

SUMMARY
Pathogen burden accelerate atherosclerosis, but the mechanisms remain unresolved. Activation of the NLRP3 inflammasome is linked to atherogenesis. Here we investigated whether Chlamydia pneumoniae (C.pn) infection engages NLRP3 in promoting atherosclerosis. C.pn potentiated hyperlipidemia-induced inflammasome activity in cultured macrophages and in foam cells in atherosclerotic lesions of Ldlr−/− mice. C.pn-induced acceleration of atherosclerosis was significantly dependent on NLRP3 and Caspase-1. We discovered that C.pn-induced extracellular IL-1β triggers a negative feedback loop to inhibit GPR109a and ABCA1 expression and cholesterol efflux leading to accumulation of intracellular cholesterol and foam cell formation. Gpr109a and Abca1 were both upregulated in plaque lesions in Nlrp3−/− mice in both hyperlipidemic and C.pn infection models. Mature IL-1β and cholesterol may compete for access to the ABCA1 transporter to be exported from macrophages. C.pn exploits this metabolic-immune cross talk, which can be modulated by NLRP3 inhibitors to alleviate atherosclerosis.
Keywords: atherosclerosis, macrophage, foam cells, C. pneumoniae, oxLDL, Nlrp3, ABCA1, Gpr109a, cholesterol efflux, niacin, Interleukin-1 beta, aspartate
eTOC
Infections can accelerate atherosclerosis, but the mechanisms remain unresolved. Tumurkhuu et al. show that C.pn infection-induced IL-1β institutes negative feedback to inhibit Gpr109a, ABCA1 expression, and cholesterol efflux leading to accumulation of intracellular cholesterol. Mature IL-1β can use ABCA1 for secretion from macrophages at the detriment of cholesterol efflux.
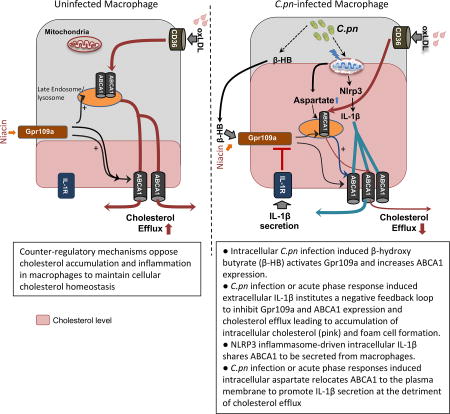
INTRODUCTION
Chronic inflammation of the arterial wall is a key process in the pathogenesis of atherosclerosis (Libby, 2002). Foam cells, myeloid cells that accumulate cholesterol in the arterial wall, are characteristic of atherosclerotic plaques and play important roles in the progression of atherosclerosis (Moore and Tabas, 2011). The lipid modification and deposition in plaques are thought to be a major source of the continuous inflammatory stimulus and earlier studies have implicated pathogen influence in this process.
The innate immune system plays a key role in the inflammatory processes implicated in atherosclerotic progression through Toll-Like receptors (TLRs) and the nucleotide binding domain and leucine-rich repeat (NLR) pyrin domain containing 3 (NLRP3) inflammasome (Broderick et al., 2015). We and others were reported the role of TLRs in atherosclerosis (Xu et al., 2001) and that TLR4/MyD88 signaling plays a critical proatherogenic role in high fat diet-induced atherosclerosis (Michelsen et al., 2004). Recent studies have shown that activation of NLRP3 inflammasome enhances atherogenesis in mice (Duewell et al., 2010)). Aberrant inflammasome activation is implicated in human atherosclerotic disease (Paramel Varghese et al., 2016)). Importantly, cholesterol crystals have been shown to activate the production of Interleukin (IL-1β and IL-1α) in both human and mouse macrophages (Duewell et al., 2010), linking cholesterol and sterile inflammation, a characteristic of atherosclerosis. Furthermore, IL-1β inhibition reduced atherosclerosis in ApoE-deficient mice (Elhage et al., 1998), while elevated IL-1β levels associate with an increased risk of atherosclerosis in humans (Olofsson et al., 2009). Abnormal inflammasome activation and the consequent increase in the circulating IL-1β and IL-18 levels correlate with more macrophage recruitment to lesions, accelerated foam cell formation and plaque progression (Duewell et al., 2010). These studies all support the concept that the NLRP3-generated inflammatory cytokines, IL-1β and IL-18, are central to lesion progression. However, a separate study using a more aggressive mouse model of atherosclerosis with exaggerated circulating cholesterol (ApoE-deficient mice) that is known to induce a strong atherogenic phenotype, failed to demonstrate a role for NLRP3 in the progression of atherosclerosis (Menu et al., 2011). These conflicting outcomes are likely due to the use of different animal models and possible redundant pathways as discussed in a prior review (De Nardo and Latz, 2011). The recently published CANTOS trial reported that neutralizing IL-1β led to a modest but significantly lower rate of recurrent cardiovascular events in patients with previous myocardial infarction (Ridker et al., 2017), and has helped strengthen the case for the inflammatory basis of human coronary artery disease. However, the exact mechanisms by which circulating IL-1β ablation benefited these patients are not completely understood. Whether these anti-IL-1β or anti-NLRP3 approaches can also benefit infection-induced acceleration of atherosclerosis is also unknown.
A large body of evidence in mice and humans suggests that an infectious agent, Chlamydia pneumonia (C.pn), which has been identified in human atherosclerotic plaques, promotes progression/exacerbation of atherosclerotic processes, but the molecular mechanisms engaged by C.pn remain unclear (Campbell and Kuo, 2004; Chen et al., 2010; Naiki et al., 2008; Rosenfeld and Campbell, 2011). Live C.pn (He et al., 2010; Shimada et al., 2011) and excessive intracellular cholesterol (Sheedy et al., 2013) also activate the NLRP3 inflammasome. Whether C.pn-induced Nlrp3 inflammasome activation and subsequent IL-1β secretion plays a role in C.pn infection-accelerated atherogenesis is not known.
As important as it is for acute and chronic inflammatory diseases, the secretory routes of mature IL-1β are still not fully understood. It was reported that a cholesterol efflux regulatory protein, ATP-binding cassette transporter A1 (ABCA1), can also transport IL-1β (Hamon et al., 1997; Marty et al., 2005; Zhou et al., 2002). By promoting reverse cholesterol transport, ABCA1 provides protection from atherosclerosis (Oram and Heinecke, 2005). Taken together, these studies suggest that ABCA1 expression by macrophages has pleiotropic mechanisms of action and may be involved in cross-talk between metabolic and inflammatory pathways.
G-protein-coupled receptor (Gpr)109a is an inducer of ABCA1 and is expressed in both adipocytes and immune cells. Gpr109a is a receptor for niacin and the ketone body, 3-hydroxybutyrate (β-HB) (also known as niacin receptor 1 (Niacr1) or hydroxycarboxylic acid (HCA) receptor 2 (HCA2)) (Tunaru et al., 2003). Activation of Gpr109a reduces the atherogenic lipoproteins LDL-c and VLDL-c, and raise plasma HDL-c by inducing the reverse cholesterol transport apparatus (Guyton, 2007), as well as induces anti-inflammatory effects in myeloid cells reducing progression of atherosclerosis (Lukasova et al., 2011).
In this study, we found that the NLRP3 inflammasome plays an important role in C.pn-induced acceleration of atherosclerosis. Our data show that Nlrp3 inflammasome-induced IL-1β suppresses ABCA1-mediated cholesterol efflux by downregulating the Gpr109a receptor, which controls ABCA1 expression. We also show that C.pn-induced IL-1β secretion can occur through the ABCA1 transporter, which negatively impacts cholesterol efflux due to competition for this shared transporter. This involves C.pn-induced increase in intracellular aspartate levels, which diverts ABCA1 to the plasma membrane and facilitates IL-1β secretion at the expense of cholesterol efflux. As a consequence of these C.pn-induced cellular mechanisms cholesterol accumulates inside the cells to enhance foam cell formation. These pathways are activated in both diet-induced atherosclerosis and in C.pn infection-induced acceleration of atherosclerosis, demonstrating how a pathogen exploits this novel metabolic-immune cross talk.
RESULTS
Nlrp3 inflammasome plays an important role in C.pn infection-accelerated atherosclerosis
To study the role of Nlrp3 inflammasome in C.pn-accelerated atherogenesis, Nlrp3−/−, Ldlr−/−Nlrp3−/−, Casp1−/−Ldlr−/−, and control Ldlr−/− mice were fed a Western Diet (WD) for 16 weeks (Figure S1A). In some groups, mice were infected with a sublethal dose of C.pn once a week for the first three consecutive weeks of WD (Naiki et al., 2008). We observed no differences in body weight and plasma lipid profiles between Nlrp3−/−Ldlr−/−, Casp1−/−Ldlr−/− and Ldlr−/− mice, indicating that NLRP3 inflammasome-deficiency does not have a major impact on weight gain or lipid metabolism (Figure S1B and Table S1).
After 16 weeks on WD, quantification of lesion areas in the aortic roots revealed significantly smaller lesions Nlrp3−/−Ldlr−/− and Casp1−/−Ldlr−/− mice compared to Ldlr−/− mice (Figure 1A), similar to previous studies (Duewell et al., 2010; Gage et al., 2012). With C.pn infection, lesion areas increased in all genotypes, but these were significantly smaller in both Nlrp3−/−Ldlr−/− and Casp1−/−Ldlr−/− mice (Figure 1A, right panel). En face aorta analysis also showed lesion area was significantly smaller in both Nlrp3−/−Ldlr−/− and Casp1−/−Ldlr−/− mice compared with Ldlr−/− mice on WD alone (Figure 1B). By this analysis, we observed that C.pn infection resulted in a significant increase in lesion size in Ldlr−/− mice, but not in Nlrp3−/−Ldlr−/− or Casp1−/−Ldlr−/− mice (Figure 1B, right panel). Furthermore, necrotic core area and lesion macrophage content were markedly reduced in C.pn-infected Nlrp3−/−Ldlr−/− and Casp1−/−Ldlr−/− mice versus C.pninfected Ldlr−/− mice (Figures 1C-1D). While the C.pn infected Nlrp3−/− and Casp1−/− mice still show small but significant increase in aortic root plaque size (Figure 1A), the extent of atherosclerosis in these mice is now reduced back to the levels seen in the uninfected wild-type mice. These data suggest that while the NLRP3 and IL-1β axis is a significant pathway in C.pn-induced acceleration of atherosclerosis, it was not the only pathway, as additional pathways that are NLRP3-independent may also play some residual role (Chen et al., 2018; Naiki et al., 2008).
Figure 1. Nlrp3 inflammasome is required for both diet-induced and C.pn infection-accelerated atherosclerosis in Ldlr−/− mice.

(A) Nlrp3−/−Ldlr−/−, Casp1−/−Ldlr−/− and control Ldlr−/− mice were fed WD for 16 weeks and mice were infected with or without intra-nasal C.pn (5 × 104 IFU/mouse) weekly for a total of three times (beginning with the onset of WD) (n=13–15). Representative pictures and quantification of aortic root lesion area stained with Oil Red O are shown.
(B) Representative images and quantification of aorta en face stained with Oil Red O. (n=13–15).
(C) Necrotic core area (H&E) in aortic root (n=12 per group).
(D) Macrophage content determined by staining with anti-MOMA-2 (green) in aortic root (n=10 per group). Scale bar = 50 μm.
(E) VCAM positivity in aortic root (n=9 per group)
All representative pictures are from the infected mice with WD.
All data are mean ± SD. Significance was determined using two-way ANOVA with Bonneferroni's post-test (A and B), or one-way ANOVA with Tukey’s post-hoc test (C-E). *p<0.05, **p<0.01, ***p<0.001. Both male and female mice were used.
We observed that the lesion area positive for vascular cell adhesion protein-1 (VCAM-1), an endothelial adhesion molecule, was significantly lower in the C.pn-infected Nlrp3−/−Ldlr−/− and Casp1−/−Ldlr−/− mice compared with C.pn-infected Ldlr−/− mice, consistent with the reduction in macrophage content (Figure 1E). Accompanying the reduction in lesion size, we observed reduced levels of monocyte chemotaxis protein-1 (MCP-1) in the serum of Nlrp3−/−Ldlr−/− compared with Ldlr−/− mice (Figure S1C). C.pn infection led to a significant increase in MCP-1 in Ldlr−/−, but not in Nlrp3−/−Ldlr−/− mice, indicating that the increase in MCP-1 was NLRP3 inflammasome-dependent. In contrast, serum concentrations of IL-12p70 and IL-6 were not altered by C.pn infection or between the genotypes, suggesting that C.pn infection does not impact these other pro-atherogenic cytokines that are produced independent of inflammasome activity. Most importantly, our findings show that C.pn infection causes a marked acceleration and increase in atherosclerosis that is significantly dependent on NLRP3 inflammasome activation, while an NLRP3-independent pathway also exists.
Hematopoietic Nlrp3 inflammasome activity is required for C.pn infection-accelerated atherogenesis
We next assessed the impact of C.pn infection on Nlrp3 inflammasome activity in the atherosclerotic lesions from mice by quantifying active caspase-1 (stained with Fluorochrome Labeled Inhibitors of Caspases (FLICA) in macrophage-rich (MOMA-2 positive) areas. As seen in Figure 2A and B, active caspase-1 was detected in lesion macrophages in the aortic roots, and C.pn infection resulted in significantly more active caspase-1 in the plaque macrophages. These findings indicate that C.pn infection leads to NLRP3 inflammasome activation in plaque macrophages. We next evaluated how Nlrp3- or Caspase 1-deficiency in hematopoietic cells impacts C.pn infection-augmented atherogenesis. We created three groups of bone marrow chimeric (BMC) mice (Donor → Recipient): wild type (WT)→Ldlr−/−, Nlrp3−/− →Ldlr−/− and Casp1−/− →Ldlr−/−. After six weeks of reconstitution, BMC mice were fed with WD for twelve weeks and during this time the mice were infected with C.pn once a week for three consecutive weeks (Figure 2C). In comparison to WT BMC mice, aortic root and whole aorta lesion areas (Figure 2D-F) were significantly reduced in Nlrp3−/− or Casp1−/− BMC mice, despite similar cholesterol levels (data not shown). MOMA-2 staining revealed less macrophage accumulation in the aortic root lesions in Nlrp3−/− or Casp1−/− BMC versus WT BMC mice (Figure 2G). These data suggest that the Nlrp3 inflammasome in hematopoietic cells plays an important role in C.pn infection-accelerated atherosclerosis.
Figure 2. Nlrp3-deficiency in hematopoietic cells prevents atherosclerosis acceleration by C.pn infection.

(A) Caspase-1 activity was assessed by FLICA (green) and in macrophages (MOMA-2; red) in atherosclerotic lesions of Ldlr−/− mice fed a WD for 16 weeks with and without C.pn infection (n=6). Representative images for Caspase-1 positivity in lesion macrophages are shown. Scale bar = 50 μm.
(B) Quantification of active caspase-1+ cells in lesion macrophages.
(C) BM from WT, Nlrp3−/−, or Casp1−/− mice were transplanted to irradiated Ldlr−/− mice. 6-weeks after reconstitution, mice were put on WD (12-weeks). All groups were infected with intra-nasal C. pn (5 × 104 IFU/mouse) weekly for a total of three times (beginning at the onset of WD). (n=11–12).
(D) Representative Oil Red O staining of aortic root plaques.
(E) Quantification of aortic root lesion area (n= 11 per group)
(F) Quantification of aortic en face lesion area (n=12 per group).
(G) Quantification of macrophage content in the aortic root lesions (n=10 per group).
All data are mean±SD. Significance was determined using Student’s t-test (B) or One-Way ANOVA with Tukey’s post-hoc test (E-G). *p<0.05, **p<0.01, ***p<0.001. Both male and female mice were used.
Nlrp3-generated IL-1β downregulates ABCA1 expression in macrophages, suppressing cholesterol efflux and promoting foam cell formation
Accumulation of cholesterol in macrophages during early stages of the atherosclerotic plaque formation is a critical step during the development of this disease (Tabas and Bornfeldt, 2016), and C.pn infection accelerates foam cell formation in vitro (Chen et al., 2008). We next explored the mechanism by which NLRP3 inflammasome activity contributes to C.pn infection-accelerated atherosclerosis. We first investigated if NLRP3 inflammasome activation augments C.pn-induced foam cell formation in vitro. Co-treatment of peritoneal macrophages from WT mice with C.pn (multiplicity of infection (MOI)=5) and oxidized LDL (oxLDL) (25μg/ml) simultaneously for 24 hours led to large amounts of IL-1β and tumor necrosis factor-α (TNF-α) production, whereas NLRP3-deficient cells only secreted TNF-α (Figure S2A and S2B). NLRP3-deficiency also resulted in significantly attenuated foam cell formation as measured by Oil red-O staining (Figure 3A,B) and with a fluorometric intracellular cholesterol assay (Figure 3C). Experiments with Caspase1-deficient macrophages yielded similar results (data not shown).
Figure 3. IL-1 signaling suppresses ABCA1-cholesterol efflux in C.pn-infected macrophages and promotes foam cell formation.

(A) Representative pictures of foam cells stained with Oil red O are shown.
(B) Foam cell formation has been quantified from the peritoneal macrophages obtained from WT and Nlrp3−/− mice and stimulated with C.pn infection and oxLDL for 24hours as indicated in the figure. The intracellular lipids were stained with Oil Red O and foam cells were expressed as the percentage of positive cells.
(C) Intracellular cholesterol was measured by a fluorometric assay kit. Cholesterol measurements were normalized to total cellular protein content.
(D) Cholesterol efflux was measured using cholesterol efflux assay kit in WT and Nlrp3−/− peritoneal macrophages that were treated with C.pn and oxLDL as indicated.
(E) Fold change of ABCA1 and ABCG1 mRNA levels in WT and Nlrp3−/− peritoneal macrophages that were treated with C.pn and oxLDL are reported.
(F) Western blot (WB) and band densitometric analysis of ABCA1 and ABCG1 protein in lysates from WT and Nlrp3−/− peritoneal macrophages that were treated with C.pn and oxLDL as indicated.
(G) Peritoneal macrophages from WT and Nlrp3−/− mice were treated with C.pn and oxLDL for 24 hours were also treated with rIL-1β (5 ng/ml) or vehicle. ABCA1 protein was detected from cell lysates by WB and band densitometric analysis.
(H) Macrophages from WT and Il1r1−/− were treated with C.pn and oxLDL as indicated.
ABCA1 protein was determined from cell lysates by WB and band densitometric analysis.
A typical picture of three separate experiments is shown in A and F-H. All data represent mean ± SD; n≥3, Statistical significance was determined using Student’s t-test (D) and One-Way ANOVA with Tukey’s post-hoc test (B-E), denoted as *p<0.05, **p<0.01, ***p<0.001. Both male and female mice were used.
C.pn infection of multiple cell types has been associated with decreased cholesterol efflux (Samanta et al., 2017). While several prior studies have suggested that IL-1β inhibits the expression of ABCA1, a critical transporter for cholesterol efflux, the mechanisms for these observations remained elusive and underappreciated (Chen et al., 2007). We first asked if the C.pn-enhanced cholesterol accumulation in macrophages was related to reduced cholesterol efflux as a result of IL-1β production stimulated by the infection. For this purpose, peritoneal macrophages from WT and Nlrp3−/− mice were infected with C.pn and simultaneously loaded with oxLDL for 24 hours before measuring cholesterol efflux. C.pn and oxLDL co-treated WT macrophages displayed significantly less cholesterol efflux compared to Nlrp3−/− macrophages (Figure 3D). Addition of recombinant IL-1β (rIL-1β) to Nlrp3−/− macrophages reduced cholesterol efflux back to WT levels (Figure 3D), indicating a key role for IL-1β in limiting cholesterol efflux. Furthermore, Abca1 mRNA and protein levels, but not Abcg1 expression were induced significantly more in Nlrp3−/− macrophages when compared to WT macrophages upon simultaneous treatment of C.pn and oxLDL (Figure 3E-3F). In addition, rIL-1β treatment significantly downregulated ABCA1 expression in peritoneal macrophages both from WT and Nlrp3−/− mice (Figure 3G). Consistent with these findings, macrophages from Il1r1−/− mice that were co-treated with C.pn and oxLDL expressed higher ABCA1 protein compared to WT macrophages (Figure 3H). Collectively, these results demonstrate that C.pn infection-induced IL-1β suppresses the ABCA1-cholesterol efflux pathway, which in turn, drives foam cell formation. Because lipid uptake is also critical for foam cell formation, we also assessed the role of cluster of differentiation 36 (CD36), an important lipid influx protein and a central regulator of inflammasome activation in sterile inflammation (Sheedy et al., 2013), in C.pninduced foam cell formation in macrophages. The levels of Cd36 mRNA and CD36 protein were significantly but equally upregulated in both WT and Nlrp3−/− macrophages after simultaneous co-treatment of C.pn-infection and oxLDL, with no discernable difference between the genotypes (Figure S2C-S2D). Also, uptake of Dil-labeled oxidized LDL (DiloxLDL), were similar between the two genotypes in non-treated and C.pn plus oxLDL co-treated conditions (Figure S2E). These data suggest that Nlrp3-generated IL-1β mainly induces foam cell formation predominantly by reducing cholesterol efflux via downregulation of ABCA1 expression in macrophages.
IL-1β signaling inhibits ABCA1 expression by downregulating the GPR109a receptor
To identify the molecular mechanism underlying IL-1β-mediated inhibition of ABCA1 expression during C.pn-induced foam cell formation, we next compared the transcriptomes of WT and Nlrp3−/− peritoneal macrophages before and after simultaneous co-treatment of C.pn and oxLDL. The patterns and levels of gene expression were similar between untreated (PBS) WT and Nlrp3−/− peritoneal macrophages. Following treatment, we found 1794 genes were similarly expressed between genotypes while 1588 genes were differentially expressed (cut off > 1.5 fold, WT: 245 and 316 unique genes up- and down-regulated respectively, Nlrp3−/−: 436 and 591 unique genes up- and down-regulated respectively) (Figure 4A). Focusing on genes known to be either positively or negatively associated with ABCA1 expression, we noted that the biggest difference with C.pn and oxLDL co-treatment was in the expression of Lipase G (LIPG) and Gpr109a (Niacr1) genes that were significantly higher in Nlrp3−/− macrophages compared with WT cells (Figure 4B). We chose the Gpr109a (Niacr1, also known as Hcar2) gene to investigate further as it has a defined role in atherosclerosis and has been investigated in the context of cholesterol efflux and atherosclerosis. Furthermore, we noted that ABCG1 was only minimally changed by C.pn and oxLDL co-treatment in Nlrp3−/− macrophages compared with WT cells.
Figure 4. Gpr109a-ABCA1 pathway is upregulated by β-hydroxybutyrate produced by C.pn-infected macrophages.

(A) Venn diagram from a microarray compares the number of genes differentially expressed (1.5 fold or greater) between PBS and C.pn + oxLDL treatments in WT and Nlrp3−/− macrophages. The number of gene numbers changes associated with each genotype have been depicted in respectively-named areas. Red and blue numbers indicate upregulated or downregulated genes, respectively.
(B) The differentially expressed (1.5 fold or greater) genes between PBS and C.pn + oxLDL treated WT and Nlrp3−/− macrophages are shown. Heat maps depict relative gene expression (Z-score, left panel; Relative expression :Blue=low, Green=high) and fold change (by C.pn + oxLDL/PBS co-treatment, middle panel; Fold change, Blue=reduced expression, Red= increased expression) and the genes associated with ABCA1 for both WT and Nlrp3−/− macrophages are highlighted (right panel).
(C) Gpr109a (Niacr1) mRNA was measured by RT-PCR in peritoneal macrophages that were stimulated with C.pn and oxLDL. GAPDH gene served as a reference gene.
(D) Gpr109a protein level was detected by WB from cell lysates of WT and Nlrp3−/− macrophages treated with C.pn + oxLDL for 24 hours.
(E) Nlrp3−/− peritoneal macrophages were stimulated with C.pn and oxLDL with or without 5 ng/ml rIL-1β for 16h. Gpr109a protein was determined by WB and densitometric analysis.
(F) β-HB production was measured by a colorimetric assay in the conditioned cell culture medium from WT and Nlrp3−/− macrophages that were treated with C.pn+oxLDL or UV C.pn + oxLDL.
(G) Hmgcl1 and Bdh1 mRNA fold change was determined by RT-PCR in WT macrophages treated with C.pn + oxLDL for 24 hours.
(H) Peritoneal macrophages from Il1r1−/− mice were stimulated with UV C.pn or LPS with or without exogenous β-HB as indicated in the figure. ABCA1 and GAPDH protein was measured from the cell lysates by WB.
(I) Conditioned medium was prepared from WT and Nlrp3−/− macrophages that were infected with C.pn (MOI=5) for 24 hours. Il1r1−/− macrophages were treated with the indicated conditioned media. ABCA1 and GAPDH protein was measured from cell lysates of the Il1r1−/− macrophages by WB.
(J) Conditioned medium was prepared from Nlrp3−/− macrophages that were infected with C.pn (MOI=5) for 24 hours. WT and Gpr109a−/− macrophages were treated with the indicated conditioned media. ABCA1, Gpr109a, and GAPDH protein was measured from cell lysates of the recipient macrophages by WB.
A typical picture of three separate experiments is shown in D, E, and H-J.
All data represent mean ± SD; n≥3 . Statistical significance was determined using One-Way ANOVA with Tukey’s post-hoc test (C, F and G). *p<0.05, **p<0.01, ***p<0.001. Both male and female mice were used.
We next confirmed that GPR109a mRNA (Figure 4C) and protein levels (Figure 4D) were markedly upregulated by simultaneous co-treatment of C.pn and oxLDL in Nlrp3−/− macrophages. Additionally, treatment with exogenous rIL-1β greatly reduced the increased expression of the Gpr109a receptor that we observed in Nlrp3−/− macrophages (Figure 4E). These data suggest that IL-1β downregulates GPR109a expression in C.pn-induced foam cell formation. Gpr109a is a receptor in immune cells known for its key role in mediating niacin’s anti-atherosclerotic and anti-inflammatory effects (Digby et al., 2012; Guyton, 2007). GPR109a activation induces ABCA1 expression (Wu and Zhao, 2009). Our results therefore implicate a novel feedback regulation mechanism of the Gpr109a-ABCA1 axis in C.pn-induced and IL-1β-mediated reduction of cholesterol efflux and enhanced foam cell formation in macrophages.
β-hydroxybutyrate signals via Gpr109a to upregulate ABCA1-mediated cholesterol efflux in C.pn infected macrophages
Gpr109a receptor can bind and be activated by both of its ligands, niacin and the ketone body, β-hydroxybutyrate (β-HB) (Taggart et al., 2005). β-HB has anti-inflammatory effects (Singh et al., 2014) but its role in foam cell formation is unclear. It is known that Mycobacterium tuberculosis (M.tb)-infected macrophages produce β-HB (Singh et al., 2012). Similar to M.tb, C.pn is an obligate intracellular pathogen that requires intacellular cholesterol for its own growth and manipulates host cell cholesterol trafficking pathways, specifically by downregulating ABCA1-mediated cholesterol efflux, to actively acquire host cholesterol (Samanta et al., 2017). Therefore, we hypothesized that C.pn may also induce the production of β-HB, a Gpr109a ligand, in macrophages. Indeed, we observed that co-treatment with C.pn and oxLDL of WT and Nlrp3−/− macrophages induced the production and secretion of β-HB to a similar degree (Figure 4F). However, ultraviolet-killed Chlamydia (UV C.pn) did not induce β-HB production (Figure 4F), suggesting that live infection is required. Additionally, 3-hydroxymethyl-3-methylglutaryl-CoA lyase (HMGCL1) and 3-hydroxybutyrate dehydrogenase type 1 (BDH1), key enzymes involved in ketone body production, were also upregulated with simultaneous co-treatment of C.pn and oxLDL (Figure 4G). To further investigate the role of β-HB in ABCA1 expression, we primed WT macrophages with UV killed C.pn or LPS and then stimulated them with 1 mM butyric acid for 24 hours. Exogenous β-HB significantly upregulated ABCA1 expression in primed macrophages (Figure 4H, S3A). Moreover, conditioned media from C.pn and oxLDL co-treated (for 12 hours) WT or Nlrp3−/− macrophages induced ABCA1 protein levels in recipient Il1r1−/− macrophages (Figure 4I, S3B). Because similar levels of ABCA1 upregulation occurred in Il1r1−/− macrophages whether they received conditioned media from WT or Nlrp3−/− macrophages, the critical component in the conditioned media was independent of the inflammasome activity in the donor cells (Figure 4I). Finally, we repeated the same experiment, transferring conditioned medium from Nlrp3−/− macrophages onto WT or Gpr109a−/− macrophages. Upregulation of ABCA1 was only observed in WT recipients (Figure 4J, S3C), indicating that Gpr109a is the required receptor for this activity. Taken together, these data suggest that C.pn infection of macrophages leads to enhanced production of β-HB, which then engages its cognate receptor Gpr109a and induces ABCA1-mediated cholesterol efflux. Consequently, this sequence of events induced by C.pn infection would be expected to lead to a reduction in foam cell formation. On the contrary, because C.pn infection also induces the NLRP3 inflammasome and leads to secretion of IL-1β, we observed a feed-back suppression of Gpr109a and reduced downstream ABCA1-mediated cholesterol efflux. Inadvertently, through this mechanism, chronic C.pn infection-induced IL-1β secretion can promote foam cell formation. This auto-regulatory IL-1β feedback loop mechanism is summarized in the schematic shown in Figure S4.
In order to establish whether this regulatory pathway also exists in human cells, we checked if GPR109A expression in human macrophages was also regulated by IL-1β. Both GPR109A and ABCA1 were upregulated in human monocyte-derived macrophages stimulated with C.pn and oxLDL (Figure S3D). Importantly, the addition of an IL-1R antagonist (anakinra) led to an even higher increase of both GPR109A and ABCA1, indicating that similar to mouse macrophages, IL-1β signaling downregulates Gpr109a and ABCA1 and inhibiting IL-1β increases ABCA1 expression in human macrophages. These observations may have translational implications given the recently published CANTOS trial where inhibition of IL-1β provided a modest but significant beneficial effect in preventing subsequent cardiovascular events in patients with atherosclerosis (Ridker et al., 2017).
Nlrp3 inflammasome activity suppresses Gpr109a in atherosclerotic plaque macrophages in-vivo
The atheroprotective properties of Vitamin B3 (niacin), which binds and activates Gpr109a, have been investigated extensively (Guyton, 2007). Diet-induced obesity in rodents results in marked reduction in Gpr109a expression (Wanders et al., 2012). Additionally, the expression of Gpr109a is significantly reduced in vivo in macrophages in human carotid plaques and in vitro in macrophage-derived foam cells (Chai et al., 2013). These reports together with our data showing that IL-1β suppresses Gpr109a expression in cultured macrophages, prompted us to investigate the role of Nlrp3 inflammasome in regulating Gpr109a expression in vivo during atherogenesis. We observed that Gpr109a and Abca1, but not Abcg1 nor Cd36 mRNA levels were substantially higher in the aortic arches from Nlrp3−/−Ldlr−/− versus Ldlr−/− mice infected with C.pn and fed with WD for 16 weeks (Figure 5A). Furthermore, the expression of Gpr109a in MOMA-2 positive macrophages of atherosclerotic lesions was increased in Nlrp3−/−Ldlr−/− versus Ldlr−/− mice fed a WD with or without C.pn infection (Figure 5B-C). Similar results were obtained with Casp1−/−Ldlr−/− mice (data not shown). Taken together, these results demonstrate C.pn infection and hyperlipidemia-induced NLRP3 inflammasome activation leads to suppression of macrophage Gpr109a expression in lesions as well as ABCA1 expression in aortic arch lesions confirming our earlier observations in cultured macrophages, and demonstrating the in vivo relevance of this novel feedback mechanism.
Figure 5. Nlrp3 inflammasome signaling can suppress Gpr109a in plaque macrophages.

(A) The expression of Gpr109a , Abca1, Abcg1 and Cd36 mRNA was measured by RT-PCR from the pooled two aortic arches of Ldlr−/− and Nlrp3−/−Ldlr−/− mice (fed with WD and infected with C.pn). GAPDH gene served as a reference gene. Values are expressed as means ± SD; n=3 experiments performed each in triplicate.
(B) Representative images for immunofluorescent staining of Gpr109a in aortic root plaques. Macrophage marker MOMA-2 (green), Nuclei (blue) and Gpr109a (red). (n=9). Scale bar = 50 μm.
(C) Quantification of Gpr109a-stained macrophages in aortic root lesions from Ldlr−/− and Nlrp3−/−Ldlr−/− mice (fed a WD with and without infection with C.pn) (n=9).
All data represent mean ± SD. Statistical significance was determined using Student’s t-test or One-Way ANOVA with Tukey’s post-hoc test. *p<0.05, **p<0.01. Both male and female mice were used.
Accessibility of cholesterol to ABCA1 is decreased by IL-1β secretion through the same transporter: a role for intracellular aspartate
The data presented thus far show that extracellular IL-1β signaling inhibits ABCA1-mediated cholesterol efflux by down regulating the expression of GPR109a receptor in macrophages. Nlrp3−/− and Il1r1−/− macrophages displayed decreased foam cell formation compared with WT cells (Figure 6A), presumably because of increased ABCA1 protein expression in the absence of IL-1β-induced ABCA1 downregulation. However, we consistently observed that the foam cell formation was modestly but significantly decreased in Nlrp3−/− macrophages when compared with Il1r1−/− macrophages (Figure 6A). In contrast to Nlrp3−/− macrophages, IL-1β secretion is intact in Il1r1−/− macrophages, and consistent with the feedback mechanism, we observed that Il1r1−/− macrophages secreted significantly more IL-1β than WT macrophages after C.pn infection (Figure 6B). Therefore, we sought to uncover the underlying mechanism linking these observations. We asked whether intracellular mature IL-1β production and secretion may impact foam cell formation. IL-1β is produced without a signal sequence and secreted by various non-conventional secretory mechanisms (Eder, 2009; Heilig et al., 2017). Interestingly, earlier reports suggested that ABCA1 transporters, in addition to cholesterol transport, may also be involved in the secretion of leaderless proteins, including IL-1β (Hamon et al., 1997; Marty et al., 2005). We hypothesized that if IL-1β shares the ABCA1 transporter with cholesterol for exiting the cells, this could abrogate cholesterol efflux in C.pn-infected macrophages where significant amounts of IL-1β is produced and secreted. Recombinant mature IL-1β protein transfected into Il1r1−/− macrophages resulted in significantly more intracellular cholesterol retention when cells were incubated with oxLDL (Figure 6C). Notably, IL-1β secretion was comparable between untreated and oxLDL treated rIL-1β transfected cells (Figure 6D). We next observed that secreted IL-1β protein was significantly reduced after ATP treatment in LPS primed Abca1fl/fl LysmCre macrophages when compared with Abca1fl/fl control macrophages measured by ELISA (Figure 6E), without any change in TNF-α secretion (Figure S5A). Furthermore, the ratio of mature secreted IL-1β over intracellular pro-IL-1β was significantly reduced after ATP treatment in LPS primed Abca1fl/fl LysmCre macrophages when compared with Abca1fl/fl control macrophage by western blotting (Figure 6F and G). Additionally, we show that glyburide, a known chemical inhibitor of ABCA1 transporter, inhibited Cpn-induced IL-1β, but not TNF-α release in peritoneal macrophages (Figure S5B and C). While the mechanism by which IL-1β secretion through ABCA1 is still not well understood, sodium aspartate (NaAsp)-mediated chloride flux was shown to alter the anion exchanger function of ABCA1, leading to potentiation of IL-1β secretion through ABCA1 that traffics to the plasma membrane (Marty et al., 2005). Furthermore, chloride flux blockers, such as glybenclamide and 4,4’-diisothiocyanostylbene-1,2’-disulfonic acid (DIDS), strongly reduce IL-1β secretion via modulating ABCA1 (Domingo-Fernandez et al., 2017). To better understand the mechanism by which IL-1β secretion can also occur through ABCA1 transporter, we transfected WT and ABCA1-deficient macrophages with mature rIL-1β protein and followed by stimulation with NaAsp. We observed that NaAsp-mediated rIL-1β secretion was highly dependent on ABCA1. NaAsp was able to induce the secretion of transfected mature rIL-1β protein in WT macrophages but not in ABCA1-deficient macrophages (Figure 6H, S5D). Furthermore, NaAsp significantly enhanced IL-1β secretion induced by ATP (in LPS-primed macrophages) as well as in C.pn infected macrophages, without altering TNF-α production (Figure 6I and S5E). We also observed that after NaAsp treatment of macrophages, IL-1β increasingly co-localized with ABCA1 at the plasma membrane of macrophages by immunofluorescent analysis (Figure 6J).
Figure 6. IL-1β secretion through ABCA1 decreases cholesterol efflux and promotes foam cell formation.

(A) Macrophages from WT, Nlrp3 −/− and Il1r1−/− mice were co-stimulated with C.pn and oxLDL for 24h. Foam cell formation was determined by Oil Red O staining.
(B) Macrophages from WT, Nlrp3 −/− and Il1r1−/− mice were stimulated with C.pn for 24h and secreted IL-1β was measured by ELISA.
(C) Il1r1−/− macrophages were transfected with recombinant mature rIL-1β protein for 3h using Pro-ject transfection reagent. Some cells were stimulated with 50 ug/ml oxLDL. Total intracellular cholesterol level was measured by a fluorometric cholesterol assay.
(D) Il1r1−/− macrophages were stimulated as mentioned in Figure 6C. Secreted IL-1β was measured from the conditioned medium by ELISA.
(E) WT (Abca1fl/fl) and Abca1fl/fl LysMCre BMDM cells were stimulated with 400 ng/ml LPS for 4h plus 5mM ATP for last 30 min. Secreted IL-1β was measured by ELISA.
(F) The cells were stimulated as mentioned in Figure 6E. Pro and mature IL-1β was detected in supernatant and lysate by western blot.
(G) Band densitometry analysis for WB shown in Figure 6E, the ratio of secreted vs intracellular IL-1β normalized by GAPDH.
(H) BMDM of Abca1fl/fl and Abca1fl/fl LysMCre mice were transfected with mature rIL-1β for 3h then incubated with 10 mM NaAsp for indicated time points. IL-1β protein determined by WB in supernatant and lysate.
(I) Peritoneal macrophages of WT mice were primed with 400 ng/ml LPS for 4h plus 5 mM ATP for last 30 min; some cells were infected with C.pn for 24h. Exogenous 10 mM NaAsp were added as indicated. Secreted IL-1β was measured by ELISA.
(J) Representative fluorescence microscopy imaging of IL-1β (green), ABCA1 (red) and nuclei (blue) in peritoneal macrophages isolated from Il1r1−/− mice (left panel). Scale bar= 50 μm. Il1r1−/− macrophages were transfected with recombinant IL-1β as mentioned above, then treated with 10 mM NaAsp for 30 min. Colocalization (merge, yellow) efficiency of the ABCA1 and IL-1β is shown (left panel). The quantification of Pearson’s colocalization coefficient (R) between ABCA1 and IL-1β is shown in right panel. The arrows show membrane localization. (n=3 biological replicas, using the average of technical duplicate for each).
Values are expressed as means ± SD; n=3 performed each in triplicate (A-D; G and I). Statistical significance was determined using One-Way ANOVA with Tukey’s post-hoc test. *p<0.05, **p<0.01, ***p<0.001. Both male and female mice were used.
C.pn increases intracellular aspartate, which diverts ABCA1 to the membrane and facilitates IL-1β secretion at the expense of cholesterol efflux
Since sodium aspartate (NaAsp)-induced chloride flux was shown to traffic ABCA1 to the cell membrane and preferentially promote IL-1β secretion (Marty et al., 2005), we set out to determine the intracellular aspartate levels in our experimental conditions during C.pn infection of macrophages and other NLRP3 inflammasome-inducing stimuli which are associated with mitochondrial dysfunction (Shimada et al., 2012; Shimada et al., 2011). In our experimental conditions, addition of NaAsp to macrophages significantly increased intracellular aspartate level (Figure 7A). Strikingly, we also observed that NLRP3 inflammasome activators, including LPS plus ATP and C.pn alone or C.pn plus oxLDL treatment also increased intracellular aspartate levels in macrophages (Figure 7B) without altering intracellular alanine levels (Figure S5F).
Figure 7. C.pn increases intracellular aspartate and diverts ABCA1 to the membrane at the expense of cholesterol efflux.

(A) WT macrophages exogenously treated with 10 mM NaAsp for 30 min. Intracellular aspartate level was determined by aspartate assay.
(B) Peritoneal macrophages of WT were primed with 400 ng/ml LPS and stimulated with 5 mM ATP for 30 min; infected with C.pn and C.pn plus oxLDL for 24h. Intracellular aspartate level was determined by aspartate assay.
(C) Representative fluorescence microscopy imaging of BODIPY stained lipid droplets (green), ABCA1 (red) and nuclei (blue) in peritoneal macrophages isolated from Il1r1−/− mice. Scale bar = 50 μm. Il1r1−/− macrophages were infected with C.pn for 24h, and treated with BODIPY cholesterol for 2h and further incubated for 4h with 10 ug/ml ApoA-1. Colocalization (merge, yellow) efficiency of the BODIPY and ABCA1 is shown. The arrows show membrane localization.
(D) The quantification of Pearson’s colocalization coefficient (R) between ABCA1 and BODIPY is shown. (n=3 biological replicas, using the average of technical duplicate for each).
(E) Proposed scheme of the mechanism of competition of IL-1β secretion and cholesterol efflux through ABCA1 transporter.
Values are expressed as means ± SD; n=3 performed each in triplicate (A, B). Statistical significance was determined using One-Way ANOVA with Tukey’s post-hoc test. *p<0.05, **p<0.01, ***p<0.001. Both male and female mice were used.
We next investigated the co-localization of ABCA1 and cholesterol in IL-1R-deficient macrophages in the presence or absence of C.pn infection or NaAsp. Immunofluorescent staining data clearly showed colocalization of BODIPY-cholesterol with ABCA1 in the macrophages treated with only cholesterol (Figure 7C, top panel). However, C.pn infection translocated ABCA1 to the plasma membrane and reduced its co-localization with BODIPY-cholesterol resulting in greater intracellular cholesterol accumulation (Figure 7C, middle panel and 7D). Exogenous NaAsp treatment also induced ABCA1 relocation to the plasma membrane with reduction in colocalization with BODIPY-cholesterol and increase in intracellular cholesterol accumulation (Figure 7C, bottom panel and 7D). Furthermore, we measured the intracellular BODIPY-cholesterol accumulation in macrophages by flow cytometry and found that it was higher following stimulation with NaAsp (Figure S5G). In order to confirm our imaging data, we assessed the localization of ABCA1 to the outer membrane by Western blot analysis and observed increased ABCA1 protein in plasma membrane following C.pn infection (Figure S5H). Therefore, our findings demonstrate that by increasing intracellular aspartate, C.pn infection re-positions ABCA1 to the plasma membrane for more effective secretion of IL-1β at the expense of cholesterol efflux, leading to increased intracellular cholesterol accumulation. This represents an additional mechanism by which infection-mediated IL-1β secretion can interfere with cholesterol efflux by using the ABCA1 transporter (Figure 7E).
DISCUSSION
The lifetime pathogen burden is generally considered to play an important role in various chronic inflammatory diseases, including atherosclerosis (Elkind, 2010). C.pn, an obligate intracellular bacteria, lacks the machinery to synthesize cholesterol and hijacks host signaling and cholesterol trafficking pathways to acquire cholesterol for growth and persistence (Samanta et al., 2017). C.pn infection decreases ABCA1 experssion and induces foam cell formation in macrophages (Zhao et al., 2014), but the exact mechanism of this regulation and how C.pn promotes atherogenesis is not clear.
Here, we identified a novel mechanism by which C.pn-induced IL-1β signaling establishes a negative feedback loop that inhibits cholesterol efflux leading to foam cell formation but amplifies inflammatory responses. Gpr109a, the niacin receptor, is downregulated in atherosclerotic plaques or macrophages in both humans and mice, while mice defective in NLRP3 inflammasome activity express more Gpr109a. Our data depicts a new mechanism by which IL-1β signaling can suppress Gpr109a-induced expression of ABCA1 through its ability to meditate reverse cholesterol transport (Bi et al., 2015). We show that reduced ABCA1-mediated cholesterol efflux occurs as a consequence of IL-1β signaling and leads to more foam cell formation. However, we did not observe CD36 regulation by C.pn infection suggesting that lipid uptake is likely not a contributing factor in C.pn-accelerated foam cell formation. In summary, our findings depict a novel mechanism of C.pn-induced and IL-1β-mediated Gpr109a-ABCA1 axis can suppress cholesterol efflux and enhance foam cell formation in macrophages, accelerating atherosclerosis.
Some intracellular pathogens promote a ‘‘foamy’’ phenotype in macrophages along with the production of the ketone body known as β-HB, a ligand for Gpr109a, activating its signaling pathway (Mehrotra et al., 2014; Newman and Verdin, 2014; Singh et al., 2012). We show that C.pn-infected macrophages produce β-HB and upregulate ABCA1 expression in a Gpr109a-dependent manner. One reason for the observed ABCA1 upregulation could be to facilitate more efficient IL-1β secretion in response to infection. Consistent with other reports showing mature IL-1β can also be secreted by ABCA1 transporter (Hamon et al., 1997; Marty et al., 2005), our data also suggest that IL-1β secretion may compete with cholesterol efflux by shared use of ABCA1. Indeed, our data suggest that IL-1β signals back through its receptor (IL-1R), downregulating Gpr109a, and subsequently reducing ABCA1 in a negative auto-feedback loop. Furthermore, the in vivo relevance of this novel feedback mechanism was confirmed in Nlrp3−/−Ldlr−/− mice infected with C.pn and fed WD diet, where Abca1 and Gpr109a mRNA expression in aortic arch lesions and Gpr109a in lesion macrophages were significantly diminished.
In this study we found that C.pn-induced IL-1β secretion can occur through ABCA1, which negatively impacts cholesterol efflux by this shared transporter. Prior studies found that intracellular chloride channels and chloride ion efflux are required to induce NLRP3 inflammasome activation and mature IL-1β release (Domingo-Fernandez et al., 2017). ABCA1 is also regulated by chloride fluxes (Becq et al., 1997). Hamon et al. reported that glybenclamide, a sulfonylurea compound that is an antidiabetic and ABCA1 blocker, inhibited both the anion-exchanger function of ABCA1 and IL-1β release, suggesting IL-1β secretion was dependent on chloride fluxes (Hamon et al., 1997). ABCA1, driven to the plasma membrane by chloride fluxes, was shown to be required for IL-1β secretion in mouse Schwann cells (Marty et al., 2005). While ABCG1 is found primarily in endosomes, and cycles between endosomes, intracellular vesicles, and possibly the plasma membrane (Neufeld et al., 2001; Tarling and Edwards, 2011), ABCA1 is expressed on plasma membrane and on early and late endosomes, but the mechanisms of its intracellular trafficking and stability are still not fully defined (Santamarina-Fojo et al., 2001). Interestingly, the addition of aspartate leads to significant increase in chloride flux and plasma membrane localization of ABCA1 along with increased IL-1β secretion in mouse Schwann cells (Marty et al., 2005). In this study, we too observed that C.pn infection or ATP increased intracellular aspartate, which diverts ABCA1 to the plasma membrane and facilitates IL-1β secretion at the expense of cholesterol efflux, promoting foam cell formation in macrophages. This finding represents an additional mechanism by which either C.pn infection or acute phase response (APR)-mediated secretion of mature IL-1β can interfere with cholesterol efflux through the ABCA1 transporter. While several studies have reported that Abca1-deficient macrophages or mice display a pro-inflammatory phenotype, this is most likely due to the increased cholesterol accumulation and feedforward amplification, but not due to enhanced IL-1β secretion (Yvan-Charvet et al., 2008; Zhu et al., 2008). Indeed Abca1−/− macrophages secrete less IL-1β (Yvan-Charvet et al., 2008), consistent with our data.
As reported previously (Liu et al., 2014; Sheedy et al., 2013; Shimada et al., 2011), both oxLDL and C.pn activate NLRP3 inflammasome and produce mature IL-1β. Additional data support a causal association between IL-1β signaling and atherosclerosis (Lawler et al., 2016). We provide direct evidence showing C.pn potentiates hyperlipidemia-induced caspase-1 activity in lesion macrophages and promotes larger plaques. Conversely, genetic deficiency in either Nlrp3 or Casp1 significantly abrogates the C.pn infection-induced progression of atherosclerosis. These pro-inflammatory pathways play critical roles in not only in hyperlipidemia-induced atherosclerosis, but also in pathogen-rooted, accelerated atherosclerosis.
Our findings carry important therapeutic implications. For example, neutralization of exogenous IL-1β and inhibition of IL-1 signaling do not necessarily interfere with ongoing IL-1β production that could continue to compete for access to ABCA1 at the detriment of cholesterol efflux and sustain foam cell formation. Hence, direct therapeutic targeting of IL-1β production by the NLRP3 inflammasome could be more effective for treating atherosclerosis than just neutralizing or inhibiting exogenous IL-1β, because this will not eliminate the ongoing production of IL-1β. Small molecule inhibitors directly targeting the Nlrp3 inflammasome that prevent the maturation and release of IL-1β have been developed for various inflammatory disease conditions (Cook et al., 2010)). These NLRP3 inhibitors may also have the additional benefit of preventing the significantly increased risk of infections, including fatal infections, reported with IL-1β neutralizing strategies (LaRock et al., 2016) such as observed in the CANTOS trial, where 10,061 patients with previous myocardial infarction received a monoclonal anti-IL-1β antibody (Ridker et al., 2017). Consistently, a recent study shows that suppressing IL-1β maturation via blocking NLRP3 inflammasome does not carry the same risk of infections as do the strategies that block IL-1β R signaling (LaRock et al., 2016).
Mounting evidence indicates that Gpr109a plays an important role in modulating inflammation, and as an anti-dyslipidemic, niacin has been used for decades to prevent atherosclerosis (Guyton, 2007). Niacin was also improved long-term survival after myocardial infarction and reduce cardiovascular disease in the Coronary Drug Projectin the Coronary Drug Project (Canner et al., 1986). However, despite the beneficial effects, two recent randomized clinical studies failed to demonstrate an additional value for niacin therapy combined to statins in reducing cardiovascular events (Group, 2013; Group et al., 2014; Investigators et al., 2011). Taking into consideration our current data, which show that IL-1β signaling can suppress the niacin receptor (Gpr109a), exogenous IL-1β induced by infections or APR might reduce the efficacy of vitamin B3 (niacin) treatment. Therefore, a combination therapy comprised of an inflammasome inhibitor or anti-IL-1β agent and niacin might achieve synergistic highs in atheroprotection and improve niacin’s efficacy.
In summary, the findings in this study demonstrate that NLRP3-generated IL-1β can significantly increase foam cell formation by suppressing Gpr109a-ABCA1 pathway in macrophages. An important consequence of C.pn infection is enhancing IL-1β controlled auto-regulatory loop and effecting a net reduction in ABCA1, a key molecule for cholesterol efflux in macrophages. Innate inflammatory pathways and cholesterol metabolism are intimately linked, partly by the Liver X Receptors (LXRs) that orchestrate body cholesterol homeostasis (Castrillo et al., 2003; Chen et al., 2008). Studies have shown that innate immune activation by microbial components and the APR can suppress LXR and its target genes, including ABCA1 (Chen et al., 2008) and may explain, in part, infection-induced acceleration of atherosclerosis. Importantly, our current study now adds two additional mechanisms linking innate immune response and cholesterol metabolism. The first mechanism involves the exogenous IL1β-induced negative feedback loop that inhibits GPR109a and ABCA1 expression and the second involves potential competition for ABCA1 between intracellular mature IL1β secretion and cholesterol efflux. Our data underscore how pathogens are able to exploit signaling at the immune-metabolic interface. Understanding the modulation of the immune-metabolic interface by pathogens in detail can be guiding for future therapeutic strategies to manage atherosclerosis and other complex metbolic diseases. The mechanistic insights gained through our study may help explain some of the potential mechanisms by which blocking exogenous IL-1β can be beneficial (as in the CANTOS clinical trial), and warrant future studies using small molecule inhibitors for NLRP3 to inhibit the production of IL-1β for more effective anti-atherosclerosis therapies.
LIMITATIONS OF STUDY
Further work is needed to determine the exact mechanisms by which C.pn infection and ATP induced mitochondrial injury and NLRP3 inflammasome activation leads to increase in intracellular aspartate levels and surface translocation of ABCA1, allowing IL-1β release. Finally, the relevance of these findings to human disease needs to be assessed in future studies on cardiovascular patients.
STAR⋆Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Moshe Arditi (Moshe.Arditi@cshs.org).
Experimental Model and Subject Details
Mouse models
We have crossed for at least 8 generations and established Nlrp3−/−Ldlr−/− and Casp1−/−Ldlr−/− DKO mice. Mice used in this study were both male and female in C57BL/6 background. Age and sex matched controls were used for all of our studies. The mice were housed in a 12-hour light: dark cycle and given ad-libitum access to food and water for the duration of the study at 22 – 25 °C temperature with relative humidity of 50–70 percent.
Starting at 8 weeks of age, mice were fed a western diet (WD) (59% calories from fat, 42% from carbohydrate, 15% from protein) for 16 weeks and were infected intranasally with C. pn (5 × 104 IFU/mouse) (CM-1, American Type Culture Collection, Manassas, VA) three times (one week apart for first three consecutive weeks of WD) (Naiki et al, 2008). For bone marrow (BM) transplantation, BM from WT, Casp1−/−, and Nlrp3−/− mice was transplanted into irradiated Ldlr−/− mice. After recovery (6 weeks), chimeric mice were placed on a WD for 12 weeks and infected them as described above (n= 12/group). Whole blood was collected to confirm the efficiency of the BM transplant. Mice were housed under pathogen-free conditions at Cedars-Sinai Medical Center. All animal experiments were performed under protocols that had been approved by the Institutional Animal Care and Use Committee at our facility.
Culture and stimulation of peritoneal macrophages
Resident peritoneal macrophages were collected from both male and female mice and the pooled cells were incubated overnight in RPMI 1640 medium (Cellgro) with 10% FBS at 37°C in 5% CO2 and non-adherent cells were rinsed away. The macrophages were infected with C.pn CM-1 strain (American Type Culture Collection) at a multiplicity of infection (MOI) of 5 in the presence or absence of oxLDL (25 μg/ml; Biomedical technologies, MA, USA) for 24 h. In some experiments, macrophages were cultured with the TLR4 ligand, Escherichia coli LPS (200 ng/ml) or UV killed C.pn (MOI=5) prepared as described previously (Chen et al, 2008). Trypan blue staining showed that none of the treatments affected macrophage viability at 24 h (>95% of the macrophages were viable).
Analysis of human peripheral blood mononuclear cell (PBMC) signaling
Human blood was obtained from healthy volunteers (both men and women; 30–45 years of age) and all procedures were carried out in accordance with approved IRB protocols, including informed consent form. PBMCs were isolated by Ficoll Paque (GE Healthcare) density centrifugation. To differentiate PBMC into monocyte-derived macrophages (MDM), the cells were plated in complete RPMI1640 media. After 24 h, non-adherent cells were removed, and adherent cells were cultured in complete RPMI with M-CSF (50ng/ml) and medium changed every 2 days until the cells differentiated into macrophages by day 8–9.
Method details
Assessment of Atherosclerotic Lesions in the Aorta and Aortic Sinus
The aortas were dissected and the adherent (adventitial) fat was gently removed. Whole aortas were opened longitudinally from the aortic arch to the iliac bifurcation, mounted en face, and stained for lipids with Oil Red O. Hearts were embedded in OCT (Tissue-Tek; Sakura, Torrance, CA) and serial 7 μm–thick cryosections from the aortic sinus were mounted and stained with Oil Red O and hematoxylin. Image analysis was performed by a trained observer blinded to the genotype of the mice. Representative images were obtained and lesion areas were quantified with Image analysis software using a BZ-X710 microscope (Keyence, Itasca, IL). The lesion area in the aorta en face preparations was expressed as a percent of the aortic surface area, as previously reported (Tumurkhuu et al., 2016).
Assessment of foam cell formation by Oil Red O staining
Peritoneal macrophages were seeded on 24-well tissue culture dishes and stimulated with C.pn in the presence or absence of oxLDL for 24h, except described specifically in some experiments. The cells were washed twice with PBS, paraformaldehyde fixed, and stained for 30 min in 1% Oil Red O (in 60% isopropanol; Sigma-Aldrich). Following washing with PBS three times, the coverslips were mounted on glass slides and examined under light microscopy by trained observers blinded to the conditions (original magnification: ×40; BZ-X710 microscope (Keyence, Itasca, IL). Eight digital images were taken of representative microscopic fields for each condition in each experiment. The foam cells were expressed as the percentage of positive Oil Red O cells to total cells (Chen et al, 2008).
Immunofluorescence Staining and Image Acquisition
For immunohistochemical staining for frozen sections, fixing and antigen blocking was performed using immunoglobulin from the species of the secondary antibodies. Next, the sections were incubated with primary antibodies overnight at 4’C, followed by incubation with appropriate secondary antibodies conjugated with fluorescent dyes. For assessment of macrophage content, cells were detected using anti-MOMA-2 antibody and for co-localization staining, anti-Gpr109a antibody was used; nuclei were counterstained with DAPI. Caspase-1 activity was detected by FLICA staining, VCAM expression was detected using anti-VCAM antibody and visualized by DAB staining according to the manufacturer’s protocol. Images were captured using BZ-9000 microscope (Keyence, Itasca, IL) and analyzed by BZ analyzer software. A list of antibodies used can be found in a Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-ABCA1 | Abcam | Cat # ab18180 |
|
| ||
| Anti-Gpr109a | Biorbyt | Cat # orb13445 |
|
| ||
| Anti-MOMA2 | Abcam | Cat # 33451 |
|
| ||
| Anti-VCAM-1 | BD Pharmingen | Cat# 550547 |
|
| ||
| Anti-GAPDH | Santa Cruz | Cat # SC-47724 |
|
| ||
| Anti-6xHis | Genscript | Cat # A00174-40 |
|
| ||
| Anti-IL-1beta/IF2 | R&D system | Cat# AF-401-NA |
|
| ||
| Anti-ABCA1-DyLight 555 | ThermoFisher | Cat#PA5-22910 |
|
| ||
| Anti-ABCG1 | Abca | Cat# ab52617 |
|
| ||
| Bacterial and Virus Strains | ||
|
| ||
| Chlamydia pneumonia | CM-1 strain | American Type Culture Collection, Manassas, VA |
|
| ||
| Biological Samples | ||
|
| ||
| Healthy adult peripheral blood | ||
|
| ||
| Mouse heart | ||
|
| ||
| Mouse aorta | ||
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Recombinant IL-1b mouse | R&D | Cat # 401-ML-025 |
|
| ||
| oxLDL | Alfa Aesar | Cat # J65591 |
|
| ||
| Dil-oxLDL | Alfa Aesar | Cat # BT-920 |
|
| ||
| Recombinant Human M-CSF Protein | R&D | Cat # 216-MC-025 |
|
| ||
| Pro-ject™ protein transfection reagent kit | Thermo scientific | Cat # 89850 |
|
| ||
| L-aspartic acid sodium salt | Sigma-Aldrich | Cat#A6683 |
|
| ||
| TopFluor Cholesterol | Avanti Polar Lipids | Cat#810255P |
|
| ||
| ApoA-I | Sigma-Aldrich | Cat# A0722 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| Beta-hydroxybutyrate assay | Abcam | Cat # ab83390 |
|
| ||
| Total cholesterol assay | Cell Biolabs, CA | Cat # STA-384 |
|
| ||
| Cholesterol efflux assay | Abcam | Cat # ab196985 |
|
| ||
| Mouse IL-1b ELISA | eBioscience | Cat # 88-7013-22 |
|
| ||
| Aspartate assay | Abcam | Cat# ab102512 |
|
| ||
| Plasma membrane protein isolation kit | Abcam | Cat # 65400 |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| Ldlr −/− | Jackson Lab | stock 002207 |
|
| ||
| C57BL/6J | Jackson Lab | stock 000664 |
|
| ||
| Nlrp3−/− | University of Massachusetts Medical School, Worcester, MA | Dr. K. A. Fitzgerald |
|
| ||
| Caspase1−/− | Yale University, New Haven, CT | Dr. R. A. Flavell |
|
| ||
| Nlrp3−/−Ldlr−/− | Cedars Sinai Medical Center | Dr. Shuang Chen |
|
| ||
| Caspase1−/−Ldlr−/− | Cedars Sinai Medical Center | Dr. Shuang Chen |
|
| ||
| Gpr109a −/− | Medical College of Georgia, Augusta University | Dr. Nagendra Singh |
|
| ||
| Abca1flox/flox LysMCre | UCLA | Dr. Elizabeth Tarling |
|
| ||
| Abca1flox/flox | UCLA | Dr. Elizabeth Tarling |
|
| ||
| Oligonucleotides: PCR primers | ||
|
| ||
| Abcg1 Fw -GGTCCTGACACATCTGCGAA | This study | N/A |
| Abcg1 Rv- CAGGACCTTCTTGGCTTCGT | ||
|
| ||
| Gpr109a Fw – ATGACCAAAAATGGCGAGGC | This study | N/A |
| Gpr109a Rv - CATAGCATCGTGCCACCTGA | ||
|
| ||
| Abca1 Fw- AAAACCGCAGACATCCTTCAG | This study | N/A |
| Abca1 Rv- CATACCGAAACTCGTTCACCC | ||
|
| ||
| Cd36 Fw- GTGCTCTCCCTTGATTCTGC | This study | N/A |
| Cd36 Rv- CTGCACCAATAACAGCTCCA | ||
|
| ||
| Deposited Data | ||
|
| ||
| Foam cell microarray | This study | GEO: GSE114565 |
|
| ||
| Software and Algorithms | ||
|
| ||
| Ingenuity® Pathway Analysis | Qiagen | www.qiagen.com/ingenuity |
|
| ||
| Prism6.0 | GraphPad Software | https://www.graphpad.com |
|
| ||
| Image Lab | Bio-Rad Laboratories | http://www.bio-rad.com/en-cn/product/image-lab-software |
|
| ||
| BZ-X analyzer | Keyence | https://www.keyence.com/ss/products/microscope/bz-x700/ |
|
| ||
| Affymetrix Microarray System | Thermo Fisher Scientific | https://www.thermofisher.com/us/en/home/life-science/microarray-analysis/affymetrix.html |
|
| ||
| Other | ||
|
| ||
| High fat diet | HARLAN TEKLAD | TD88137 |
Quantitative measurement of Intracellular cholesterol
After stimulation for 24h macrophages were washed twice in PBS, intracellular lipids were extracted in chloroform/methanol (2:1) mix and dried under vacuum, and the total cholesterol (TC) were measured by Total Cholesterol Assay Kit (Cell Biolabs, CA) and normalized by total cell protein.
Total RNA isolation, microarray and real-time quantitative PCR
Peritoneal macrophages, pooled from three mice, were stimulated C.pn with or without oxLDL for 8h and RNA was purified using the RNeasy kit (QIAGEN, Valencia, CA), including treatment with DNase. RNA was analyzed using Affymetrix gene expression arrays (Affymetrix, California), which contain over 35 000 transcripts that completely cover the whole mouse genome, according to the manufacturer’s recommended protocols in the genomics core at the Cedars Sinai Medical Center. QIAGEN’s Ingenuity® Pathway Analysis (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity) was utilized to identify genes involved in the regulation of ABCA1. Heatmaps of relative gene expression and fold change were generated by Heatmapper.
Real-time PCR (qRT-PCR)
Total RNA was used as a template for RT with a High-Capacity cDNA RT kit from ABI (Warrington, UK). Real-time RT-PCR was performed in CFX96 (Bio rad) using SYBER green dye (Clontech, CA). GADPH was used as an internal control.
Cytokine assay
Cell culture supernatants were assayed using commercially available ELISA kits for murine IL-1β and TNF-α (eBioscience; San Diego, CA) according to the manufacturer’s instructions.
Western blot analysis
Whole-cell lysates were prepared with lysis buffer, separated by SDS-PAGE, and transferred to ImmobilonP membranes (Millipore). The following antibodies were used: anti-Gpr109a, anti-ABCA1, and anti-Actin (Cell Signaling Technology).
β-hydroxybutyrate (β-HB) quantification
After stimulation with C.pn with oxLDL for 24h, the levels of β-HB in the culture supernatant were measured using the beta HB Assay kit (Abcam), according to the manufacturer’s protocols.
Quantification and Statistical Analysis
All statistical analysis was performed using GraphPad Prism 6.0. Data are reported as mean ± SD. For selection of appropriate statistical tests, recorded experimental data were subjected to the D'Agostino-Pearson omnibus test to detect normal, Gaussian distribution. After verifying the Gaussian distribution, statistical differences were assessed by Student’s t test between two groups and one-way ANOVA for three or more groups (Tukey's post-test), and values of p < 0.05 were considered significant. For data with two independent variables two-way ANOVA was used with Bonferonni's post-test. Statistical parameters can be found in the figure legends.
Data and Software Availability
The accession number for the microarray-based gene expression data reported in this paper is GEO: GSE114565.
Supplementary Material
Highlights.
Nlrp3 inflammasome plays an important role in C.pn-accelerated atherosclerosis
IL-1β induces a negative feedback inhibiting Gpr109a-ABCA1 and cholesterol efflux
C.pn and aspartate divert ABCA1 to plasma membrane for IL-1β secretion
IL-1β can be exported by ABCA1 in macrophages competing with cholesterol efflux
Acknowledgments
This work has been supported by the National Institutes of Health grant HL111483 (to S.C.), AI105845 (to M.A.), and HL066436 (to M.A.). We also thank W Zhang, G. Huang and P. Sun for excellent technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
G.T, J.D, and S.C conducted the experiments. G.T, J.D, T.R.C, K.S, E.E., E.J.T., M.A, and S.C designed the experiments. G.T, T.R.C, R.A.P, K.S, E.E., E.J.T, M.A, and S.C interpreted the data. G.T, T.R.C, E.E, E.J.T., M.A, and S.C wrote the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests .
References
- Becq F, Hamon Y, Bajetto A, Gola M, Verrier B, Chimini G. ABC1, an ATP binding cassette transporter required for phagocytosis of apoptotic cells, generates a regulated anion flux after expression in Xenopus laevis oocytes. J Biol Chem. 1997;272:2695–2699. doi: 10.1074/jbc.272.5.2695. [DOI] [PubMed] [Google Scholar]
- Bi X, Vitali C, Cuchel M. ABCA1 and Inflammation: From Animal Models to Humans. Arteriosclerosis, thrombosis, and vascular biology. 2015;35:1551–1553. doi: 10.1161/ATVBAHA.115.305547. [DOI] [PubMed] [Google Scholar]
- Broderick L, De Nardo D, Franklin BS, Hoffman HM, Latz E. The inflammasomes and autoinflammatory syndromes. Annu Rev Pathol. 2015;10:395–424. doi: 10.1146/annurev-pathol-012414-040431. [DOI] [PubMed] [Google Scholar]
- Campbell LA, Kuo CC. Chlamydia pneumoniae--an infectious risk factor for atherosclerosis? Nature reviews Microbiology. 2004;2:23–32. doi: 10.1038/nrmicro796. [DOI] [PubMed] [Google Scholar]
- Canner PL, Berge KG, Wenger NK, Stamler J, Friedman L, Prineas RJ, Friedewald W. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. J Am Coll Cardiol. 1986;8:1245–1255. doi: 10.1016/s0735-1097(86)80293-5. [DOI] [PubMed] [Google Scholar]
- Castrillo A, Joseph SB, Vaidya SA, Haberland M, Fogelman AM, Cheng G, Tontonoz P. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol Cell. 2003;12:805–816. doi: 10.1016/s1097-2765(03)00384-8. [DOI] [PubMed] [Google Scholar]
- Chai J, Digby J, Ruparelia N, Jefferson A, Handa A, Choudhury R. 167 Nicotinic Acid Receptor Gpr109a Is down-Regulated in Human Macrophage-Derived Foam Cells. Heart. 2013;99:A97.91–A97. doi: 10.1371/journal.pone.0062934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Li W, Wang N, Zhu Y, Wang X. ROS and NF-kappaB but not LXR mediate IL-1beta signaling for the downregulation of ATP-binding cassette transporter A1. Am J Physiol Cell Physiol. 2007;292:C1493–1501. doi: 10.1152/ajpcell.00016.2006. [DOI] [PubMed] [Google Scholar]
- Chen S, Shimada K, Crother TR, Erbay E, Shah PK, Arditi M. Chlamydia and Lipids Engage a Common Signaling Pathway That Promotes Atherogenesis. J Am Coll Cardiol. 2018;71:1553–1570. doi: 10.1016/j.jacc.2018.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Shimada K, Zhang W, Huang G, Crother TR, Arditi M. IL-17A is proatherogenic in high-fat diet-induced and Chlamydia pneumoniae infection-accelerated atherosclerosis in mice. Journal of immunology (Baltimore, Md : 1950) 2010;185:5619–5627. doi: 10.4049/jimmunol.1001879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Sorrentino R, Shimada K, Bulut Y, Doherty TM, Crother TR, Arditi M. Chlamydia pneumoniae-induced foam cell formation requires MyD88-dependent and - independent signaling and is reciprocally modulated by liver X receptor activation. Journal of immunology (Baltimore, Md: 1950) 2008;181:7186–7193. doi: 10.4049/jimmunol.181.10.7186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook GP, Savic S, Wittmann M, McDermott MF. The NLRP3 inflammasome, a target for therapy in diverse disease states. Eur J Immunol. 2010;40:631–634. doi: 10.1002/eji.200940162. [DOI] [PubMed] [Google Scholar]
- De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends in immunology. 2011;32:373–379. doi: 10.1016/j.it.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Digby JE, Martinez F, Jefferson A, Ruparelia N, Chai J, Wamil M, Greaves DR, Choudhury RP. Anti-inflammatory effects of nicotinic acid in human monocytes are mediated by GPR109A dependent mechanisms. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:669–676. doi: 10.1161/ATVBAHA.111.241836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo-Fernandez R, Coll RC, Kearney J, Breit S, O'Neill LAJ. The intracellular chloride channel proteins CLIC1 and CLIC4 induce IL-1beta transcription and activate the NLRP3 inflammasome. J Biol Chem. 2017;292:12077–12087. doi: 10.1074/jbc.M117.797126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder C. Mechanisms of interleukin-1beta release. Immunobiology. 2009;214:543–553. doi: 10.1016/j.imbio.2008.11.007. [DOI] [PubMed] [Google Scholar]
- Elhage R, Maret A, Pieraggi MT, Thiers JC, Arnal JF, Bayard F. Differential effects of interleukin-1 receptor antagonist and tumor necrosis factor binding protein on fatty-streak formation in apolipoprotein E-deficient mice. Circulation. 1998;97:242–244. doi: 10.1161/01.cir.97.3.242. [DOI] [PubMed] [Google Scholar]
- Elkind MSV. Infectious burden: a new risk factor and treatment target for atherosclerosis. Infect Disord Drug Targets. 2010;10:84–90. doi: 10.2174/187152610790963519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage J, Hasu M, Thabet M, Whitman SC. Caspase-1 deficiency decreases atherosclerosis in apolipoprotein E-null mice. Can J Cardiol. 2012;28:222–229. doi: 10.1016/j.cjca.2011.10.013. [DOI] [PubMed] [Google Scholar]
- Group HTC. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J. 2013;34:1279–1291. doi: 10.1093/eurheartj/eht055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Group HTC, Landray MJ, Haynes R, Hopewell JC, Parish S, Aung T, Tomson J, Wallendszus K, Craig M, Jiang L, et al. Effects of extended-release niacin with laropiprant in high-risk patients. The New England journal of medicine. 2014;371:203–212. doi: 10.1056/NEJMoa1300955. [DOI] [PubMed] [Google Scholar]
- Guyton JR. Niacin in cardiovascular prevention: mechanisms, efficacy, and safety. Curr Opin Lipidol. 2007;18:415–420. doi: 10.1097/MOL.0b013e3282364add. [DOI] [PubMed] [Google Scholar]
- Hamon Y, Luciani MF, Becq F, Verrier B, Rubartelli A, Chimini G. Interleukin-1beta secretion is impaired by inhibitors of the Atp binding cassette transporter, ABC1. Blood. 1997;90:2911–2915. [PubMed] [Google Scholar]
- He X, Mekasha S, Mavrogiorgos N, Fitzgerald KA, Lien E, Ingalls RR. Inflammation and fibrosis during Chlamydia pneumoniae infection is regulated by IL-1 and the NLRP3/ASC inflammasome. Journal of immunology (Baltimore, Md: 1950) 2010;184:5743–5754. doi: 10.4049/jimmunol.0903937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, Broz P. The Gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur J Immunol. 2017 doi: 10.1002/eji.201747404. [DOI] [PubMed] [Google Scholar]
- Investigators A-H, Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. The New England journal of medicine. 2011;365:2255–2267. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- LaRock CN, Todd J, LaRock DL, Olson J, O'Donoghue AJ, Robertson AA, Cooper MA, Hoffman HM, Nizet V. IL-1beta is an innate immune sensor of microbial proteolysis. Sci Immunol. 2016;1 doi: 10.1126/sciimmunol.aah3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawler PR, Akinkuolie AO, Chandler PD, Moorthy MV, Vandenburgh MJ, Schaumberg DA, Lee IM, Glynn RJ, Ridker PM, Buring JE, et al. Circulating N-Linked Glycoprotein Acetyls and Longitudinal Mortality Risk. Circ Res. 2016;118:1106–1115. doi: 10.1161/CIRCRESAHA.115.308078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- Liu W, Yin Y, Zhou Z, He M, Dai Y. OxLDL-induced IL-1 beta secretion promoting foam cells formation was mainly via CD36 mediated ROS production leading to NLRP3 inflammasome activation. Inflammation research : official journal of the European Histamine Research Society [et al] 2014;63:33–43. doi: 10.1007/s00011-013-0667-3. [DOI] [PubMed] [Google Scholar]
- Lukasova M, Malaval C, Gille A, Kero J, Offermanns S. Nicotinic acid inhibits progression of atherosclerosis in mice through its receptor GPR109A expressed by immune cells. The Journal of clinical investigation. 2011;121:1163–1173. doi: 10.1172/JCI41651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty V, Medina C, Combe C, Parnet P, Amedee T. ATP binding cassette transporter ABC1 is required for the release of interleukin-1beta by P2X7-stimulated and lipopolysaccharide-primed mouse Schwann cells. Glia. 2005;49:511–519. doi: 10.1002/glia.20138. [DOI] [PubMed] [Google Scholar]
- Mehrotra P, Jamwal SV, Saquib N, Sinha N, Siddiqui Z, Manivel V, Chatterjee S, Rao KV. Pathogenicity of Mycobacterium tuberculosis is expressed by regulating metabolic thresholds of the host macrophage. PLoS Pathog. 2014;10:e1004265. doi: 10.1371/journal.ppat.1004265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menu P, Pellegrin M, Aubert JF, Bouzourene K, Tardivel A, Mazzolai L, Tschopp J. Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. 2011;2:e137. doi: 10.1038/cddis.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, Arditi M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:10679–10684. doi: 10.1073/pnas.0403249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naiki Y, Sorrentino R, Wong MH, Michelsen KS, Shimada K, Chen S, Yilmaz A, Slepenkin A, Schroder NW, Crother TR, et al. TLR/MyD88 and liver X receptor alpha signaling pathways reciprocally control Chlamydia pneumoniae-induced acceleration of atherosclerosis. Journal of immunology (Baltimore, Md: 1950) 2008;181:7176–7185. doi: 10.4049/jimmunol.181.10.7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld EB, Remaley AT, Demosky SJ, Stonik JA, Cooney AM, Comly M, Dwyer NK, Zhang M, Blanchette-Mackie J, Santamarina-Fojo S, et al. Cellular localization and trafficking of the human ABCA1 transporter. J Biol Chem. 2001;276:27584–27590. doi: 10.1074/jbc.M103264200. [DOI] [PubMed] [Google Scholar]
- Newman JC, Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab. 2014;25:42–52. doi: 10.1016/j.tem.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olofsson PS, Sheikine Y, Jatta K, Ghaderi M, Samnegard A, Eriksson P, Sirsjo A. A functional interleukin-1 receptor antagonist polymorphism influences atherosclerosis development. The interleukin-1beta:interleukin-1 receptor antagonist balance in atherosclerosis. Circ J. 2009;73:1531–1536. doi: 10.1253/circj.cj-08-1150. [DOI] [PubMed] [Google Scholar]
- Oram JF, Heinecke JW. ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiol Rev. 2005;85:1343–1372. doi: 10.1152/physrev.00005.2005. [DOI] [PubMed] [Google Scholar]
- Paramel Varghese G, Folkersen L, Strawbridge RJ, Halvorsen B, Yndestad A, Ranheim T, Krohg-Sorensen K, Skjelland M, Espevik T, Aukrust P, et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J Am Heart Assoc. 2016;5 doi: 10.1161/JAHA.115.003031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. The New England journal of medicine. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- Rosenfeld ME, Campbell LA. Pathogens and atherosclerosis: update on the potential contribution of multiple infectious organisms to the pathogenesis of atherosclerosis. Thromb Haemost. 2011;106:858–867. doi: 10.1160/TH11-06-0392. [DOI] [PubMed] [Google Scholar]
- Samanta D, Mulye M, Clemente TM, Justis AV, Gilk SD. Manipulation of Host Cholesterol by Obligate Intracellular Bacteria. Frontiers in cellular and infection microbiology. 2017;7:165. doi: 10.3389/fcimb.2017.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santamarina-Fojo S, Remaley AT, Neufeld EB, Brewer HB., Jr Regulation and intracellular trafficking of the ABCA1 transporter. J Lipid Res. 2001;42:1339–1345. [PubMed] [Google Scholar]
- Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, Becker CE, Ediriweera HN, Mullick AE, Golenbock DT, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nature immunology. 2013;14:812–820. doi: 10.1038/ni.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Crother TR, Arditi M. Innate immune responses to Chlamydia pneumoniae infection: role of TLRs, NLRs, and the inflammasome. Microbes and infection / Institut Pasteur. 2012;14:1301–1307. doi: 10.1016/j.micinf.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Crother TR, Karlin J, Chen S, Chiba N, Ramanujan VK, Vergnes L, Ojcius DM, Arditi M. Caspase-1 dependent IL-1beta secretion is critical for host defense in a mouse model of Chlamydia pneumoniae lung infection. PloS one. 2011;6:e21477. doi: 10.1371/journal.pone.0021477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, Thangaraju M, Prasad PD, Manicassamy S, Munn DH, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40:128–139. doi: 10.1016/j.immuni.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V, Jamwal S, Jain R, Verma P, Gokhale R, Rao KV. Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe. 2012;12:669–681. doi: 10.1016/j.chom.2012.09.012. [DOI] [PubMed] [Google Scholar]
- Tabas I, Bornfeldt KE. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circulation research. 2016;118:653–667. doi: 10.1161/CIRCRESAHA.115.306256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taggart AK, Kero J, Gan X, Cai TQ, Cheng K, Ippolito M, Ren N, Kaplan R, Wu K, Wu TJ, et al. (D)-beta-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J Biol Chem. 2005;280:26649–26652. doi: 10.1074/jbc.C500213200. [DOI] [PubMed] [Google Scholar]
- Tarling EJ, Edwards PA. ATP binding cassette transporter G1 (ABCG1) is an intracellular sterol transporter. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:19719–19724. doi: 10.1073/pnas.1113021108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumurkhuu G, Shimada K, Dagvadorj J, Crother TR, Zhang W, Luthringer D, Gottlieb RA, Chen S, Arditi M. Ogg1-Dependent DNA Repair Regulates NLRP3 Inflammasome and Prevents Atherosclerosis. Circulation research. 2016;119:e76–90. doi: 10.1161/CIRCRESAHA.116.308362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, Pfeffer K, Offermanns S. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med. 2003;9:352–355. doi: 10.1038/nm824. [DOI] [PubMed] [Google Scholar]
- Wanders D, Graff EC, Judd RL. Effects of high fat diet on GPR109A and GPR81 gene expression. Biochemical and biophysical research communications. 2012;425:278–283. doi: 10.1016/j.bbrc.2012.07.082. [DOI] [PubMed] [Google Scholar]
- Wu ZH, Zhao SP. Niacin promotes cholesterol efflux through stimulation of the PPARgamma-LXRalpha-ABCA1 pathway in 3T3-L1 adipocytes. Pharmacology. 2009;84:282–287. doi: 10.1159/000242999. [DOI] [PubMed] [Google Scholar]
- Xu XH, Shah PK, Faure E, Equils O, Thomas L, Fishbein MC, Luthringer D, Xu XP, Rajavashisth TB, Yano J, et al. Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. 2001;104:3103–3108. doi: 10.1161/hc5001.100631. [DOI] [PubMed] [Google Scholar]
- Yvan-Charvet L, Welch C, Pagler TA, Ranalletta M, Lamkanfi M, Han S, Ishibashi M, Li R, Wang N, Tall AR. Increased inflammatory gene expression in ABC transporter-deficient macrophages: free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation. 2008;118:1837–1847. doi: 10.1161/CIRCULATIONAHA.108.793869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao GJ, Mo ZC, Tang SL, Ouyang XP, He PP, Lv YC, Yao F, Tan YL, Xie W, Shi JF, et al. Chlamydia pneumoniae negatively regulates ABCA1 expression via TLR2-Nuclear factor-kappa B and miR-33 pathways in THP-1 macrophage-derived foam cells. Atherosclerosis. 2014;235:519–525. doi: 10.1016/j.atherosclerosis.2014.05.943. [DOI] [PubMed] [Google Scholar]
- Zhou X, Engel T, Goepfert C, Erren M, Assmann G, von Eckardstein A. The ATP binding cassette transporter A1 contributes to the secretion of interleukin 1beta from macrophages but not from monocytes. Biochemical and biophysical research communications. 2002;291:598–604. doi: 10.1006/bbrc.2002.6473. [DOI] [PubMed] [Google Scholar]
- Zhu X, Lee JY, Timmins JM, Brown JM, Boudyguina E, Mulya A, Gebre AK, Willingham MC, Hiltbold EM, Mishra N, et al. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J Biol Chem. 2008;283:22930–22941. doi: 10.1074/jbc.M801408200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.