

Abstract
Increased expression of cytochrome P450 CYP2C9, together with elevated levels of its products epoxyeicosatrienoic acids (EETs), is associated with aggressiveness in cancer. Cytochrome P450 variants CYP2C9*2 and CYP2C9*3 encode proteins with reduced enzymatic activity, and individuals carrying these variants metabolize drugs more slowly than individuals with wild-type CYP2C9*1, potentially impacting their response to drugs and altering their risk of disease. Although genetic differences in CYP2C9-dependent oxidation of arachidonic acid (AA) have been reported, the roles of CYP2C9*2 and CYP2C9*3 in EET biosynthesis and their relevance to disease are unknown. Here we report that CYP2C9*2 and CYP2C9*3 metabolize AA less efficiently than CYP2C9*1 and that they play a role in the progression of non-small cell lung cancer (NSCLC) via impaired EET biosynthesis. When injected into mice, NSCLC cells expressing CYP2C9*2 and CYP2C9*3 produced lower levels of EETs and developed fewer, smaller, and less vascularized tumors than cells expressing CYP2C9*1. Moreover, endothelial cells expressing these two variants proliferated and migrated less than cells expressing CYP2C*1. Purified CYP2C9*2 and CYP2C9*3 exhibited attenuated catalytic efficiency in producing EETs, primarily due to impaired reduction of these two variants by NADPH-P450 reductase. Loss-of-function SNPs within CYP2C9*2 and CYP2C9*3 were associated with improved survival in female cases of non-small-cell lung cancer. Thus, decreased EET biosynthesis represents a novel mechanism whereby CYPC29*2 and CYP2C9*3 exert a direct protective role in NSCLC development.
Introduction
CYP2C9 is a human microsomal cytochrome P450 (CYP) enzyme that metabolizes several xenobiotics and endogenous compounds, including arachidonic acid (AA) (1). Two polymorphic variants in this gene, CYP2C9*2 (R144C) and CYP2C9*3 (I359L), have significantly reduced enzymatic activity (2, 3). Individuals with two of these alleles are called “poor metabolizers” of CYP2C9 substrates, as they oxidize drugs slower than individuals carrying wild type CYP2C9*1. Altered drug responses in these people make them either more protected or more at risk of disease, depending on the situation (4).
CYP2C9 inactivates several non-steroidal anti-inflammatory drugs (NSAIDs), including cyclooxygenase inhibitors and chemopreventive agents. High-doses of the NSAID celecoxib are more effective in preventing colorectal adenomas in CYP2C9*3 individuals (5), and the presence of CYP2C9*2 or CYP2C9*3 alleles increases the protective effect of ibuprofen against colorectal cancer (6). Thus, chemopreventive effects of NSAIDs in colorectal cancer associate with slower NSAID metabolism.
CYP2C9*2 and CYP2C9*3 variants are also positively associated with the risk of cancer. Tobacco intake results in a several fold increased risk of head and neck squamous cell carcinoma (HNSSC) in CYP2C9*2 individuals (7). Moreover, individuals with HNSSC carrying CYP2C9*2 or CYP2C9*3 variants respond poorly to cisplatin treatment (7). Thus, whether CYP2C9*2 and CYP2C9*3 protect from cancer risk and progression depends on cancer type, treatment, and etiology.
Given the role of CYP2C9 and its variants in cancer risk and outcomes, it may be a key target for cancer treatment and prevention. Members of the CYP2C subfamily, including CYP2C9, have epoxygenase activity and catalyze the oxidation of AA to epoxyeicosatrienoic acids (EETs). Besides promoting vasodilation, lowering blood pressure, and contributing to insulin sensitivity (8, 9), EETs are mitogenic and pro-angiogenic lipids (10). Specifically, human endothelial cells (ECs) express CYP2C9 (11) that promotes proliferation, tubulogenesis, and cell invasion (12). Downregulation of CYP2C9 expression by PPAR-α ligands reduces EET biosynthesis in ECs as well as proliferation and tubulogenesis (13). Also, when injected with tumor cells, Cyp2c44KO mice (that lack the functional homolog of human CYP2C9) develop smaller and less vascularized tumors than wild type mice (13). Furthermore, inhibition of EET biosynthesis in mice and reduction of circulating EET levels by PPAR-α ligands decrease tumor angiogenesis and primary and metastatic growth of non-small cell lung cancer (NSCLC) cells (14). Finally, CYP2C9 is highly expressed in the vasculature of human NSCLC (13). These studies, together with data showing that increased expression of CYP2C9 and elevated EET levels associate with aggressive human cancer (15), suggest that inhibition of CYP2C9-based EET biosynthesis may reduce cancer growth and progression.
Although CYP2C9 genetic variation can alter the oxidation of AA (16), the contributions of CYP2C9*2 and CYP2C9*3 to EET biosynthesis and their relevance to disease have not been fully described. Here, we determined if the CYP2C9*2 and CYP2C9*3 variants metabolize AA less efficiently than CYP2C9*1 and whether they protect from NSCLC cancer development due to reduced ability to generate EETs. We show that human NSCLC cells expressing CYP2C9*2 and CYP2C9*3 produce significantly lower levels of EETs and develop fewer, smaller, and less vascularized tumors than tumor cells expressing wild-type CYP2C9*1 when injected into mice. Moreover, ECs expressing these two variants proliferate and migrate less than cells expressing CYP2C9*1. Importantly, the loss-of-function CYP2C9 SNPs rs1799853 (CYP2C9*2), and rs1057910 (CYP2C9*3) are associated with improved survival in women with NSCLC adjusted for cancer stage, surgical resection, and chemotherapy. Thus, decreased biogenesis of pro-angiogenic EETs appears to represent a novel mechanism whereby CYP2C9*2 and CYP2C9*3 exert a direct protective role in NSCLC cancer development.
Materials and Methods
Materials
All chemicals were purchased from Sigma-Aldrich (St Louis, MO) unless specified otherwise. 14C-Arachidonic acid (AA) was purchased from PerkinElmer (Waltham, MA). The EET analog EET-A (14) and the EET inhibitor 14,15-epoxyeicosa-5(Z)-enoic acid (14,15-EEZE) (17) were generated by JRF.
Site-directed mutagenesis
The human CYP2C9 construct in the vector pCW was a gift from Dr. Eric F. Johnson (The Scripps Research Institute, La Jolla, CA) (18). This construct lacks the first 22 N-terminal residues, with the coding sequence starting at residue 23 and the first 5 residues being MAKKT. The C-terminal valine was replaced with an isoleucine followed by a 6-histine tag to facilitate purification. Mutagenesis of the wild type enzyme (CYP2C9*1) to CYP2C9*2 (R144C) and CYP2C9*3 (I359L) was performed using the QuikChange II XL Site-Directed Mutagenesis kit (Agilent Technologies) using the primers and PCR conditions described in Supplementary Table S1.
Enzyme expression and purification
CYP2C9 variants were purified from Escherichia coli transformed with pCWCYP2C9*1, pCWCYP2C9*2, and pCWCYP2C9*3 constructs using the basic procedures described elsewhere (19), except that a C-terminal (His)6 tag was used. The proteins were purified using Ni2+-nitrilotriacetic and hydroxylapatite chromatography. Recombinant rat NADPH-P450 reductase was prepared from E. coli as described (20).
In vitro enzymatic activity
Rates of AA oxidation by CYP2C9*1 and its variants were measured using a mixture of 100 nM CYP2C9 protein in 100 mM Tris-HCl buffer (pH 7.4) containing 10 mM MgCl2, sodium isocitrate (2mg/ml), isocitrate dehydrogenase (0.2 IU/ml), L-α−1,2-dilauroyl-sn-glycero-3-phsphocholine (100 μg/ml), 1.0 μM NADPH-P450 reductase, 14C-AA (70 μM), and 1 mM NADPH at 37 oC. Products were quantified by radio-HPLC. kcat and Km values were calculated using the Michaelis-Menten equation with nonlinear regression (Origin Lab Software). NADPH oxidation rates were determined using the A340 decrease, in duplicate. Hydrogen peroxide formation was monitored with a xylenol orange dye method (21).
Absorbance spectra were recorded as described by Jefcoate et al. (22). The binding constant, Ks, and the extrapolated maximum spectral change (Bmax) were estimated by nonlinear regression (Origin Lab Software). Second-derivative spectra were obtained using the manufacturer’s software (OLIS, Bogart, GA), with the application of the curve smoothing program and a zero-baseline method (23).
Reduction of ferric P450 to the ferrous form was measured at 37 °C under an anaerobic CO environment using anaerobic techniques, as described in detail (24), with the concentrations of CYP2C9 proteins and NADPH-P450 reductase were 2 and 4 μM, respectively. All experiments were done in an OLIS-RSM-1000 stopped-flow apparatus at 37 °C under CO, with data collected at 450 nm (Fe2+·CO complex). Traces are presented as averages of several individual reactions (>15) (25).
Generation of adenoviruses
Full length CYP2C9*1, CYP2C9*2 and CYP2C9*3 cDNAs were cloned into pre-linearized pAdenoX vector DNA within the E1/E3-deleted replication-incompetent human adenoviral vector using in-fusion enzyme (Adeno-XTM adenoviral system 3, Clontech laboratories). Upon application into StellarTM E. coli, Pac-1 linearized recombinant adenoviral DNA was used to transfect Adeno-X 293 cell lines. Adenovirus was collected from cell lysates upon detection of cytopathic effect and purified using the Adeno-X purification kit. Upon determination of the adenovirus titer (ifu/ml) with Adeno-X rapid titer kit, adenovirus stocks were stored at −80 oC.
Cells
A549 and HUVEC cells were purchased from ATCC and cultured according to the manufacturer’s instructions (ATCC CLL-185TM, ATCC CRL-1730TM). Cyp2c44KO ECs were isolated from the lungs of Cyp2c44KO mice and cultured as described (14). A549 and HUVEC cells were used within 15 days from thawing and passaged no more than 2 times per each experiment. Cyp2c44KO ECs were used within 10 days from isolation and passaged no more than 2 times per each experiment. All cell lines were tested for mycoplasma immediately upon purchase and/or isolation. A549 and HUVEC cells were characterized and authenticated by the Vendor (e.g., cytogenic analysis, expression of specific markers). Cyp2c44KO ECs were authenticated immediately after isolation based on their cobblestone shape and expression of endothelial markers (e.g. CD31) and only population of >90% pure endothelial cells were used for experiments.
Cell proliferation
Cells (3 × 105) were plated in 6-well plates in complete medium. The following day, the cells were treated in triplicate with empty adenovirus (20 ifu/cell, vector) or adenovirus carrying CYP2C9*1 (20 ifu/cell), CYP2C9*2 (20 ifu/cell), or CYP2C9*3 (40 ifu/cell) in serum free medium. After 4 hrs, an equal volume of complete medium was added and the cells were incubated at 37 °C. After 24 hrs, the cells were trypsinized and plated in 96-well plates in complete medium (1 × 104 cells/well). The rest of the cells was pelleted and frozen at −80 °C for western blotting analysis. After 12 hrs, the cells were incubated with serum-free medium with or without AA (1–5 μM), the EET analog EET-A (1.25–5 μM) (14), the EET inhibitor 14,15-epoxyeicosa-5(Z)-enoic acid (14,15-EEZE, 1.25–5 μM) (17), or the reactive oxygen species scavenger TEMPOL (1–5 μM) and with 1 μCi 3H-thymidine. After 48 hrs, proliferation was determined as described (26). Two to three independent experiments were performed at least in triplicate.
Analysis and quantification of epoxygenase metabolites
Cells (3 × 105) were plated in 6-well plates in complete medium. After 24 hrs, the cells were infected in quadruplicate with the adenovirus constructs described above. After 72 hrs, the cells were incubated in serum-free medium for 24 hrs. Cells were incubated with AA (10 μM), and after 4–5 hrs the cells in one well were collected for analysis of CYP2C9 proteins by western blotting analysis. Cells and medium from the other three wells were used for analysis of EETs and their hydrolysis products (DHETs) using a ultra high performance HPLC/tandem mass spectrometry as described (27). The values, reported as EETs + DHETs ng/mg total cell protein or ng/ml total medium, were adjusted for CYP2C9 transfection efficiency. Two independent experiments were performed.
2D migration assay
A549 cells, infected as described above, were plated in a Culture-insert 2 Well in 35 mm μ-dish consisting of a 2-well silicone insert with a 500 μm defined cell-free gap (Ibidi, catalog 80206B). Cells (5 × 104/insert) were plated in 70 μl of complete medium for 4 hrs and then in serum-free medium for 24 hrs. The insert was subsequently removed and the cells were incubated with 2 ml medium containing 1% fetal calf serum (v/v) with or without 1 μM AA, 5 μM EET-A, 5 μM 14,15-EEZE, or 5 μM TEMPOL. Pictures were taken at 0 and 24 hrs from the removal of the insert using an Olympus IX81 (slidebook6: acquisition software). Images of the gap were acquired with a 40x lent (air) and phase contrast. Three-four images/time points were taken for each cell line and Image J software was used to analyze the area of the gap. Data are expressed as % of gap area closed relative to zero time. Two-three experiments were performed.
Cell migration
Transwells (24-well) were used with the following modifications. A549 cells (3 ×105) infected for 24 hrs as described above were plated in complete medium at the bottom of a 24-well plate. After 24 hrs, the cells were incubated in 500 μl of serum free medium with or without 5 μM AA, or 14,15-EEZE (5 μM). Transwells (8-μm pores whose undersides were precoated with 2 μg/ml matrigel from BD Pharmingen) were added to the wells containing A549 cells. In some experiments, transwells were added to wells containing serum free medium only with or without EET-A (1,25–5 μM). HUVEC or Cyp2c44KO ECs (1 × 105 cells in 200 μ serum free medium) were added to the upper wells and allowed to migrate through the Transwell for 4 hrs. ECs on the top of the filter were removed by wiping, and the filter was then fixed in 1% formaldehyde (w/v) in PBS. Migrating ECs were stained with 1% crystal violet (w/v), and 4 random fields from triplicate wells were counted at 200-fold magnification.
Animals and tumor studies
Experiments were approved by the Vanderbilt University IACUC, and NIH principles of laboratory animal care were followed. Mice were housed in an AAALAC-accredited, temperature-controlled facility with a 12 h light–dark cycle. A549 cells (3 × 105) were infected with empty adenovirus (50 ifu/well) or adenovirus carrying CYP2C9*1 (50 ifu/well), CYP2C9*2 (50 ifu/well), or CYP2C9*3 (70 ifu/well). These ifu doses were chosen based on pilot experiments showing that CYP2C9* protein expression persists up to 10 days after adenovirus infection. After 24 hrs, 1 × 106 cells in 200 μl PBS containing 20 μl Matrigel were injected s.c. into the back of 12-week old athymic nude mice (2 injection/mouse). Two weeks later, when A549 cells form measurable primary tumors (27), the mice were sacrificed and the number and volume of tumors were analyzed. Tumor volumes were calculated using the following formula: tumor volume (mm3) = (length × width2)/2.
Immunofluorescence
Frozen tumor sections (7 μm) were stained with rat anti-mouse CD31 antibody (1:400, Amersham Biosciences), followed by rhodamine isothiocyanate-conjugated goat anti-rat IgG (1:200, Jackson ImmunoResearch). The degree of vascularization, was evaluated using Image J software with one image/tumor and 8–11 mice/tumor cell line (26).
A549 cells non-infected or infected as described above were plated onto 8 well chamber slides (Ibidi) (1 × 104/well) in complete medium. After 24 hrs they were cultured in medium containing 1% FCS with or without different concentrations of the EET analog EET-A (14). After 24 hrs, the cells were fixed with 4% paraformaldehyde in PBS, permeabilized with 0.05% Triton X-100 in 3% BSA in PBS, and subsequently stained with rhodamine-phalloidin (ThermoFisher Scientific) to visualize the cytoskeleton.
Western blotting analysis
Cell lysates (10 μg/lane) from adenovirus infected cells were resolved by SDS-PAGE in 10% gels and transferred to Immobilon-P membranes (Millipore). The membranes were incubated with rabbit anti-human CYP2C9 antibody (1:100, v/v, Abcam) or anti-rabbit FAK (1:1000 Santa Cruz). Immunoreactive proteins were visualized using a peroxidase-conjugated goat anti-rabbit antibody and an ECL kit (Pierce).
Human NSCLC cases from BioVU
Clinical data and DNA for NSCLC cases were extracted BioVU a biorepository consisting of DNA derived from discarded blood samples linked to the de-identified electronic medical records (EMR) at Vanderbilt University Medical Center (28). Cases were identified based on ICD-9 codes for NSCLC at least three times in the EMR which also provided date of diagnosis and NSCLC histology. Subsequent manual review confirmed case and ever-smoking status. Where the BioVU algorithm (29) and manual review were discordant for smoking status, the manual review of smoking status was used. Samples with ambiguous smoking history or never smokers were excluded from subsequent analyses. Other relevant data were extracted by manual review. Staging was determined using the American Joint Committee on Cancer TNM System staging guidelines at the time of diagnosis. Local (stage I), regional (stage II and III), and distant (stage IV) stages were specified (30). Cancer-specific treatments, including chemotherapy, radiation, and resection, along with dates of either last known follow-up or death, were obtained from the EMR. A total of 398 NSCLC cases with staging data were available, with 114 observed deaths in the EMR.
Genotyping
Tag single nucleotide polymorphisms (SNPs) were selected for CYP2C9 including ± 10 kb from the coding region. The criteria for tagging were: 1) minor allele frequency (MAF) > 0.1, and 2) linkage disequilibrium (LD) between SNPs of r2 < 0.8 in the HapMap CEU population, as the BioVU case-control study was entirely European American. Nine tag SNPs were included for genotyping along with functional SNPs CYP2C9*2 (rs1799853), and CYP2C9*3 (rs1057910) (Supplementary Table S2). Genotyping was performed on a Sequenom (n=10) or Taqman (n=1) platform.
Quality control prior to analyses including the following criteria: 1) only samples and SNPs with genotyping efficiency greater than 95%; and 2) Hardy-Weinberg equilibrium p-values > 0.01 (Supplementary Table S3). No samples or SNPs were excluded. Quality control was performed with PLINK (31).
NSCLC survival analyses
Non-genetic variables including age at diagnosis, body mass index (BMI), sex, NSCLC stage and histology, surgical resection, chemotherapy, and radiation therapy were examined for an association with NSCLC survival using the Kaplan-Meir (KM) estimator. Variables that associated significantly with outcome were adjusted for all other associating variables and tested for independent association in a multivariate Cox proportional hazard model.
The effect of SNPs was first assessed using the KM estimator using an additive model. For SNPs with fewer than five homozygotes of the minor allele, homozygous individuals were collapsed with heterozygotes for association analyses using a dominant model (rs17057291). Because both loss of function CYP2C9 variants reduced enzyme activity, they were analyzed together in a dominant model by pooling into a single class.
Genetic variants associating with survival with a KM p-value < 0.1 were further modeled with Cox proportional hazard multivariate models. The effect of these SNPs was adjusted for NSCLC staging, resection, chemotherapy, and sex. In addition, sex-stratified analyses were performed with adjustment for the variables noted above. All the above statistical analyses were conducted using STATA 12.0 (College Station, TX). A p ≤ 0.05 value was considered statistically significant.
Statistical analysis
In vitro and in mouse in vivo studies: To test for differences between the CYP2C9* proteins in regards to tumor volume and vascularization as well as EET biosynthesis and effects of cell functions, we first tested whether the variances differed between the two groups with an F-test. To compare means between any two groups, the two-sampled t-test or the Welch’s t-test was used for groups with equal variances and unequal variances, respectively. All the above statistical analyses were conducted using STATA 12.0 (College Station, TX). A p value < 0.05 was considered to be statistically significant.
Results
Decreased tumorigenesis of A549 cells expressing CYP2C9*2 and CYP2C9*3
We investigated whether the loss-of-function proteins CYP2C9*2 and CYP2C9*3 play a protective role in cancer due to potentially impaired ability to generate EETs. We chose NSCLC because it is a highly vascularized cancer with upregulated expression of CYP2C9 in the endothelium (13). We generated A549 NSCLC cells expressing empty adenovirus (A549-V) or adenovirus carrying CYP2C9*1 (A549–2C9*1), CYP29C*2 (A549–2C9*2), or CYP29C*3 (A549–2C9*3) cDNAs (Fig. 1A). We used A549 cells because they: 1) form tumors upon subcutaneous (s.c.) injection in athymic nude mice (27); 2) do not express endogenous CYP2C9 (14); and 3) produce low levels of EETs (see below and (14)). Two weeks after s.c. injection, tumor number and volume were evaluated. Most mice developed tumors (Fig. 1B), although tumors derived from A549–2C9*1 cells were more numerous, bigger, and more vascularized than tumors derived from A549-V, −2C9*2, and −2C9*3 cells (Fig. 1C-F). No significant differences in tumor number, size, or vascularization were observed in mice injected with A549-V cells compared to those injected with A549–2C9*2 and −2C9*3 cells (Fig. 1C-F). Thus, CYP2C9*2 and CYP2C9*3 are associated with reduced tumorigenesis compared to CYP2C9*1.
Figure 1.
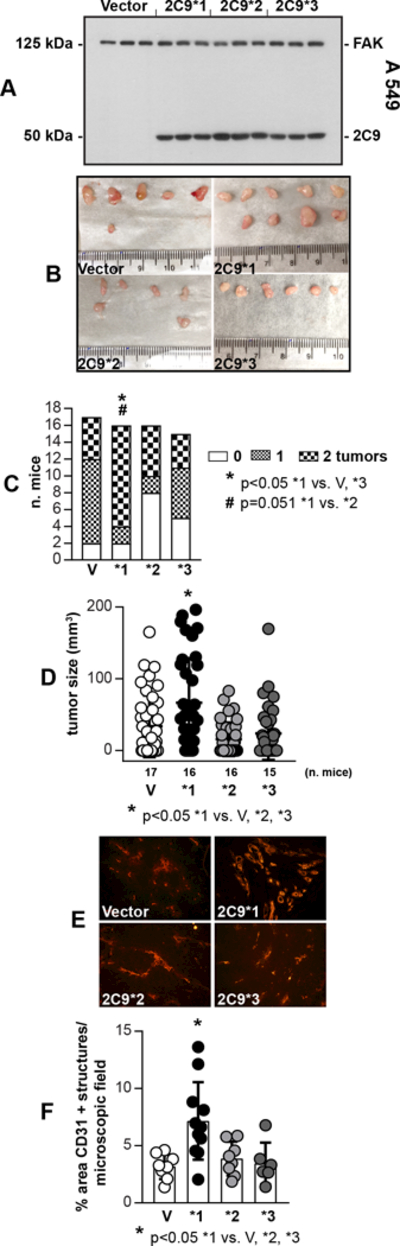
CYP2C9*1 promotes tumorigenesis. A, Cell lysates from A549 cells infected with empty adenovirus (Vector), or adenovirus carrying CYP2C9*1, CYP2C9*2, or CYP2C9*3 cDNAs were analyzed by Western blotting for expression of CYP2C9 and focal adhesion kinase (FAK, used as loading control) (n=3 infections). B, Images of tumors isolated from mice 14 days after receiving two s.c. injections of A549-Vector, −2C9*1, −2C9*2, or −2C9*3 cells. C, Tumor uptake was evaluated by counting the number of tumors in each mouse. The mice were divided into three groups: no tumors (0% uptake), one tumor (50% uptake), and two tumors (100% uptake). D, Tumor volume was evaluated with a caliper. Circles show individual tumors, while bars and errors show mean values and SD E, F, Tumor frozen sections were stained with anti-mouse CD31 antibody and vascularization was quantified as described in the Methods. Circles show individual mice, while bars and errors show mean values and SD.
Reduced EET production in A549 cells expressing CYP2C9*2 and CYP2C9*3
To determine the mechanisms whereby CYP2C9*2 and CYP2C9*3 affect tumorigenesis, we utilized a GC/MS method (32) to measure the levels of EETs and their hydrolysis products, DHETs, in A549-V, −2C9*1, −2C9*2 and −2C9*3 cells incubated with exogenous AA. Intracellular and secreted EETs and DHETs were detected in A549 cells transfected with vector only or expressing all CYP2C9* variants, although they were significantly higher in A549–2C9*1 cells (~1000–2000 ng/mg total proteins), followed by A549–2C9*2 cells (~750–950 ng/mg total proteins) and with the lowest levels in A549-V and A549–2C9*3 cells (~40–300 ng/mg total proteins) (Fig. 2A, 2B). We detected 8,9-EET, 11,12-EET, 14,15-EET and their corresponding DHETs in A549-V cells incubated with AA, with 11,12-EET/DHET representing the highest regioisomeric product (Fig. 2C). Expression of CYP2C9*1 and CYP2C9*2, but not CYP2C9*3, resulted in increased production of all three EET+DHET products compared to A549-V cells (Fig. 2C). However, 11,12-EET/DHET levels were still significantly higher than the other two products and overall higher in A549–2C9*1 than A549–2C9*2 cells (Fig. 2C). Thus, CYP2C9*1 is the most metabolically active epoxygenase.
Figure 2.
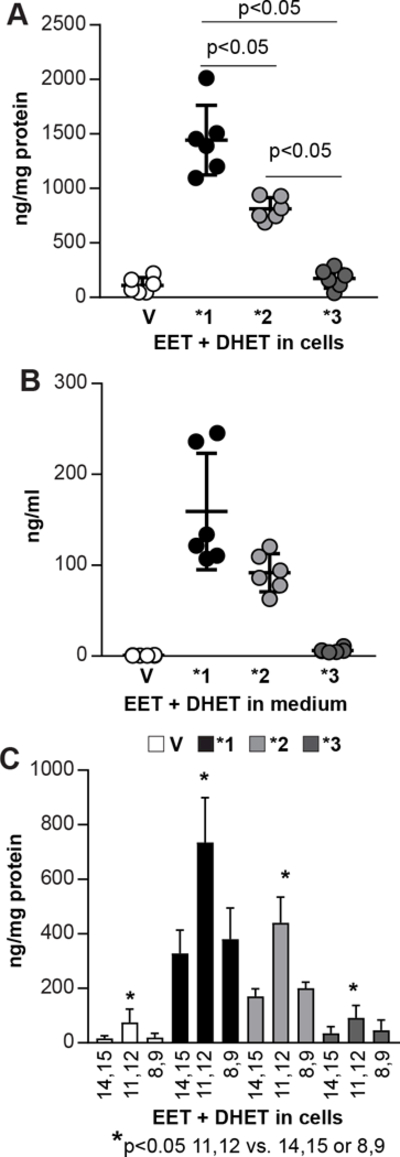
Analysis of EET production in A549 cells expressing CYP2C9* variant proteins. A, B, A549-vector (V), −2C9*1 (*1), −2C9*2 (*2), or −2C9*3 (*3) cells were incubated in serum-free medium with AA. After 4–5 hrs, cells and medium were collected, and the EETs and DHETs were quantified as described (32). Circles indicate individual cell plates, and bars and errors show mean values and SD. The values in A and B are reported as ng/mg total cell protein or ng/ml total medium of EET + DHET (n=2 experiments). C, Levels of individual 8,9-, 11,12-, and 14,15- EET regioisomers and their DHET metabolites detected in A549 infected and analyzed as indicated above. Values are reported as ng/mg total protein (n=2 experiments).
EETs produced by A549 cells expressing CYP2C9* variants influence cell migration, but not proliferation
Because EETs exert mitogenic action (33), we examined the proliferation of A549 cells expressing empty adenovirus or the CYP2C9* variants with or without AA. No differences in proliferation were observed among the various cell lines, independent of the addition of AA (Fig. 3A). Thus, EETs do not promote A549 proliferation.
Figure 3.
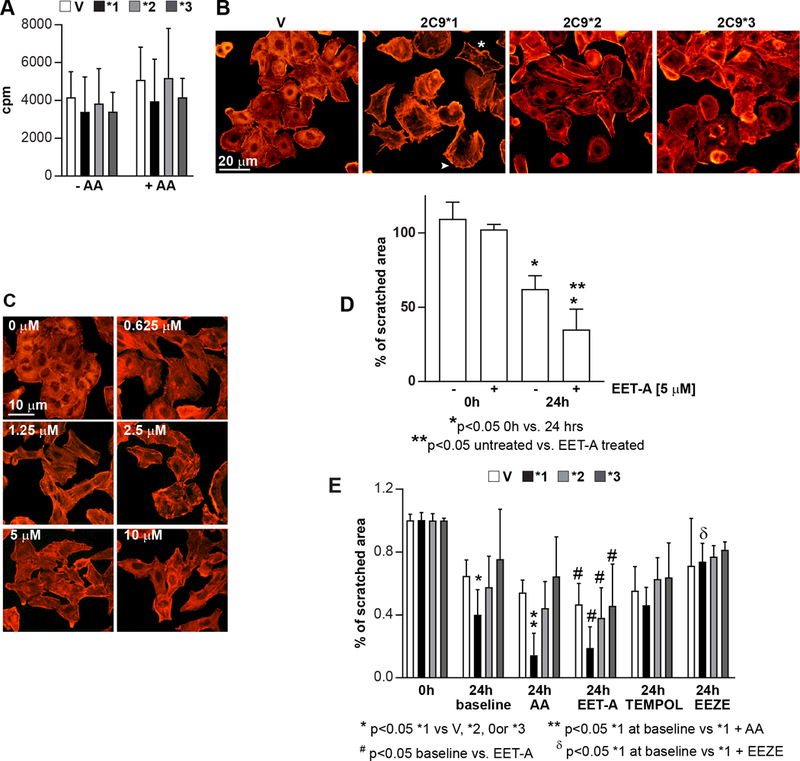
Effects of CYP2C9* variants on A549 proliferation, migration, and morphology. A, A549 cells infected as described in Fig. 2A were plated in 96-well plates in complete medium. After 12 hrs, the cells were incubated with serum-free medium with or without AA and with 3H-thymidine. After 48 hrs, proliferation was determined as described (26). Values represent the mean ± SD of two experiments performed in triplicate. B, A549 cells (infected as described in Fig. 2A) were plated onto chamber slides in complete medium. After 12 hrs the cells were serum starved and after 24 hrs they were stained with rhodamine-phalloidin. A549-CYP2C9*1 cells show a pro-migratory phenotype, with elongated shape (*) and lamellopodia-like protrusions (arrowhead). C, A549 cells were plated onto chamber slides in complete medium. After 12 hrs the cells were incubated in serum containing 1% FCS with or without different concentrations of the EET analog EET-A. After 24 hrs they were stained with rhodamine-phalloidin. D, Scratch wound assays were performed on A549 cells, untreated or treated with the EET analog EET-A as described in the Methods. Values are the mean ± SD of a representative experiment with four images/cell type. (E) Scratch wound assays was performed on A549 cells infected with the various CYP2C9* variants. Expression of CYP2C9*1 significantly increased the migration of A549 cells incubated with or without AA for 24 hrs. Addition of EET-A significantly increased migration of all cell types, while addition of EEZE, but not TEMPOL, inhibited migration of A549-CYP2C9*1 cell only. Values are the mean ± SD of 2–3 independent experiments with 4 images/cell type.
EETs also promote cell migration and invasion. Analysis of A549 expressing CYP2C9* variants showed that the morphology of A549–2C9*2 and −2C9*3 cells resembled that of A549-V cells with typical epithelial morphology (Fig. 3B). In contrast, A549–2C9*1 cells presented a migratory-like phenotype with an elongated shape, the presence of lamellopodia-like protrusions, and they grew more dispersed (Fig. 3B). To ensure that the effects on cell morphology observed in A549–2C9*1 cells were due to high levels of EETs, we treated A549 cells with the EET analog EET-A (14). Similar to A549–2C9*1 cells, EET-A-treated A549 cells showed a migratory-like phenotype (Fig. 3C). Using a 2-dimentional modified ‘scratch assay’, we examined the ability of A549 cells to migrate for 24 hrs towards a defined cell-free gap with or without EET-A. Without EET-A, A549 cells partially migrated towards the cell free gap and their migration was significantly enhanced by treatment with EET-A in a dose-dependent manner (Fig. 3D). Next, we evaluated the migratory capacity of A549 cells expressing the various CYP2C9* variants at baseline or following treatment with AA. Without any treatment, all cells partially migrated towards the cell free gap with the highest migration observed for A549–2C9*1 cells and the lowest migration detected for A549–2C9*3 cells (Fig. 3E). When AA was added, the gap was significantly smaller for A549–2C9*1 cells, while the other three cell types still failed to migrate (Fig. 3E).
CYP2Cs produce reactive oxygen species (ROS) (34) which promote cell proliferation and migration (35). To further analyze the contribution of EETs in mediating migration of A549–2C9*1 cells, we performed a ‘scratch assay’ in the presence of the EET analog EET-A (5 μM), the EET antagonist 14,15-EEZE (5 μM) (17), or the ROS scavenger TEMPOL (5 μM). Addition of EET-A significantly increased the migration of all four cell types, with the highest migration still detected for A549–2C9*1 cells (Fig. 3E). Addition of TEMPOL did not exert any effect on cell migration, while treatment with 14,15-EEZE significantly impaired migration of A549–2C9*1 cells only (Fig. 3E). All together, these results indicate that EET analogs promote A549 cell migration independently of the CYPC9* variants they express, while EET antagonists, but not ROS scavenger, inhibit migration of A549 cells expressing the most metabolically active CYP2C9*1 isoform.
ECs expressing CYP2C9*2 and CYP2C9*3 show impaired cell migration and proliferation
Since EETs are pro-angiogenic lipids (10), we examined the effects of CYP2C9* variants on EC function by generating HUVECs and murine Cyp2c44KO ECs expressing CYP2C9*1, CYP2C9*2, and CYP2C9*3 (Fig. 4A, 4B). We selected HUVECs as they express undetectable basal levels of CYP2C9 (Fig. 4A). HUVECs or Cyp2c44KO ECs expressing CYP2C9*1 proliferated significantly more than cells infected with empty adenovirus, CYP2C9*2 or CYP2C9*3 cDNA (Fig. 4C). In the presence of AA, only ECs expressing CYP2C9*1 grew significantly more than cells grown in the absence of AA (Fig. 4C). However, no increase in proliferation was observed in ECs infected with empty adenovirus or CYP2C9*2 or CYP2C9*3 (Fig. 4C). To determine whether the differences in proliferation were due to EETs, we treated ECs expressing vector or the various CYP2C9* variants with EET-A (1,25–5uM). Compared to untreated cells, treatment with EET-A significantly stimulated the proliferation of both HUVEC and Cyp2c44KO ECs and this effect was evident in cells expressing vector as well as any of the CYP2C9* variants (Fig. 4D). To confirm that the increased proliferation observed in EC expressing CYP2C9*1 is due to EET production only, we treated ECs expressing the various CYP2C9* variants with 14,15-EEZE or with TEMPOL alone or in combination with EET-A. Treatment with the EET antagonist significantly reduced the proliferation of HUVEC and Cyp2c44KO ECs expressing CYP2C9*1 only, while it did not affect the proliferation of ECs expressing vector, CYP2C9*2, or CYP2C9*3 (Fig. 4E). Importantly, treatment with TEMPOL did not affect the basal or EET-A-induced EC proliferation (Fig, 4F), further confirming that EETs and not CYP2C-derived ROS are responsible for promoting EC proliferation.
Figure 4.
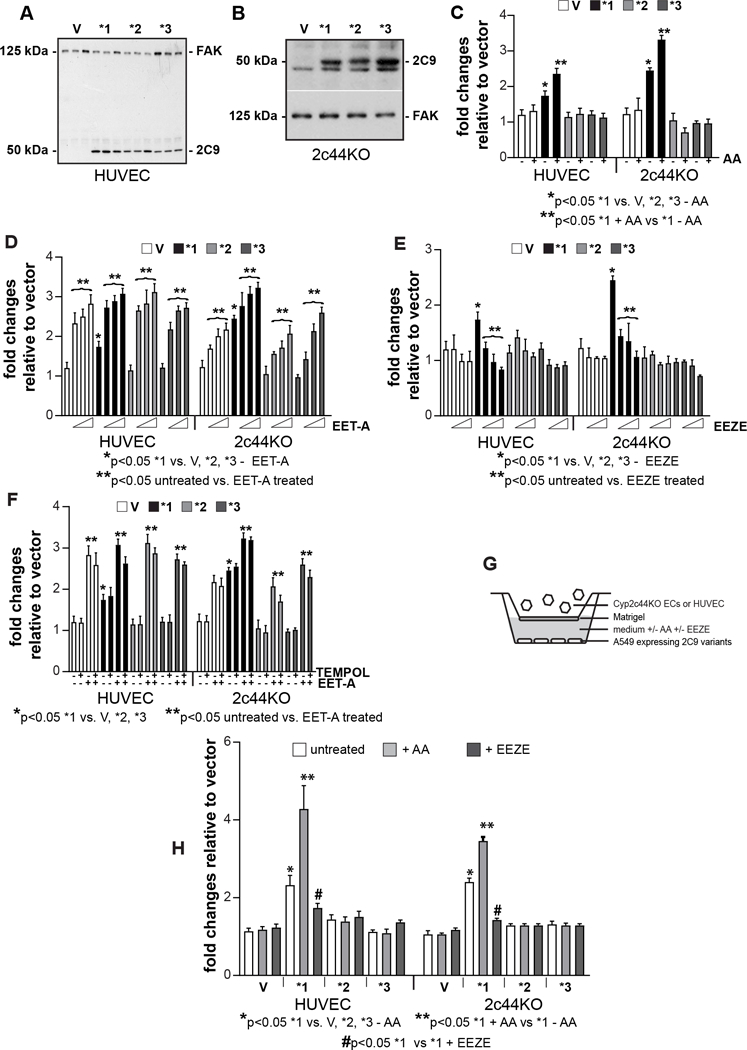
Effects of CYP2C9* variants on EC proliferation and migration. A, B, Cell lysates from HUVEC (A) or Cyp2c44KO (B) ECs infected with empty adenovirus or adenovirus carrying CYP2C9*1, CYP2C9*2, or CYP2C9*3 cDNAs were analyzed by Western blotting for expression of CYP2C9 proteins and FAK. C-F, HUVEC or Cyp2c44KO ECs infected as described above were plated in 96-well plates in complete medium. After 12 hrs, the cells were incubated with serum-free medium with 3H-thymidine with or without AA (1 uM) (C), EET-A (1.25–5 μM) (D), 14,15-EEZE (1.25–5 μM) (E), or TEMPOL (4 μM) alone or in combination with EET-A (5 μM) (F). After 48 hrs, proliferation was determined as described (26). Values represent the mean ± SD of two experiments performed at least in triplicate. G, Schematic representation of the modified Boyden chamber assay used to evaluate migration of HUVEC or Cyp2c44KO ECs. The bottom well contains A549 cells infected with vector or the various CYP2C9* variants incubated with serum free medium that contained or not AA (1 uM) or 14,15-EEZE (5 uM). H, HUVEC or Cyp2c44KO ECs, added to the upper wells of Boyden chamber as described in G, were allowed to migrate for 4 hrs. The number of migrated cells was counted and expressed as the number of cells per microscopic field. Values are the mean ± SD of two experiments with at least eight microscopic fields evaluated.
Migration, using a modified Boyden chamber assay, of HUVEC and Cyp2c44KO ECs exposed to conditioned medium produced by A549 cells expressing vector or the various CYP2C9* variants was also examined (Fig. 4G). EC migration was detected in all the experimental conditions; however, it was greatest in wells containing A549–2C9*1 cells (Fig. 4H). Addition of AA significantly increased, while addition of 14,15-EEZE significantly decreased HUVEC and Cyp2c44KO EC migration, but only in wells containing A549-C9*1 cells (Fig. 4H). In contrast, treatment with TEMPOL did not affect EC migration,, suggesting that CYP2C-derived ROS do not regulate EC migration (Fig. 4H). Thus expression of CYP2C9* variants differentially affects EC proliferation and migration, with expression of CYP2C9*1 stimulating EC proliferation and migration.
CYP2C9*2 and CYP2C9*3 metabolize AA less efficiently than CYP2C9*1
Purified bacterial CYP2C9* variant proteins expressed in bacteria (Fig. 5A) were assessed for their integrity by analyzing their spectra for the presence of a peak at ~450 nm (visible by reducing the ferric form of enzyme with Na2S2O4 in the presence of CO) and the absence of a peak at ~420nm (present upon protein denaturation or misfolding). The presence of a peak at 452 nm and the absence of a peak at 420 nm were visible and comparable among the three variants, suggesting that the single amino acid mutations did not denature or cause misfolding of the protein (Supplementary Fig. S1A).
Figure 5.
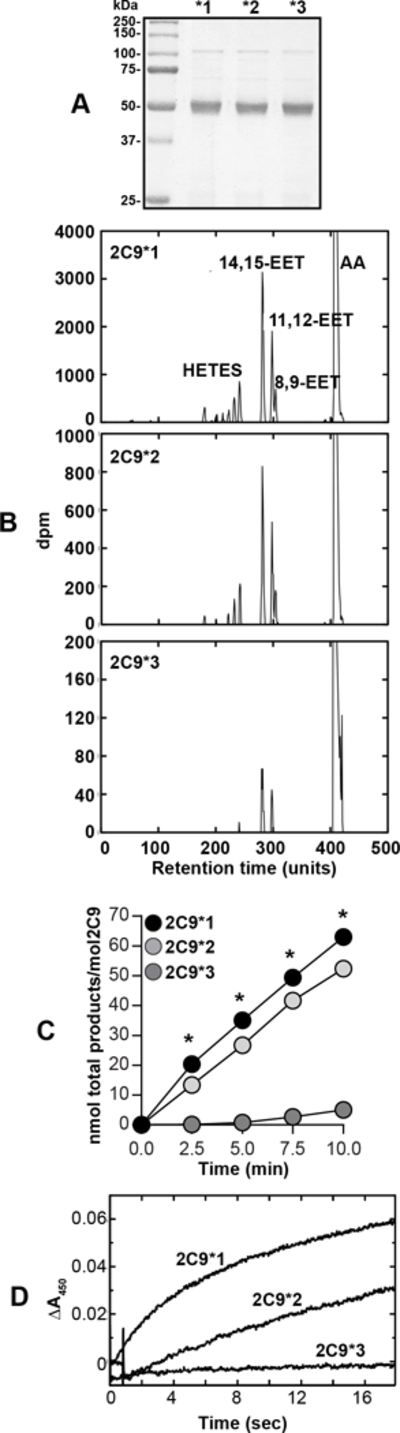
Oxidation of arachidonic acid by CYP2C9* variants. A, Coomassie staining of purified CYP2C9* variant proteins at the expected molecular weight of 50 kDa. B, CYP2C9*1, CYP2C9*2, and CYP2C9*3 were incubated with AA and the products were separated by HPLC after an incubation period of 7.5 min. Retention times are presented as units (100 units = 12 min). C, CYP2C9* proteins were incubated with AA and the total products (EETs and DHETs) were calculated at the time points indicated. D, Reduction of CYP2C9* variants. Rates of reduction (ferric-to-ferrous) for CYP2C9* variants were measured under anaerobic conditions as described in the Methods. Rates of reduction were estimated by global fitting of the 430–470 nm data points (from 10–17 individual reductions, ± SD) to first-order plots in the OLIS software: CYP2C9*1, 9.0 ± 2.3 min−1; CYP2C9*2, 2.9 ± 0.5 min−1; CYP2C9*3, 0.010 ± 0.003 min−1 (the high CYP2C9*3 SD reflects the slow rate).
We incubated purified CYP2C9* variant proteins with AA and followed the formation of EETs by HPLC. CYP2C9*1 efficiently generated EETs (6.5 ± 0.5 pmol EETs/min/pmol P450), while CYP2C9*2 and CYP2C9*3 had significantly reduced catalytic activities (5.0 ± 0.7 pmol vs. 0.57 ± 0.03 pmol EETs/min/pmol P450, respectively) (Fig. 5B, 5C). CYP2C9*1 had a Km value of 14 ± 1 μM, and CYP2C9*2 a Km value of 11 ± 1 μM. A Km value for CYP2C9*3 could not be calculated due to the very low activity at the substrate concentrations measured.
The patterns of products were qualitatively similar among the CYP2C9 variants (Fig. 5B), with 11,12-EETs and 14,15-EET dominating, with minor amounts of hydroxy eicosatetraenoic acids (HETEs) and 8,9-EET formed. The profiles are nearly superimposable and differ only quantitatively.
Mutations in CYP2C9 do not affect the coupling and shunting during AA metabolism
The formation of H2O2 and NADPH consumption were measured during the course of enzyme catalysis. CYP2C9*1 and CYP2C9*2 consumed 17 ± 2 and 23 ± 1 pmol NADPH/min/pmol P450, respectively, while CYP2C9*3 consumed barely detectable amounts of NADPH (~ 1 ± 0.6 pmol NADPH/min/pmol P450). H2O2 formation was 15 ± 3 and 13 ± 3 pmol H2O2 formed min/pmol P450 for CYP2C9*1 and CYP2C9*2. CYP2C9*3 produced 0.7 ± 0.6 pmol H2O2 formed/min/pmol P450. Thus, CYP2C9*2 showed similar coupling as CYP2C9*1, while the rates for the weak CYP2C9*3 could not be calculated accurately.
Mutations in CYP2C9 do not affect the ability to bind AA
The reduced catalytic activity of CYP2C9*2 and CYP2C9*3 could be due to the fact that the binding of AA does not induce a change of the enzyme from a low-spin state to high-spin state. The amplitude change in the CYP2C9*2 spectra was almost twice that of CYP2C9*1, and the amplitude change in CYP2C9*3 was similar to that of CYP2C9*1 (Supplementary Fig. S1B). The binding constants (Ks) were comparable among the three proteins, with CYP2C9*1, CYP2C9*2, and CYP2C9*3 having KS values of 5.7 ± 0.3, 5.3 ± 0.4, and 9.1 ± 0.5 μM, respectively. A major difference in catalytic activity was seen among the variants even at a substrate concentration of 70 μM, which should be high enough to saturate binding of all the variants. Thus, the lower activity of CYP2C9*2 and CYP2C9*3 cannot be relegated to a loss of substrate binding.
We recorded absolute spectra to determine if the mutation affects the spin state. Addition of AA to any of the CYP2C9* variants increased the fraction of high-spin P450, but the overall spectra of all were largely low-spin (>90%) even in the presence of AA (Supplementary Fig. S1B).
The CYP2C9*2 and CYP2C9*3 variants have decreased rates of reduction of ferric P450
To determine if the mutations affected the reduction of ferric P450 to the ferrous form, we measured rates of reduction. CYP2C9*2 and CYP2C9*3 showed dramatic decreases in the reduction rate compared to CYP2C9*1 (Fig. 5D). Thus, the I359L mutation somehow affects the interaction between P450 and its partner NADPH-P450 reductase, disrupting the flow of electrons to the iron. Further, rates of reduction were similar to overall rates of formation of EETs, indicating that the reduction step is rate-limiting.
Loss-of-function CYP2C9 SNPs associate with improved survival in females NSCLC cases
We conducted a retrospective case-only study assessing the role of CYP2C9 SNPs in NSCLC survival. Our NSCLC cases were mostly male, consistent with the higher prevalence of cigarette smoking in American men (Supplementary Table S4A). At clinical presentation, regional disease was the most common stage (Supplementary Table S4A), as was adenocarcinoma histology (Supplementary Table S4A). Treatments consisted of surgical resection, chemotherapy, and/or radiation (Supplementary Table S4A). The most common treatment was resection only, followed by both chemotherapy and radiation (Supplementary Table S4B). Median survival time was not estimated because more than 50% of patients survived longer than five years or were lost to follow-up (Supplementary Table S4A; Fig. 6A).
Figure 6.
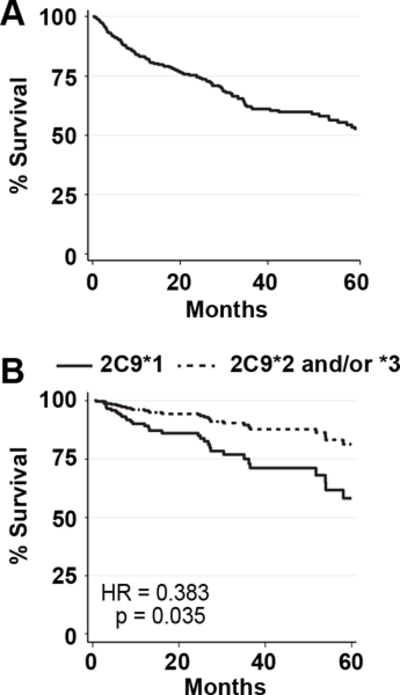
CYP2C9*2 and/or CYP2C9*3 genotypes associate with decreased hazard in female NSCLC patients. A, Five year survival curve is shown for all NSCLC cases (n = 398). Survival was over 50% at 5 years after NSCLC diagnosis. B, A Cox proportional hazard was used to assess the association of loss of function mutations CYP2C9*2 and CYP2C9*3 with improved survival adjusted for NSCLC stage, resection, and chemotherapy. The number of cases were 120 for females with CYP2C9*1 and 54 for CYP2C9*2 and/or YP2C9*3.
Overall, the average follow-up was 23.7 months, with 114 deaths, 46 individuals censored at 5-years, and 238 individuals lost to follow-up (Supplementary Table S4A). Among non-genetic factors, sex, NSCLC stage, tumor resection, chemotherapy treatment, radiation treatment, adenocarcinoma histology, and squamous histology all associated with survival in univariate analysis (Supplementary Tables S5A, S5B). Chemotherapy associated with decreased NSCLC survival, despite the improvements in survival with its use in patients with NSCLC stage 2 or greater, but this result was not adjusted for NSCLC stage (Supplementary Table S5A).
NSCLC stage, sex, resection, and chemotherapy continued to associate significantly with NSCLC mortality in a multivariate model (Supplementary Table S5B). Chemotherapy, which associated with decreased survival in univariate analysis, was protective when adjusted for the other prognostic variables (Supplementary Table S5B). This is consistent with chemotherapy being protective; however, chemotherapy associated with higher stages of disease thereby associating with risk of death in univariate analysis (Supplementary Table S5A).
Because CYP2C9*2 and CYP2C9*3 alleles reduce EET biosynthesis (Figs. 2, 5) and have low MAFs, these alleles were combined in our analyses, allowing us to test for reduced function versus no reduced function association with mortality. Having reduced CYP2C9 function associated with improved NSCLC survival (p = 0.015; Supplementary Table S6). No other SNPs associated with survival including CYP2C9*2 and CYP2C9*3 when individually considered, although both showed trends towards protection (Supplementary Table S6). An adjusted Cox proportional hazard model was tested, including stage, sex, resection, and chemotherapy as covariates. In the adjusted model, having either CYP2C9*2 or CYP2C9*3 did not associate with survival (HR = 0.675, p-value = 0.077; Table 1). However, based on the improved survival in women with NSCLC, sex was controlled by stratifying based on sex. Presence of at least one loss-of-function CYP2C9 allele associated with reduced risk in women when adjusted for other prognostic variables, but not in men (HR = 0.383, p-value = 0.035; HR = 0.854, p-value = 0.538, respectively; Table 1 and Fig. 6B). In addition, the association of the hypomorphic CYP2C9 alleles was examined stratified by platinum chemotherapy status, a commonly used chemotherapy in NSCLC. In patients receiving platinum chemotherapy, loss-of-function CYP2C9 variants associated with longer survival; whereas in patients not receiving platinum chemotherapy, no difference in survival was seen based on CYP2C9 functional alleles (Supplementary Fig. S2A, S2B).
Table 1.
Loss-of-function CYP2C9 alleles *2 and *3 associate with improved survival in NSCLC patients. Collapsed CYP2C9*2/*3 was associated with improved survival when adjusted for NSCLC stage, resection, and chemotherapy. To adjust for the effect of sex, sex was added to the model and a stratified Cox proportional hazard model was used. The sex stratified Cox proportional hazard models indicated the association of collapsed CYP2C9*2/*3 with NSCLC survival is limited to women.
| Collapsed CYP2C9*2 or *3 model | HR |
95% CI | p-value |
|---|---|---|---|
| No sex adjustment | 0.644 | 0.417–0.994 | 0.047 |
| Sex adjusted | 0.675 | 0.437–1.044 | 0.077 |
| Within men only | 0.854 | 0.516–1.412 | 0.538 |
| Within women only | 0.383 | 0.157–0.935 | 0.035 |
Discussion
Two variants of CYP2C9, CYP2C9*2 (R144C) and CYP2C9*3 (I359L), have significantly reduced enzyme activity. Because these two variants metabolize carcinogens and chemopreventive agents less efficiently than CYP2C9*1, they may affect risk and treatment response in seemingly unpredictable and opposite directions. Given the observed inconsistency of effect directions, we assessed how specific variation impacts biochemical function and the progression of one type of cancer, NSCLC. We show that CYP2C9*2 and CYP2C9*3 variants metabolize AA less efficiently than CYP2C9*1 and this is associated with decreased growth and vascularization of tumors derived from human NSCLC cells expressing these two variants. Reduced function alleles also associated with improved survival in women of NSCLC.
CYP2C9*2 and CYP2C9*3 have attenuated AA catalytic efficiency, and our results implicate reduction of ferric P450 as the basis for this finding. The rate of reduction appears to be rate-limiting in CYP2C9-catalyzed AA oxidation, in that it was similar to the overall substrate oxidation for all three variants, which varied considerably. That is, the lower activities of the variants are due to the decrease reduction rates. This phenomenon has been seen with some other P450s (36, 37) but it is not the general case, in that C-H bond-breaking is sometimes at least partially rate-limiting, as judged by kinetic deuterium isotope effects (38). The rates of reduction of CYP2C9*2 and CYP2C9*3 were very slow compared with CYP2C9*1 in the presence of some other substrates (39), corresponding to the rates measured in the absence of substrates (39). The slow reduction rates may be related to the limited spin-state change observed in the presence of AA. Spectral changes were clearly seen upon the addition of AA, but all three CYP2C9 variants were predominantly low-spin iron.
The effect on coding-region variations on P450 activity varies with substrate and reaction (40), making it difficult to extrapolate. However, the general *1/*2/*3 patterns with AA are similar to those for (S)-warfarin 7-hydroxylation, (S)-fluorbiprofen 4´-hydroxylation, and diclofenac 4´-hydroxylation (41). In considering different substrates, sometimes either a kcat difference or a Km difference is seen (40).
Several mechanisms could account for the attenuated catalytic activities of CYP2C9 variants, including poor interactions with NADPH-P450 reductase (42, 43). The evidence is however based upon the ability of the oxygen surrogate cumene hydroperoxide or high concentrations of NADPH-P450 reductase to abrogate the differences in activity of the variants. In a study with the substrate (S)-naproxen (which showed a pattern similar to AA), no differences in the two CYP2C9* variants in substrate binding or in NADPH-P450 reductase interactions were observed (44) and the attenuated catalytic activity was attributed to coupling of electron flow (44). Rates of P450 reduction associated with altered catalytic activity in AA metabolism. The details of the attenuated reduction remain to be elucidated, but are unrelated to differences in substrate binding or the iron spin state.
A study performed with CYP2C9*1, CYP2C9*2, and CYP2C9*3 purified from yeast showed that *2, but not *3, had reduced AA metabolism (16). However, the activities with the yeast expressed enzymes were low, more than 25-fold lower than ours, and the Km values were very high, so the comparisons of the activities of the variants are difficult.
CYP2C9* reduced-function variants differentially affect proliferation and migration of ECs. These results agree with the observation that EETs are pro-angiogenic and promote EC proliferation and migration in vitro and in vivo (10). ECs expressing CYP2C9*1 exhibited the greatest cell proliferation and migration. This effect could be due to the ability of this variant to generate the highest levels of EETs, or produce high levels of reactive oxygen species (ROS) (34) which promote endothelial proliferation, migration and angiogenesis (35). Our finding that treatment with the EET antagonist 14,15-EEZE, but not the ROS scavenger TEMPOL, significantly decreases EC proliferation and migration only in cells expressing the CYP2C9*1, suggests that CYP2C9*1-derived EET and not CYP2C9*1-derived ROS are the major driver of EC function in our model.
Unlike our data with purified proteins, ECs expressing CYP2C9*2 and CYP2C9*3 proliferate and migrate similarly to ECs transfected with vector only. One explanation is that ECs generate EETs through other epoxygenases, thus promoting baseline cell function. While the levels of EETs derived from overexpression of CYP2C9*1 are sufficient to significantly enhance EC functions, EETs produced by overexpressed CYP2C9*2 and CYP2C9*3 are insufficient to induce EC functions above baseline. Another difference is the limited difference in the in vitro catalytic activity between purified CYP2C9*1 and CYP2C9*2 compared with the highest proliferative or tumorigenic capacity of CYP2C9*1. This discrepancy may be a reflection of performing some of the assays with purified enzymes under optimized conditions, (e.g., with high reductase, AA, and NADPH concentrations) that may not have been reflected in the work with the cells and mice. Finally, we provide evidence that major EETs produced by the purified CYP2C9 enzymes and in the transfected cells are 14,15-, 11,12-, and 8,9-EETs. The 8,9-EETs were formed least in both systems but the orders of the 14,15- and 11,12-EETs were reversed in the two enzymatic systems. However, the analysis is each case was done only at a single AA concentration, and the Km values for formation of the different EETs may vary. Alternatively, the ratio of NADPH-P450 reductase (POR) to P450 may influence the regioselectivity of epoxidation. In the cells, we measured the sums of the EETs and their hydrolysis products, the DHETs, so presumably the difference cannot be attributed to the presence of cellular epoxide hydrolases.
We provide evidence that A549–2C9*1 cells generated more vascularized tumors than A549–2C9*2 or A549–2C9*3 cells. A549-V cells form tumors similar to those derived from A549–2C9*2 or A549–2C9*3 cells, despite the fact that A549–2C9*2 cells produce produced significantly higher level of EETs than A549-Vector cells. This result indicates that other factors, such as growth factors or other AA-derived lipids, mediate the baseline in vivo growth of NSCLC cells, but only with CYP2C9*1 and concomitant high levels of EETs is tumor growth potentiated. CYP2C9 protein expression is upregulated in some human tumors particularly in their vasculature (13), indicating that this enzyme could contribute to tumor growth by promoting EET-mediated angiogenesis. In addition to an autocrine effect, we suggest that EETs produced by tumor cells could also potentiate EC function via paracrine effects.
To translate our findings to a more clinically relevant setting, we analyzed the effects of CYP2C9* variants in NSCLC patients. Sex-based differences in the association of CYP2C9 SNPs with NSCLC survival were found, consistent with sex-based differences in this enzyme. CYP2C9*2 or CYP2C9*3 significantly associated with improved NSCLC survival in women only. Based on our model that disruption of CYP2C9 function reduces production of pro-angiogenic EETs, it was expected that the hypomorphic alleles would associate with improved survival in both sexes and the results are consistent with this trend as the hazard ratio is less than one in men as well, albeit insignificantly. However, the limitation of this association to women suggests loss of one wild-type CYP2C9 allele fails to inhibit tumor progression in men with NSCLC. Higher CYP2C9 activity in men at baseline could account for this, since elimination of one functional allele may be insufficient to exert a protective effect.
The existence of sexual dimorphism in CYP2C9 expression and/or activity is controversial. Several studies have investigated sex based CYP2C9 differences but have found discordant directions of effect (45–47), while other studies have found no sex based difference (48, 49). Thus, it is unclear whether sexual dimorphism in CYP2C9 expression and/or activity exists. In addition, these studies were performed based on activity in the liver only, limiting their generalizability. Our results suggest the need to perform explicit analysis in more cancer relevant tissues/cells.
In addition, there are certain limitations and caveats concerning the human studies. First, EMRs inadequately capture smoking exposure, and we were limited to assessing ever-smoking status, which is insufficient to assess lung cancer prognosis as active smoking associates with NSCLC prognosis (50). Second, the 5-year survival in the BioVU NSCLC population was unusually high (~ 50%). The high 5-year survival probably reflects differences in stage of disease as distant disease only accounts for only 27% of our cases, whereas distant disease typically accounts for 57% of lung cancer cases (Table 1). This is probably due to identifying many cases through the National Lung Screening Trial. Given the excess of early disease, the association of hypomorphic CYP2C9 SNPs with survival in female NSCLC cases may be limited to local and/or regional disease.
In conclusion, we propose that the presence of CYP2C9*2 and CYP2C9*3 plays a beneficial effect in limiting NSCLC cancer growth and improving survival by impaired production of EETs from endogenous pools of AA. These novel findings together with our previous work that downregulation of CYP2C9 expression in human ECs leads to reduced EET biosynthesis, cell migration and cell proliferation (13), clearly indicate that inhibition of CYP2C9 might be viewed as a valid tool to slow and ideally halt the production of pro-angiogenic lipids.
Supplementary Material
Acknowledgments
This work was in part supported by Veterans Affairs Merit Reviews 1I01BX002025–01 (A. Pozzi) and 1I01BX002196–01 (R. Zent); National Institutes of Health grants R01-CA162433 (A. Pozzi), R01-DK095761 (A. Pozzi), PO1-DK38226 (J.H. Capdevila, J.R. Falck, A. Pozzi), R01-DK069221 (R. Zent), R01-DK083187 (R. Zent), HL111392 (J.R. Falck); DK081662 (J.M. Luther), R37-CA090426 and R01-GM118122 (F.P. Guengerich); and the Robert A. Welch Foundation (I-0011) (J.R. Falck).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U. S. C. Section1734 solely to indicate this fact.
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed
REFERENCES
- 1.Shahabi P, Siest G, Meyer UA, Visvikis-Siest S. Human cytochrome P450 epoxygenases: variability in expression and role in inflammation-related disorders. Pharmacol Ther. 2014;144:134–61. [DOI] [PubMed] [Google Scholar]
- 2.Steward DJ, Haining RL, Henne KR, Davis G, Rushmore TH, Trager WF, et al. Genetic association between sensitivity to warfarin and expression of CYP2C9*3. Pharmacogenetics. 1997;7:361–7. [DOI] [PubMed] [Google Scholar]
- 3.Rettie AE, Wienkers LC, Gonzalez FJ, Trager WF, Korzekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allelic variant of CYP2C9. Pharmacogenetics. 1994;4:39–42. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein JA. Clinical relevance of genetic polymorphisms in the human CYP2C subfamily. Br J Clin Pharmacol. 2001;52:349–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan AT, Zauber AG, Hsu M, Breazna A, Hunter DJ, Rosenstein RB, et al. Cytochrome P450 2C9 variants influence response to celecoxib for prevention of colorectal adenoma. Gastroenterology. 2009;136:2127–36 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samowitz WS, Wolff RK, Curtin K, Sweeney C, Ma KN, Andersen K, et al. Interactions between CYP2C9 and UGT1A6 polymorphisms and nonsteroidal anti-inflammatory drugs in colorectal cancer prevention. Clinical gastroenterology and hepatology : the official clinical practice journal of the American Gastroenterological Association. 2006;4:894–901. [DOI] [PubMed] [Google Scholar]
- 7.Yadav SS, Seth S, Khan AJ, Maurya SS, Dhawan A, Pant S, et al. Association of polymorphism in cytochrome P450 2C9 with susceptibility to head and neck cancer and treatment outcome. Applied & translational genomics. 2014;3:8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sacerdoti D, Pesce P, Di Pascoli M, Bolognesi M. EETs and HO-1 cross-talk. Prostaglandins Other Lipid Mediat. 2016;125:65–79. [DOI] [PubMed] [Google Scholar]
- 9.Xu X, Li R, Chen G, Hoopes SL, Zeldin DC, Wang DW. The Role of Cytochrome P450 Epoxygenases, Soluble Epoxide Hydrolase, and Epoxyeicosatrienoic Acids in Metabolic Diseases. Advances in nutrition. 2016;7:1122–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pozzi A, Macias-Perez I, Abair T, Wei S, Su Y, Zent R, et al. Characterization of 5,6- and 8,9-epoxyeicosatrienoic acids (5,6- and 8,9-EET) as potent in vivo angiogenic lipids. J Biol Chem. 2005;280:27138–46. [DOI] [PubMed] [Google Scholar]
- 11.Daikh BE, Lasker JM, Raucy JL, Koop DR. Regio- and stereoselective epoxidation of arachidonic acid by human cytochromes P450 2C8 and 2C9. J Pharmacol Exp Ther. 1994;271:1427–33. [PubMed] [Google Scholar]
- 12.Michaelis UR, Fisslthaler B, Barbosa-Sicard E, Falck JR, Fleming I, Busse R. Cytochrome P450 epoxygenases 2C8 and 2C9 are implicated in hypoxia-induced endothelial cell migration and angiogenesis. J Cell Sci. 2005;118:5489–98. [DOI] [PubMed] [Google Scholar]
- 13.Pozzi A, Popescu V, Yang S, Mei S, Shi M, Puolitaival SM, et al. The anti-tumorigenic properties of peroxisomal proliferator-activated receptor alpha are arachidonic acid epoxygenase-mediated. J Biol Chem. 2010;285:12840–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skrypnyk N, Chen X, Hu W, Su Y, Mont S, Yang S, et al. PPARalpha Activation Can Help Prevent and Treat Non-Small Cell Lung Cancer. Cancer Res. 2014;74:621–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei X, Zhang D, Dou X, Niu N, Huang W, Bai J, et al. Elevated 14,15- epoxyeicosatrienoic acid by increasing of cytochrome P450 2C8, 2C9 and 2J2 and decreasing of soluble epoxide hydrolase associated with aggressiveness of human breast cancer. BMC Cancer. 2014;14:841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lundblad MS, Stark K, Eliasson E, Oliw E, Rane A. Biosynthesis of epoxyeicosatrienoic acids varies between polymorphic CYP2C enzymes. Biochem Biophys Res Commun. 2005;327:1052–7. [DOI] [PubMed] [Google Scholar]
- 17.Dimitropoulou C, West L, Field MB, White RE, Reddy LM, Falck JR, et al. Protein phosphatase 2A and Ca2+-activated K+ channels contribute to 11,12-epoxyeicosatrienoic acid analog mediated mesenteric arterial relaxation. Prostaglandins Other Lipid Mediat. 2007;83:50–61. [DOI] [PubMed] [Google Scholar]
- 18.Wester MR, Yano JK, Schoch GA, Yang C, Griffin KJ, Stout CD, et al. The structure of human cytochrome P450 2C9 complexed with flurbiprofen at 2.0-A resolution. J Biol Chem. 2004;279:35630–7. [DOI] [PubMed] [Google Scholar]
- 19.Sandhu P, Baba T, Guengerich FP. Expression of modified cytochrome P450 2C10 (2C9) in Escherichia coli, purification, and reconstitution of catalytic activity. Arch Biochem Biophys. 1993;306:443–50. [DOI] [PubMed] [Google Scholar]
- 20.Hanna IH, Teiber JF, Kokones KL, Hollenberg PF. Role of the alanine at position 363 of cytochrome P450 2B2 in influencing the NADPH- and hydroperoxide-supported activities. Arch Biochem Biophys. 1998;350:324–32. [DOI] [PubMed] [Google Scholar]
- 21.Jiang ZY, Woollard AC, Wolff SP. Hydrogen peroxide production during experimental protein glycation. FEBS Lett. 1990;268:69–71. [DOI] [PubMed] [Google Scholar]
- 22.Jefcoate CR. Measurement of substrate and inhibitor binding to microsomal cytochrome P-450 by optical-difference spectroscopy. Methods Enzymol. 1978;52:258–79. [DOI] [PubMed] [Google Scholar]
- 23.Guengerich FP. Oxidation-reduction properties of rat liver cytochromes P-450 and NADPH-cytochrome p-450 reductase related to catalysis in reconstituted systems. Biochemistry. 1983;22:2811–20. [DOI] [PubMed] [Google Scholar]
- 24.Druguet M, Pepys MB. Enumeration of lymphocyte populations in whole peripheral blood with alkaline phosphatase-labelled reagents. A method for routine clinical use. Clin Exp Immunol. 1977;29:162–7. [PMC free article] [PubMed] [Google Scholar]
- 25.Guengerich FP, Krauser JA, Johnson WW. Rate-limiting steps in oxidations catalyzed by rabbit cytochrome P450 1A2. Biochemistry. 2004;43:10775–88. [DOI] [PubMed] [Google Scholar]
- 26.Pozzi A, Moberg PE, Miles LA, Wagner S, Soloway P, Gardner HA. Elevated matrix metalloprotease and angiostatin levels in integrin alpha 1 knockout mice cause reduced tumor vascularization. Proc Natl Acad Sci U S A. 2000;97:2202–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pozzi A, Ibanez MR, Gatica AE, Yang S, Wei S, Mei S, et al. Peroxisomal proliferator-activated receptor-alpha-dependent inhibition of endothelial cell proliferation and tumorigenesis. J Biol Chem. 2007;282:17685–95. [DOI] [PubMed] [Google Scholar]
- 28.Roden DM, Pulley JM, Basford MA, Bernard GR, Clayton EW, Balser JR, et al. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther. 2008;84:362–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wiley LK, Shah A, Xu H, Bush WS. ICD-9 tobacco use codes are effective identifiers of smoking status. Journal of the American Medical Informatics Association : JAMIA. 2013;20:652–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beadsmoore CJ, Screaton NJ. Classification, staging and prognosis of lung cancer. Eur J Radiol. 2003;45:8–17. [DOI] [PubMed] [Google Scholar]
- 31.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Capdevila JH, Dishman E, Karara A, Falck JR. Cytochrome P450 arachidonic acid epoxygenase: stereochemical characterization of epoxyeicosatrienoic acids. Methods Enzymol. 1991;206:441–53. [DOI] [PubMed] [Google Scholar]
- 33.Spector AA, Norris AW. Action of epoxyeicosatrienoic acids on cellular function. Am J Physiol Cell Physiol. 2007;292:C996–1012. [DOI] [PubMed] [Google Scholar]
- 34.Fleming I, Michaelis UR, Bredenkotter D, Fisslthaler B, Dehghani F, Brandes RP, et al. Endothelium-derived hyperpolarizing factor synthase (Cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ Res. 2001;88:44–51. [DOI] [PubMed] [Google Scholar]
- 35.Ushio-Fukai M, Alexander RW. Reactive oxygen species as mediators of angiogenesis signaling: role of NAD(P)H oxidase. Mol Cell Biochem. 2004;264:85–97. [DOI] [PubMed] [Google Scholar]
- 36.Shinkyo R, Guengerich FP. Cytochrome P450 7A1 cholesterol 7alpha-hydroxylation: individual reaction steps in the catalytic cycle and rate-limiting ferric iron reduction. J Biol Chem. 2011;286:4632–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson KM, Phan TTN, Albertolle ME, Guengerich FP. Human mitochondrial cytochrome P450 27C1 is localized in skin and preferentially desaturates trans-retinol to 3,4-dehydroretinol. J Biol Chem. 2017;292:13672–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guengerich FP. Kinetic deuterium isotope effects in cytochrome P450 oxidation reactions. Journal of labelled compounds & radiopharmaceuticals. 2013;56:428–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guengerich FP, Johnson WW. Kinetics of ferric cytochrome P450 reduction by NADPH-cytochrome P450 reductase: rapid reduction in the absence of substrate and variations among cytochrome P450 systems. Biochemistry. 1997;36:14741–50. [DOI] [PubMed] [Google Scholar]
- 40.Takanashi K, Tainaka H, Kobayashi K, Yasumori T, Hosakawa M, Chiba K. CYP2C9 Ile359 and Leu359 variants: enzyme kinetic study with seven substrates. Pharmacogenetics. 2000;10:95–104. [DOI] [PubMed] [Google Scholar]
- 41.Yamazaki H, Inoue K, Chiba K, Ozawa N, Kawai T, Suzuki Y, et al. Comparative studies on the catalytic roles of cytochrome P450 2C9 and its Cys- and Leu-variants in the oxidation of warfarin, flurbiprofen, and diclofenac by human liver microsomes. Biochem Pharmacol. 1998;56:243–51. [DOI] [PubMed] [Google Scholar]
- 42.Crespi CL, Miller VP. The R144C change in the CYP2C9*2 allele alters interaction of the cytochrome P450 with NADPH:cytochrome P450 oxidoreductase. Pharmacogenetics. 1997;7:203–10. [DOI] [PubMed] [Google Scholar]
- 43.Lee MY, Borgiani P, Johansson I, Oteri F, Mkrtchian S, Falconi M, et al. High warfarin sensitivity in carriers of CYP2C9*35 is determined by the impaired interaction with P450 oxidoreductase. Pharmacogenomics J. 2014;14:343–9. [DOI] [PubMed] [Google Scholar]
- 44.Wei L, Locuson CW, Tracy TS. Polymorphic variants of CYP2C9: mechanisms involved in reduced catalytic activity. Mol Pharmacol. 2007;72:1280–8. [DOI] [PubMed] [Google Scholar]
- 45.Rugstad HE, Hundal O, Holme I, Herland OB, Husby G, Giercksky KE. Piroxicam and naproxen plasma concentrations in patients with osteoarthritis: relation to age, sex, efficacy and adverse events. Clinical rheumatology. 1986;5:389–98. [DOI] [PubMed] [Google Scholar]
- 46.Meyer MC, Straughn AB, Mhatre RM, Shah VP, Chen ML, Williams RL, et al. Variability in the bioavailability of phenytoin capsules in males and females. Pharm Res. 2001;18:394–7. [DOI] [PubMed] [Google Scholar]
- 47.Travers RD, Reynolds EH, Gallagher BB. Variation in response to anticonvulsants in a group of epileptic patients. Archives of neurology. 1972;27:29–33. [DOI] [PubMed] [Google Scholar]
- 48.Greenblatt DJ, Shader RI, Franke K, MacLaughlin DS, Ransil BJ, Koch-Weser J. Kinetics of intravenous chlordiazepoxide: sex differences in drug distribution. Clin Pharmacol Ther. 1977;22:893–903. [DOI] [PubMed] [Google Scholar]
- 49.Yang X, Zhang B, Molony C, Chudin E, Hao K, Zhu J, et al. Systematic genetic and genomic analysis of cytochrome P450 enzyme activities in human liver. Genome Res. 2010;20:1020–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee SJ, Lee J, Park YS, Lee CH, Lee SM, Yim JJ, et al. Impact of smoking on mortality of patients with non-small cell lung cancer. Thoracic cancer. 2014;5:43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.