidines to the point that cell proliferation is inhibited. Conversely, in the comparator cell lines that have higher levels of TYMS, reduced levels of TYMS effected by fluorinated pyrimidine treatment is not sufficient to impact proliferation. Two in silico EPN-selective targets showed inconsistency in corresponding drug sensitivity, specifically MRAS and DDR2 which are targets for sorafenib and nilotinib, both of which were significantly less effective in EPN than comparator cell lines.
Target genes under-expressed in EPN were predominantly mitosis-related, including topoisomerases (TOP1, TOP2A) and DNA polymerases (POLE2, POLD1, POLE, POLA1). Collectively, the reduced expression of these would suggest decreased sensitivity to classic chemotherapeutics including DNA synthesis inhibitors. This conclusion is supported by our finding that our EPN cell lines are particularly insensitive to anthracycline DNA synthesis inhibitors in vitro (daunorubicin, idarubicin, epirubicin, etc) (Table 2A), and the failure of classic chemotherapy agents such as etoposide in EPN clinical trials(7).
Comparative dose curve analysis confirms EPN-selectivity of floxuridine, bexarotene and axitinib
We validated our top hits from in vitro and in silico screening results by generating dose curves for drugs from each of the 3 classes of drugs classes found to be EPN-selective, namely floxuridine, bexarotene and axitinib. Variation in cell line mitotic rates introduces a potential confounding factor in screening of drugs that are dependent on mitotic rate, that may have introduced bias to in vitro comparative drug screening, where all cell lines were exposed to treatment for 3 days. We therefore treated EPN and comparator pediatric brain tumor cell lines for exposure times equivalent to the doubling time for each cell line. This treatment time adjustment was made in order to account for the significantly slower growth rate of EPN cell lines (~3 days) compared to comparator cell lines (~ 1 day).
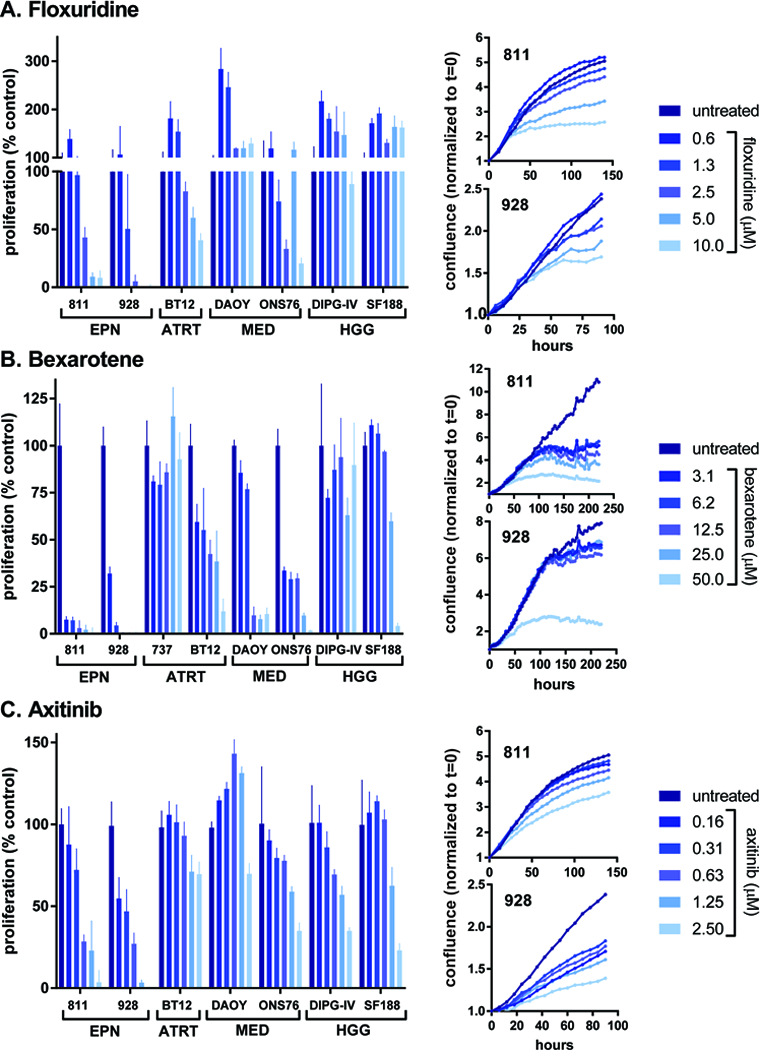
Each of these drugs was confirmed to be more potent in both EPN cell lines than any of the other pediatric brain tumor cell lines tested based on dose curve modelling (Fig. 2). Average IC-50 values for floxuridine treatment of EPN cell lines 811 and 928 were 2.3μM (95% confidence interval (CI): 1.5 to 3.4) and 1.8μM (CI: 0.4 to 7.7) respectively, whereas other pediatric brain tumor combined demonstrated higher IC-50 values (ATRT BT12, 8.0 μM (CI: 5.7 to 10.7); MED DAOY, no effect; GBM SF188, no effect; DIPG-IV, no effect) apart from MED cell line ONS-76 (2.1μM, CI: 0.6 to 4.7)(Fig. 2A). The effective drug doses in EPN established by our dose curve modeling were compared to previously published pharmacokinetic data. A high-risk colon cancer Phase I clinical trial that involved intraperitoneal administration of floxuridine at 500 mg/m2 administered for 3 consecutive days for 3 cycles resulted in a mean plasma concentration of 2.7μg/ml (11.0μM)(22). Treatment of both EPN cell lines with floxuridine at 10μM was sufficient to achieve >75% reduction in proliferation rate.
Figure 2. Dose response effect and time courses for EPN-selective drugs floxuridine, bexarotene and axitinib.
Treatment responses for (A) floxuridine, (B) bexarotene, and (C) axitinib, representing each of the 3 EPN-selective drug classes identified by FDA library screening. Bar graphs (left) represent dose response effects on proliferation normalized to untreated controls (% control), measured by tritiated thymidine incorporation, in EPN cell lines (811 and 928) and comparator cell lines from other pediatric brain tumors(ATRTs 737 and BT12; MEDs DAOY and ONS76; HGGs DIPG-IV and SF188). Treatments were performed in triplicate (error bars represent standard deviation). Growth curve time courses (right) for EPN cell lines 811 and 928 treated with a range of doses of (A) floxuridine, (B) bexarotene, and (C) axitinib, as measured by live cell imaging (IncuCyte) of confluence. Each treatment was performed in triplicate and values are given as confluence normalized to time of treatment (t=0) for each well.
Bexarotene demonstrated higher potency in both EPN cell lines (811, 440nM (CI: 319 to 583); 928, 984nM (CI: 0.74 to 1.268)) than any of the non-EPN pediatric brain tumor cell lines tested (ATRT BT12, 3.4μM (CI: 1.0 to 6.8); MED DAOY, 9.2μM (CI: 5.7 to 15.2); MED ONS-76, 3.2μM (CI: 2.5 to 4.0); GBM SF188, 35.8μM (CI: 22.9 to 57.6); DIPG-IV, no effect; ATRT 737, no effect)(Fig. 2B). Bexarotene administered orally at 300mg/m2/day for 5 days in healthy human subjects resulted in average plasma levels of 1.46μM, above the IC-50 values for either EPN cell line (23).
EPN cell lines also demonstrated lower IC-50 doses for axitinib (811, 500nM (CI: 354 to 695); 928, 150nM (CI: 110 to 203)) than any of the other pediatric brain tumor types (ATRT BT12, 5.8μM (CI: 3.7 to 10.07); MED DAOY, no effect; MED ONS-76, 1.6μM (CI: 0.8 to 2.6); GBM SF188, 2.5μM (CI: 1.3 to 4.7); DIPG-IV, 1.7μM (CI: 0.7 to 3.2))(Fig. 2C). A peak axitinib plasma concentration of 57ng/ml (147nM) was achieved in patients with advanced malignancies who were treated with 5mg orally twice daily for 14 days (24), which is the in the effective range for the EPN cell lines but not for any other pediatric brain tumor cell line.
Treated EPN cell lines were next subjected to live cell imaging (IncuCyte) to determine treatment effects on cell growth, as measured by cell monolayer confluence, over time (Fig 2). Floxuridine, bexarotene and axitinib each showed a dose-dependent decrease in growth rate in both 811 and 928. This readout also identified distinctions between the onset of treatment effects, notably that axitinib showed immediate growth inhibition, floxuridine showed a 1–2 day delay in effect, and bexarotene showed a dose-dependent delay in effect, where lower dose effects were not seen until up to 5 days post-treatment. Induction of apoptosis, as measured by caspase 3/7 activation, was not observed in floxuridine, bexarotene or axitinib (Supplementary Figure 2).
Axitinib treatment of EPN cell lines results in decreased mitosis-related gene expression, upregulation of interferon response gene expression and induction of senescence.
Small molecule RTKIs have proven effective in the treatment of a number of cancer types, with more specificity for tumors and consequently less harmful side-effects than classic chemotherapeutics(25,26). As RTKIs have not been extensively explored in EPN we chose to study the mechanism of action of axitinib, the most EPN-selective RTKI in our screen. The EPN-selective RTKIs identified in our study not only provide therapeutic leads for treatment but also insights into disease biology, potentially identifying growth factor-driven mitogenic signaling cascades specific to EPN.
As an unbiased approach to determine mechanisms of axitinib activity in EPN, we measured the transcriptomic effect of axitinib treatment in EPN cell lines. Both EPN cell lines treated with an IC-50 dose of axitinib for 24 hours and after which cells were harvested for RNA extraction. Transcriptome profiles of samples were generated using HG-U133plus2 gene expression microarrays, and transcriptomic changes between axitinib-treated and untreated controls were analyzed using GSEA to identify enrichment or depletion of MSigDB “Hallmark” genesets. We showed that the predominant transcriptomic effect common to both EPN cell lines 811 and 928 was down-regulation of mitosis-associated genes, the most depleted geneset being “hallmark_E2F_targets” (Fig. 3A, Supplementary data). Specific mitosis-related genes down-regulated by axitinib included ASF1B (3.1-fold), MKI67 (3.0), HMGA1 (2.2), BRCA2 (2.1), ESPL1 (2.1), TACC3 (2.1), CDC25A (2.0), RAD51AP1 (1.8), AURKA (1.6), BUB1B (1.6), CENPE (1.8) and HELLS (1.6). Conversely, axitinib treatment of both EPN cell lines resulted in a significant up-regulation of interferon-associated genes, most notably those included in the “hallmark_interferon_alpha_response” geneset (Fig. 3B, Supplementary data). Notable interferon genes upregulated in response to axitinib treatment included MX1 (4.1-fold), SERPING1 (3.9), IFI44L (3.3), RTP4 (3.2), IFITM1 (2.9) and VCAM1 (2.2). Similar immunomodulatory effects of axitinib have been observed in melanoma and glioma(27,28). The upregulation of “hallmark_xenobiotic_metabolism” genes in response to axitinib treatment suggests underlying drug resistance activity in EPN.
Figure 3. Investigation of mechanism of EPN-selective effect of axitinib treatment.
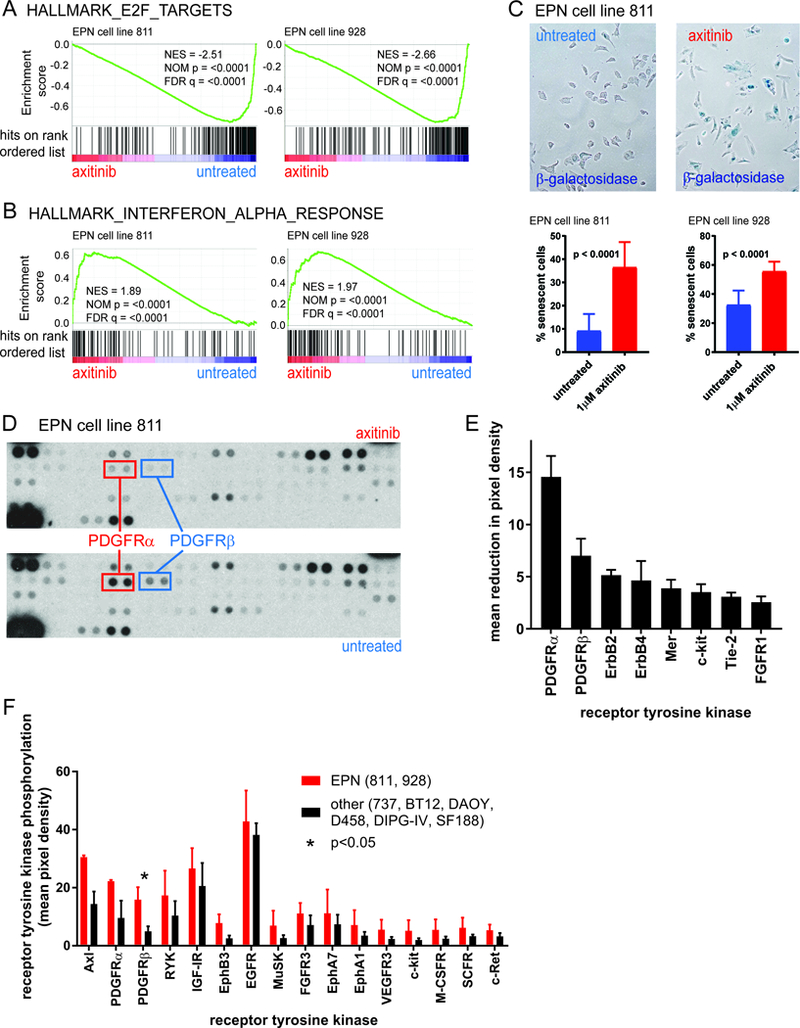
Axitinib treatment of EPN cell lines resulted in (A) depletion of mitosis-related gene expression, and (B) upregulation of IFNα response genes, as measured by GSEA of transcriptomic profiles. (C) β-galactosidase staining revealed that axitinib induced senescence in EPN cell lines (12 high power fields across treatments in triplicate). (D, E) Proteomic analysis of receptor tyrosine kinase (RTK) phosphorylation revealed that PDGFRα and PDGFRβ are the predominant targets of axitinib in EPN cell lines (E, sorted left to right by decreasing effect). Bars represent the average difference between phosphorylation of RTKs (n=2) between untreated and axitinib-treated in both EPN cell lines (811 and 928) (error bars = standard deviation) (F) RTKs phosphorylated to a greater degree in EPN cell lines versus other pediatric brain tumor cell lines (sorted left to right by decreasing difference), demonstrating increased levels of phosphorylated Axl, PDGFRα and PDGFRβ in EPN cell lines. Each phosphorylation measurement is the mean pixel density for 2 membrane spots (error bars = standard deviation).
Both EPN cell lines showed a significant increase in cellular senescence in response to axitinib treatment, as measured by β-galactosidase staining (4.0-fold and 1.7-fold respectively in 811 and 928) (Fig. 3C). This is consistent with the decreased proliferation rate in both EPN cell lines after treatment with axitinib.
PDGFRα and PDGFRβ signaling is a potentially therapeutic susceptibility in EPN that is exploited by axitinib.
Each of the EPN-selective RTKIs axitinib, imatinib and pazopanib are reported to target the same set of RTKs, namely VEGFR1–3, c-kit and PDGFRα and β suggesting that these inhibitors share a common mechanism in EPN. Tyrosine phosphorylation of RTKs is a required step in enzymatic activation and triggering of downstream signaling cascades, and is therefore used as a measure of RTK function. We therefore measured axitinib treatment effects on the phosphorylation status of 49 RTKs, including those listed above, using a Human Phospho-RTK Array Kit.
We observed consistent de-phosphorylation of PDGFRα and PDGFRβ, in both EPN cell lines after 24 hours of axitinib treatment (1μM) compared to untreated controls (Fig. 3D, E). Other RTKs that were dephosphorylated by axitinib to a lesser extent in both EPN cell lines (decrease in pixel density >2) included ErbB2, ErbB4, Mer, c-kit, Tie-2 and FGFR1 (Fig. 3E). A number of RTKs that showed baseline phosphorylation in untreated EPN cell lines but were not de-phosphorylated in response to axitinib treatment in both EPN cell lines included EGFR, IGF-IR, Axl, FGFR3, ROR2, ALK, EphB3, EphB2, ROR1, HGFR, Tie-1 and DDR1. A single RTK, RYK, demonstrated increased phosphorylation in both EPN cell lines (increase in pixel density >2). There was no detectable baseline phosphorylation of the remaining RTKs tested by the phospho-array including EphA10, EphB6, c-Ret, EphA6, EphA7, FGFR2A, EphA1, VEGFR3, EphA2, ErbB3, MuSK, MSPR, EphB4, Flt-3, M-CSFR, TrkB, EphA3, EphB1, SCFR, c-kit, TrkC, DDR2, FGFR4, VEGFR1, EphA5, TrkA, VEGFR2.
These data suggest that PDGFRα and PDGFRβ are the predominant targets of axitinib in EPN cell lines. As axitinib had been shown to be EPN-selective, we hypothesized that PDGFRα and PDGFRβ activity would be more activated in EPN cell lines than other pediatric brain tumor cell lines. We therefore compared the baseline tyrosine phosphorylation status of RTKs in EPN cell lines (811 and 928) versus other pediatric brain tumor cell lines that were used prior in in vitro and in silico screening (ATRT (737, BT12), MED (DAOY, D458) and high grade glioma (DIPG-IV, SF188)). Sixteen of 49 RTKs were phosphorylated to a greater extent in EPN versus others (increased mean pixel density >2) (Fig. 3F). Those RTKs that demonstrated the highest relative baseline phosphorylation in EPN were Axl, PDGFRα, PDGFRβ (increased mean pixel density >10). Collectively these data identify PDGFRα and PDGFRβ signaling as a potentially therapeutic susceptibility specific to EPN that is exploited by axitinib.
Discussion
The majority of EPN will recur, and treatment options in this event are limited to re-resection and re-irradiation, as there are currently no effective chemotherapeutic options. Identification of effective chemotherapies in EPN has been hampered in part by the lack of available preclinical models. Our in vitro and in silico screening of FDA-approved oncology drugs identified 3 classes of EPN-selective therapies that can be rapidly advanced to clinical trials, specifically fluorinated pyrimidines, retinoids, and a subset of RTKIs. This is the first reported large-scale comparative drug screen in human EPN cell lines. Furthermore, these cell lines were established from recurrent EPN metastases and harbor high-risk chromosomal 1q gain and characteristics of PFA(12), of particularly clinical relevance given the urgent need for novel therapies for children with recurrent high-risk EPN.
By pinpointing fluorinated pyrimidines 5-FU, carmofur and floxuridine as EPN-selective oncology drugs in our screen, our findings match those of Atkinson et al. who performed a high-throughput screen of 5303 unique compounds in a mouse model of supratentorial EPN(10). Their study identified 4 compounds with greater than 2-fold potency in EPN cells compared to neural stem cells, 3 of which were 5-FU, carmofur and floxuridine. This striking concordance with the present study supports continued investigation of fluorinated pyrimidines for the treatment of EPN. Furthermore, these similar findings in both in vivo murine supratentorial and in vitro human posterior fossa EPN models suggest that fluorinated pyrimidines have a broad anti-EPN activity across different molecular subtypes. However, an early phase clinical trial of 5-FU in EPN showed partial responses only in posterior fossa EPN and not in supratentorial cases, underscoring the need for molecular subgroup stratification in future trials.
Although this single agent 5-FU clinical trial showed limited efficacy, the validation of this agent as EPN-selective support further study as part of a combinatorial therapy. Screening for synergistic drug combinations with fluorinated pyrimidines could be achieved using the in vitro EPN screening model described in the present study. Unlike retinoids and EPN-selective RTKIs, fluorinated pyrimidines were not predicted to be EPN-selective by overexpression of putative targets TYMS and DHFR by in silico analysis, TYMS in particularly being underexpressed in EPN. Future work should focus on better understanding the role of predictive markers in the anti-tumor mechanism of fluorinated pyrimidines in EPN..
All 3 retinoids tested in the in vitro screen, tretinoin, Isotretinoin and bexarotene, demonstrated significant EPN-selective potency (Table 1). Isotretinoin is effective in the treatment of pediatric cancers such as promyelocytic leukemia and neuroblastoma(29,30). There is evidence that the anti-tumor activity of retinoids is partly due to either induction of cellular differentiation and/or inhibition of epithelial cell proliferation(31,32). This potentially explains their EPN selective effect, as EPN have an underlying epithelial phenotype. Preclinical studies of retinoids in pediatric brain tumors such as glioblastoma and MED have similarly demonstrated induction of differentiation, leading to testing of Isotretinoin in a phase III COG trial in MED (ACNS0332). Our data showed that all 3 FDA-approved retinoids have greater potency in EPN than glioblastoma, MED and other pediatric brain tumor types, and express higher levels of retinoid target RARB, and should therefore be further investigated as a treatment option for this disease.
The identification of a subset of RTKIs not only provide therapeutic leads for the clinic but also insights into disease biology. EPN-selective compounds axitinib, imatinib and pazopanib each target a common set of RTKs, namely VEGFRs, PDGFRs and c-kit, suggesting that one or more of these growth factor signaling pathways may be driving growth in EPN. Identification of PDGFR inhibition as the anti-EPN mechanism of axitinib, and potentially imatinib and pazopanib, identify PDGFR as a potential therapeutic susceptibility in EPN that warrants further investigation. In addition to our data showing that PDGFRB is overexpressed in PFA patient samples, Witt et al. identified PDGF signaling as a potential target for PFA-specific therapy in their study that first identified this EPN subgroup(3). Axitinib has a completed a Phase I pediatric trial (COG ADVL1315) in relapsed/refractory non-CNS solid tumors, which identified a maximum-tolerated dose in children and showed that the drug was not associated with significant risks of myelosuppression. Furthermore, an adult study in glioblastoma showed objective responses, which suggests blood-brain barrier penetrance(33). Together with the findings of the current preclinical study, these clinical studies support the advancement of axitinib as a therapeutic option for EPN.
Our use of existing FDA-approved compounds with known efficacy and toxicity profiles facilitates rapid translation into pediatric clinic trials. Given the continued high mortality of this tumor and the lack of specific therapies, data from this study have the potential to dramatically improve survival in children with EPN, whose therapy and outcome have remained essentially unchanged for decades.
Supplementary Material
Acknowledgments
Financial support: This work was funded by the Tanner Seebaum Foundation and the Morgan Adams Foundation. JML is supported by an elope, Inc. St. Baldrick’s Foundation Scholar Award and NIH/NCI (K08CA193982). The Genomics and Microarray Core receives direct funding support from the National Cancer Institute (P30CA046934).
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Ramaswamy V, Hielscher T, Mack SC, Lassaletta A, Lin T, Pajtler KW , et al. Therapeutic Impact of Cytoreductive Surgery and Irradiation of Posterior Fossa Ependymoma in the Molecular Era: A Retrospective Multicohort Analysis. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2016;34:2468–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marinoff AE, Ma C, Guo D, Snuderl M, Wright KD, Manley PE , et al. Rethinking childhood ependymoma: a retrospective, multi-center analysis reveals poor long-term overall survival. J Neurooncol 2017;135:201–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Witt H, Mack SC, Ryzhova M, Bender S, Sill M, Isserlin R , et al. Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell 2011;20:143–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parker M, Mohankumar KM, Punchihewa C, Weinlich R, Dalton JD, Li Y , et al. C11orf95-RELA fusions drive oncogenic NF-kappaB signalling in ependymoma. Nature 2014;506:451–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F , et al. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 2015;27:728–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bouffet E, Foreman N. Chemotherapy for intracranial ependymomas. Childs Nerv Syst 1999;15:563–70 [DOI] [PubMed] [Google Scholar]
- 7.Khatua S, Ramaswamy V, Bouffet E. Current therapy and the evolving molecular landscape of paediatric ependymoma. Eur J Cancer 2017;70:34–41 [DOI] [PubMed] [Google Scholar]
- 8.Gojo J, Lotsch D, Spiegl-Kreinecker S, Pajtler KW, Neumayer K, Korbel P , et al. Telomerase activation in posterior fossa group A ependymomas is associated with dismal prognosis and chromosome 1q gain. Neuro Oncol 2017;19:1183–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kilday JP, Mitra B, Domerg C, Ward J, Andreiuolo F, Osteso-Ibanez T , et al. Copy number gain of 1q25 predicts poor progression-free survival for pediatric intracranial ependymomas and enables patient risk stratification: a prospective European clinical trial cohort analysis on behalf of the Children's Cancer Leukaemia Group (CCLG), Societe Francaise d'Oncologie Pediatrique (SFOP), and International Society for Pediatric Oncology (SIOP). Clinical cancer research : an official journal of the American Association for Cancer Research 2012;18:2001–11 [DOI] [PubMed] [Google Scholar]
- 10.Atkinson JM, Shelat AA, Carcaboso AM, Kranenburg TA, Arnold LA, Boulos N , et al. An integrated in vitro and in vivo high-throughput screen identifies treatment leads for ependymoma. Cancer Cell 2011;20:384–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wright KD, Daryani VM, Turner DC, Onar-Thomas A, Boulos N, Orr BA , et al. Phase I study of 5-fluorouracil in children and young adults with recurrent ependymoma. Neuro Oncol 2015;17:1620–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amani V, Donson AM, Lummus SC, Prince EW, Griesinger AM, Witt DA , et al. Characterization of 2 Novel Ependymoma Cell Lines With Chromosome 1q Gain Derived From Posterior Fossa Tumors of Childhood. J Neuropathol Exp Neurol 2017;76:595–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levy JM, Thompson JC, Griesinger AM, Amani V, Donson AM, Birks DK , et al. Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer discovery 2014;4:773–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alimova I, Birks DK, Harris PS, Knipstein JA, Venkataraman S, Marquez VE , et al. Inhibition of EZH2 suppresses self-renewal and induces radiation sensitivity in atypical rhabdoid teratoid tumor cells. Neuro Oncol 2013;15:149–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amani V, Prince EW, Alimova I, Balakrishnan I, Birks D, Donson AM , et al. Polo-like Kinase 1 as a potential therapeutic target in Diffuse Intrinsic Pontine Glioma. BMC Cancer 2016;16:647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donson AM, Apps J, Griesinger AM, Amani V, Witt DA, Anderson RCE , et al. Molecular Analyses Reveal Inflammatory Mediators in the Solid Component and Cyst Fluid of Human Adamantinomatous Craniopharyngioma. J Neuropathol Exp Neurol 2017;76:779–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics 2007;23:3251–3 [DOI] [PubMed] [Google Scholar]
- 18.Rojas-Marcos I, Calvet D, Janoray P, Delattre JY. Response of recurrent anaplastic ependymoma to a combination of tamoxifen and isotretinoin. Neurology 2003;61:1019–20 [DOI] [PubMed] [Google Scholar]
- 19.Sie M, den Dunnen WF, Lourens HJ, Meeuwsen-de Boer TG, Scherpen FJ, Zomerman WW , et al. Growth-factor-driven rescue to receptor tyrosine kinase (RTK) inhibitors through Akt and Erk phosphorylation in pediatric low grade astrocytoma and ependymoma. PLoS One 2015;10:e0122555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wetmore C, Daryani VM, Billups CA, Boyett JM, Leary S, Tanos R , et al. Phase II evaluation of sunitinib in the treatment of recurrent or refractory high-grade glioma or ependymoma in children: a children's Oncology Group Study ACNS1021. Cancer Med 2016;5:1416–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M , et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016;166:740–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelsen DP, Saltz L, Cohen AM, Yao TJ, Enker W, Tong W , et al. A phase I trial of immediate postoperative intraperitoneal floxuridine and leucovorin plus systemic 5-fluorouracil and levamisole after resection of high risk colon cancer. Cancer 1994;74:2224–33 [DOI] [PubMed] [Google Scholar]
- 23.Ghosal K, Haag M, Verghese PB, West T, Veenstra T, Braunstein JB , et al. A randomized controlled study to evaluate the effect of bexarotene on amyloid-beta and apolipoprotein E metabolism in healthy subjects. Alzheimers Dement (N Y) 2016;2:110–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bruce JY, Scully PC, Carmichael LL, Eickhoff JC, Perlman SB, Kolesar JM , et al. Pharmacodynamic study of axitinib in patients with advanced malignancies assessed with (18)F-3'deoxy-3'fluoro-L-thymidine positron emission tomography/computed tomography. Cancer Chemother Pharmacol 2015;76:187–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM , et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. The New England journal of medicine 2001;344:1038–42 [DOI] [PubMed] [Google Scholar]
- 26.Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS , et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. The New England journal of medicine 2012;366:707–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du Four S, Maenhout SK, Benteyn D, De Keersmaecker B, Duerinck J, Thielemans K , et al. Disease progression in recurrent glioblastoma patients treated with the VEGFR inhibitor axitinib is associated with increased regulatory T cell numbers and T cell exhaustion. Cancer Immunol Immunother 2016;65:727–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Du Four S, Maenhout SK, De Pierre K, Renmans D, Niclou SP, Thielemans K , et al. Axitinib increases the infiltration of immune cells and reduces the suppressive capacity of monocytic MDSCs in an intracranial mouse melanoma model. Oncoimmunology 2015;4:e998107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chomienne C, Ballerini P, Balitrand N, Amar M, Bernard JF, Boivin P , et al. Retinoic acid therapy for promyelocytic leukaemia. Lancet 1989;2:746–7 [DOI] [PubMed] [Google Scholar]
- 30.Villablanca JG, Khan AA, Avramis VI, Seeger RC, Matthay KK, Ramsay NK , et al. Phase I trial of 13-cis-retinoic acid in children with neuroblastoma following bone marrow transplantation. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 1995;13:894–901 [DOI] [PubMed] [Google Scholar]
- 31.Strickland S, Mahdavi V. The induction of differentiation in teratocarcinoma stem cells by retinoic acid. Cell 1978;15:393–403 [DOI] [PubMed] [Google Scholar]
- 32.Breitman TR, Selonick SE, Collins SJ. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc Natl Acad Sci U S A 1980;77:2936–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duerinck J, Du Four S, Vandervorst F, D'Haene N, Le Mercier M, Michotte A , et al. Randomized phase II study of axitinib versus physicians best alternative choice of therapy in patients with recurrent glioblastoma. J Neurooncol 2016;128:147–55 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.