

Summary
There are around 100 varieties of outer membrane proteins in each Gram negative bacteria. All of these proteins have the same fold—an up-down βbarrel. It has been suggested that all membrane β-barrels excluding lysins are homologous. Here we suggest that β-barrels of efflux pumps have converged on this fold as well. By grouping structurally-solved outer membrane β-barrels (OMBBs) by sequence we find that the membrane environment may have led to convergent evolution of the barrel fold. Specifically, the lack of sequence linkage to other barrels coupled with distinctive structural differences, such as differences in strand tilt and barrel radius, suggest that the outer membrane factor of efflux pumps evolutionarily converged on the barrel. Rather than being related to other OMBBs, sequence and structural similarity in the periplasmic region of the outer membrane factor of efflux pumps suggests an evolutionary link to the periplasmic subunit of the same pump complex.
eTOC blurb
Although most outer membrane β-barrels (OMBBs) are related, Franklin et al. use sequences and structures to suggest that efflux-OMBBs are unrelated to other OMBBs. This leaves the prototypical-related autotransporters as the best models of primordial OMBB structure. Moreover, the efflux-OMBB structural differences support an iris-like mechanism of antibiotic efflux.
Introduction
All bacterial outer membrane proteins save one (Dong et al., 2006), are right-handed, up-down β-barrels with the N and C termini facing the periplasm. This extreme topological homogeneity has given rise to questions of outer membrane β-barrel (OMBB) evolutionary origin. Specifically, did this fold arise from divergent evolution of a single common ancestor or did multiple ancestors converge onto an identical fold required by the biological and physical constraints of the outer membrane? In support of divergent evolution, Remmert et al. propose that the strand number diversity results from amplification of an ancestral hairpin or double hairpin (Remmert et al., 2010).
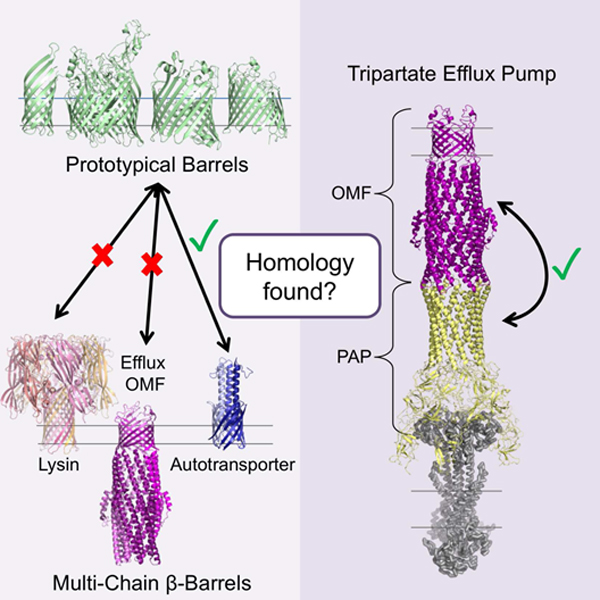
A useful counter example to the well-established hypothesis of divergence of all OMBBs from a common ancestor is the existence of membrane barrels, such as alpha hemolysin and the leukocidins, which are not localized in the outer membrane of Gram negative bacteria. Unlike the OMBBs, the lysins are exported during cellular warfare to create pores in membranes of other organisms (Menestrina et al., 1995). It has been hypothesized that the lysins have evolutionarily converged to their barrel structure separately from the divergent evolution of the other β-barrels. This hypothesis was based on the distance in sequence and on the differences in organisms that produce them (Remmert et al., 2010).
Efforts to document the homology among OMBBs have been frustrated by two factors, 1) the high sequence similarity required for the strands of β-barrels, and 2) extreme bacterial sequence variation.
First, β-barrel structure and environment collude to enforce common sequence patterns. The β-barrel structure causes half of the positions to be facing the membrane and half of the positions to face the interior which is an (often solvated) pore. Therefore, β-strand sequences organize into a pattern with a polar-nonpolar alternation of residues in each strand (Schulz, 2002). Because of this, the membrane environment may enforce sequence similarity even between non-homologous barrels.
Second, β-barrels are also subject to extreme variation through evolution. Despite the fact that prokaryotes lack exons, there is substantial complexity in their evolution. The Gram negative bacteria is likely about 3 billion years old (Battistuzzi et al., 2004), and bacterial generations are quick— generally less than an hour, though only 10% of its time is in growth phase. This rapid replication adds three orders of magnitude, resulting in at least 1012 opportunities for introducing genetic variation such as amplification, recombination, and accretion of mutations. In addition, membrane proteins are less conserved than soluble proteins, partly because they are more involved in adapting to new environments (Sojo et al., 2016). For outer membrane proteins the lipid-facing side is particularly prone to variation (Jimenez-Morales and Liang, 2011). Ultimately, these factors can result in the sequences of OMBB proteins diverging beyond recognition which means that the usual rule of thumb of E-values of less than 10−3 (Pearson, 2013) being suggestive of homology may not apply; higher E-values could also indicate homology.
To establish homology of OMBBs, previous studies created large databases of sequence similar proteins and then culled those sequences to increase the likelihood of true OMBBs (Reddy and Saier, 2016; Remmert et al., 2010). Creating these large databases of sequences is extremely useful for teasing out evolutionary relationships within proteins that are related. The benefit of larger databases is that it reveals a more connected network. However, using these large databases to understand relationships between structure and homology is harder. Without experimental structural determination, structure prediction must be used. Both detecting homology by sequence and predicting structure/topology relies on Hidden Markov Models (HMMs) – a probabilistic model that is constructed from a large set of input sequences. The dependence on sequence based HMMs is especially the case as HMMs are used in the most successful methods of OMBB structure prediction (Bagos et al., 2005). This leads to a double counting of the sequence relationships that may obscure the very thing we are trying to determine: the relationship between protein fold and protein homology.
To study OMBB homology through combined sequence and structural relationships, one must curate a specialized dataset. As structural data for bacterial OMBBs have increased, the annotated databases of protein domains have struggled with whether to place OMBBs into the same or separate groups (Tsirigos et al., 2018). In general, current classification schemes reflect the underlying homologous relationships, but the high degree of sequence divergence makes that difficult to achieve for OMBBs. For example, SCOPe (Fox et al., 2014) artificially defines a class of “membrane and cell surface proteins and peptides”. The OMBBs fall into four different folds in SCOPe, of which the fold “transmembrane β-barrels” is labeled as “not a true fold”. CATH (Sillitoe et al., 2015) strictly classifies OMBBs by overall topology and many of the more recently characterized structures do not yet have a CATH classification. In CATH with the exception of the autotransporters like HiaA, the OMBBs fall into the all-β class and then into at least five different architectures. Finally, most of the single-chain OMBBs fall into the same possibly-homologous group of outer membrane meander β-barrels in ECOD (Cheng et al., 2014). However, the multi-chain β-barrels, like the β-barrel component of the efflux pumps, autotransporters, and especially the lysins, are splintered into many different X-groups.
Here, we studied the evolutionary relationships among OmBb proteins, and present evidence that the outer membrane components of tripartite efflux pump proteins have converged upon the OMBB fold. These efflux pump proteins are responsible for expelling metals and most small hydrophobic molecules from bacterial cells. They have recently become a focus of scientific interest as they are the primary mode of Gram negative antibiotic resistance. Tripartite efflux pumps expel most classes of antibiotics and biocides including chloramphenicol, tetracycline, macrolides, fluoroquinolones, quaternary ammonium compounds, amino glycosides, triclosan, and even the pine oil used in pine scented cleaner (Liu et al., 2010; Poole, 2005). In clinical isolates, expression of tripartite efflux pumps have been found to correlate with antibiotic resistance (Swick et al., 2011). These pumps extend from the inner membrane, through the periplasm, to the outer membrane. As per their name, tripartite efflux pumps are comprised of three types of proteins, outer membrane factors (OMFs), periplasmic adaptor proteins (PAPs), and inner membrane transporters (IMTs). These efflux pumps utilize the proton motive force of the inner membrane to drive substrate transport (Zgurskaya and Nikaido, 1999).
Our study analyzes the structural connections in tandem with sequence data, in a bespoke dataset of OMBBs. This extensive analysis of structural data makes our study unique among those that search for homologous relationships; while sequence data is often used to determine evolutionary relationships, we can gain different and new perspectives on OMBB relationships. We describe results that suggest OMFs of efflux pump may have evolutionarily converged upon the OMBB fold. Ultimately, the structural insights described inform our understanding of the mechanism of antibiotic resistance.
Results
Several groups of OMBBs
Our dataset is composed of 130 structurally characterized OMBB proteins which are <85% similar to each other, including 113 which are less than 50% similar to each other. These include single OMBBs, multi-chain OMBBs, and multi-chain lysins, which are used as a control for describing lack of relationship. Our database contains structures from 39 species in 29 genera. Most proteins are encoded by Gram negative bacteria. Lysins are the only group discussed here which are made not only by Gram negative bacteria, but also by Gram positive bacteria, as well as the sea cucumber. The small group of OMBBs that do not fall into another group, labeled “Other”, include mitochondrial mouse VDAC1 and two proteins from Gram positive bacteria.
By filtering the results of the HMM alignments of our dataset, we searched for meaningful groupings of β-barrels with sequence similarity indicative of an evolutionary relationship. Our groupings are delineated by a sequence similarity score known as E-value. This E-value is the expected number of false positives in the database with a score at least as good as that of the match (Karlin and Altschul, 1990). Using a relatively high E-value threshold of <1, the HMM alignments distinguish eight, seemingly unrelated groups of OMBBs (fig. 1) – the autotranporters, Fim/Ushers, lysins, assembly proteins, OMFs of efflux pumps, LptD or the lipid assembly proteins, the injectisomes, and the large group of prototypical barrels. The members within each of the seven, smaller groups of proteins share a high degree of structural similarity and have related functions. Here we are tailoring the E-value threshold to the specific task, using large values when aiming to rule out the possibility that two proteins share a common ancestor. Later we will use a lower threshold value to argue in favor of an evolutionary link between two proteins.
Figure 1. Network map of OMBBs by sequence similarity.

A) Sequence alignments among the membrane β-barrels at E < 1, colored by component; ovals are structures for which only a monomer has been crystalized but the protein is proposed to be a full, oligomeric β-barrel. The purple group, “other”, are defined as the barrels for which no alignment to any of the other structurally characterized beta barrels was found. Our naming follows the form pdbID_chainID. B) Representative examples of each group. From left to right: lysin (pink, alpha-hemolysin, PDB ID 7ahl), prototypical (light green, NanC, PDB ID 2wjr), fim/usher (orange, PapC, PDB ID 3fip), LptD (dark green, LptD, PDB ID 5iva), assembly (yellow, BamA, PDB ID 4k3c), autotransporter (purple, Hia, PDB ID 2gr7), OMF of efflux pumps (dark pink, TolC, PDB ID 1ek9).
The seven smaller groups are at best weakly related to the prototypical group (Table 1). An interactive version of the network, allowing visual inspection of the underlying sequence and structural similarity, is available online (http://cytostruct.info/rachel/barrels/index.html). Because the majority of the barrel structure of the injectisomes is embedded in the periplasmic space and not in the outer membrane, we exclude these from further consideration even though they are described as OMBBs. Figure 1 also excludes any alignments which are exclusively in a non-barrel portion of the OMBB.
Table 1.
Minimum E-value connecting each group. An E-value of >20 indicates no connection was found.
| Prototypical | Hemolysins | Efflux | Assembly | Fim/Usher | Injectisomes | Autotransporters | LptD | |
|---|---|---|---|---|---|---|---|---|
| Prototypical | 5.6e-94 | >20 | >20 | 1.9 | 7.8 | >20 | 2.2 | 1.3 |
| Hemolysins | 1.6e-80 | >20 | >20 | >20 | >20 | >20 | >20 | |
| Efflux | 5.0e-57 | >20 | >20 | >20 | >20 | >20 | ||
| Assembly | 5.3e-63 | >20 | >20 | >20 | >20 | |||
| Fim/Usher | 4.4e-57 | >20 | >20 | >20 | ||||
| Injectisomes | 1.6e-44 | >20 | >20 | |||||
| Autotransporters | 2.5e-26 | >20 | ||||||
| LptD | 6.8e-71 |
In order to find possible common or unrelated ancestors among groups, we searched for sequence alignments between OMBBs and any other structurally solved proteins (fig. 2). An interactive version of the network, allowing visual inspection of the underlying sequence and structural similarity, is available online (http://cytostruct.info/rachel/barrels/pumps.html). In the online network the E-value threshold can be decreased, so that only some of the relationships remain. To see the widest ranges of possible paths, this assessment was done at E-value < 20. We find common relatives among all eight groups of barrels except for the lysins and the OMFs of efflux pumps, suggesting that both groups are evolutionarily distinct.
Figure 2. Network map of OMBB sequence similarity to soluble proteins.

Possible ancestors of OMBB shown through a network of alignments of non-OMBBs to OMBBs at E-value < 20. OMBBs are shown as colored nodes as in Fig 1. White nodes are non-OMBB-containing proteins. Edges represent alignments between two proteins. With a few exceptions primarily involving the lysin cluster or CsgG (discussed in the text), no white nodes align with the barrel of an OMBB. The inset structures of the multi-chain OMBBs, such as the OMFs of efflux pumps and the lysins, are colored by chain. Outer membrane factors of efflux pumps (dark pink) are shown to align with periplasmic adaptor proteins of efflux pumps in the bottom right.
Most of the lysins align with another toxin in the aerolysin family. This is unsurprising given the soluble nature of the monomeric units before barrel formation. Interestingly, the only possible relatives found for the outer membrane factors (OMFs) of efflux pumps are the periplasmic adaptor proteins (PAPs) of the same tripartite efflux pumps. Specifically, the helical portions the OMFs of the pumps aligned with the helical portions of the PAPs.
Confirming evolutionary links using proteins that have not been structurally resolved
To identify even more evolutionary relationships, we rely on a large protein sequence database and consider proteins from two groups related if we detect a trusted relationship between both proteins and an intermediary protein (of unresolved structure). For this, we cannot use the E-value threshold of 20, that we used to demonstrate a lack of an alignment; rather we use a threshold of <10−2 which suggests a meaningful alignment. pHMMER (Finn et al., 2015) was used to search the RefSeq (Pruitt et al., 2002) protein database for homologues of each pair of groups at an E-value <10−2. We searched for sequences with strong alignments between all groups of membrane beta barrels (prototypical barrels, OMFs of efflux pumps, autotransporters, LptDs, fim/ushers, lysins, injectosomes, assembly, and other un-linked barrels) and between the PAPs of efflux pumps and the β-barrels as further discussed below. We found three groups of sequences that connected other groups (Table S1): LptDs to PagP, autotransporters to prototypical barrels, and PAPs to the OMF efflux pump barrels.
Two groups of barrels distinct from the others
As discussed above we see very little sequence similarity between the prototypical barrels and any of the six smaller groups of proteins (i.e., autotransporters, LptDs, Fim/Usher proteins, assembly proteins, lysins, and the OMFs of efflux pumps). In addition to the lack of sequence similarity between the groups, we also find structural differences between lysins and the OMFs of efflux pumps to all other β-barrels (and from each other).
Lysins
The lysinsare multi-chain barrels with either seven or eight chains contributing two strands each. As previously noted, the consensus that lysins are non-homologous to the OMBBs (Remmert et al., 2010) stems from the lack of sequence similarity between the lysins and other OMBBs as well as the findings of lysin expression in other varieties of prokaryotes beyond Gram negative bacteria.
We find that the lysins are also distinct from the other groups by structure. Our structural analysis of these barrels shows that the lysins have the longest average barrel height (fig. 3A), with a mean barrel height of 54.1 Å. This is significantly different from the mean barrel height of the prototypical barrels (33.2 Å) with a p-value = 7.23 × 10−5 using a Wilcoxon rank-sum test.
Figure 3. Structural differences between families of OMBBs.

A-C) Structural characteristics of OMBBs. Groups are colored to match Figure 1. A) A kernel density estimator of the distribution of each barrel’s height. The circles along the bottom represent the data used to generate the kernel. The prototypical group itself has a bimodal distribution, explaining the two humps in the distribution for “All other barrels”. Separation by barrel height for all groups shown in fig. S1A. B) A kernel density estimator was used to show the distribution of each barrel’s average strand tilt in OMBBs. See also fig. S1. Results using an alternate metric for tilt, shear, shown in fig S2. C) Graph showing the increase in radius resulting from each additional hairpin.
Efflux pumps
Like the lysins, the OMFs of efflux pump proteins are multi-chain barrels. However, unlike the lysins, each chain of the OMFs of efflux pumps contributes four strands. We find this group to be the most structurally different from other barrels. The β-strands in the OMFs of efflux pumps are disproportionately tilted compared to those of prototypical barrels, at 56.6° compared to 41.7° (fig. 3B and fig. S1) (p-value of 1.46× 10−6 by Wilcoxon rank-sum test).
In general the addition of each hairpin to a barrel imparts an extra 1.66Å to the radius (fig. 3C). However, as a result of the large tilt, efflux pump OMBBs also have a much wider radius for the total number of strands (n=12) with an average radius of 15.7 Å while the 12-stranded prototypical barrels average 11.9 Å radius. In figure 3C the line of best fit (R2 = 0.953) excludes the efflux pump OMBBs. The predicted radius for 12 strands is 12.2 Å which is significantly different from the observed radii in the efflux pump OMBBs with a p-value = 1.235 × 10−8 by a paired t-test.
These two characteristics are correlated as the tilt angle dictates the radius. McLachlan (McLachlan, 1979) originally described a mathematical relationship between tilt and radius, , in which b is the interstrand distance (4.4 Å, (Murzin et al., 1994)), n is the number of strands, and α is the tilt in radians. We find that this equation holds true for all structures described in all clusters shown in figure 1. Therefore, with a tilt of ~57° the radius of the OMF of the efflux pumps is predicted to be ~15.5 Å. We find no significant difference between the radius predicted by this formula and the observed values (paired t-test, t = 1.16, p-value = 0.283).
Discussion
Looking at a combination of structural and sequence information allows us to better understand the different evolutionary paths to OMBBs.
Convergent evolution of efflux pump OMFs to barrel topology
It is extraordinarily difficult to prove convergence to the exclusion of divergence because bacterial membrane proteins can diverge beyond sequence recognition as described above. Moreover, it has been shown that convergent evolution is not the most likely explanation for many unrelated but structurally similar domains in bacteria (Deeds et al., 2004).
Based on differences in organism, it has previously been hypothesized that the lysins have evolutionarily converged to their barrel structure separately from the divergent evolution of the other β-barrels (Remmert et al., 2010). Here we find a difference in structure between the lysins and the OMBBs. Though other OMBBs in our database are exclusively found in Gram negative bacteria, we find one grouping of OMBBs—the OMFs of efflux pumps— which have no detectable homology to other OMBBs by our metrics. The utility of these efflux pumps which are critical for bacteria to thrive in hostile environments may have necessitated their evolution.
The OMFs of efflux pumps have more tilted strands and are wider for their strand number than all other barrels of known structure. This structural difference, along with the lack of sequence similarity, leads us to suggest convergent evolution. We anticipate that divergent evolution is less likely because it would require substitutions for change in tilt angle while under restraint to maintain the hydrogen bonding network of the β-barrel.
The proposed mechanism of an iris-like mode of opening (Andersen et al., 2002) for OMBB-containing efflux pumps may explain the difference in structure between the OMFs of efflux pumps and other OMBBs. Though originally the iris-like mechanism of opening of efflux pumps was believed to only apply to the alpha helical barrel of the OMFs, some have documented movement in the β-barrel as well (Vaccaro et al., 2008). The ability of a β-barrel to modulate its opening for the peristaltic movement of a pump is different from most OMBBs which are understood to be extremely structurally stable and rigid. Evolving a dynamic barrel would be sufficiently different that it could require a separate evolutionary origin.
The alignments between the outer membrane factors of the tripartite efflux pumps and their periplasmic adaptor proteins may be a key to understanding from where the efflux pump proteins evolved. The PAPs and the OMFs along with the inner membrane proteins are part of the same three part complex. The OMFs and the PAPs both have barrels of alpha helices that are attached to each other (Fig 4A). It is these regions that have similar sequences to each other. Because the two halves of the OMF (TolC) align with each other (indicating an early duplication event), the two helices of PAP (MecA) align twice with the OMF (TolC), once for each half. The helical barrels of PAP and OMF then continue on to β-sheet regions (Fig 4B). This relationship may be further evidence of the OMFs of the efflux pumps’ separate origins from the prototypical OMBBs. The idea that the OMFs and the PAPs have evolved from a common ancestor to create a structural palindrome in an efflux pump is appealing. Both would need to be sorted into the periplasm and neither can function without the other. Similar alignments exist between other OMFs and PAPs (Fig 2 and the online network interface http://cytostruct.info/rachel/barrels/pumps.html).
Figure 4. Alignment between TolC and MecA makes a structural palindrome.

The OMF (TolC, gold) with aligned regions in pink and red, the PAP (MecA) in light blue with aligned regions in dark blue, and inner membrane protein MecB in teal. The region where the PAP (MecA) aligns with the OMF (TolC) is shown in dark blue for the PAP (MecA) and red and pink for the OMF (TolC). Red and Pink on the OMF (TolC) each represents a different alignment. A) The tripartate MecAB-TolC efflux pump (PDB ID 5nik). B) the structural overlay of the two 60-residue sequence alignments between the PAP (MecA) and the OMF (TolC).
A pHMMER search for homologues of both efflux pump OMFs and efflux pump PAPs in the RefSeq database yielded six hits that are shared between these two groups at an E-value < 10−2. The Gene Ontology (Ashburner et al., 2000; Gene-Ontology-Consortium, 2017) annotation (obtained from Uniprot) of all six hits suggest that they are involved in efflux (Table S1). The sequence similarity and function annotation provide further support to the possibility that PAPs and OMFs are evolutionarily linked.
LptD to PagP
By searching for evolutionary connectors between different groups we found two connector proteins between 26-stranded lipid A transporter LptD and 8-stranded lipid A palmitoyltransferase PagP (Table S1). We find this linkage especially intriguing given their similarity of function and we anticipate that future investigation will further explore the evolutionary path between these two proteins responsible for membrane maintenance.
Primordial OMBB
OMBBs are understood to be repeat proteins (Neuwald et al., 1995) with the repeat unit of a double hairpin (Remmert et al., 2010). Since most repeat proteins like OMBBs do not function as individual domains, it has been hypothesized that repeat proteins originated as homo-oligomers. Homo-oligomers would then evolve into the monomeric repeat proteins through genetic duplication (Ponting and Russell, 2000). This perspective favors the view that if there are convergently evolved OMBBs they would be homo-oligomeric, as multi-chain barrels would be the ancestor of a single-chain barrel. This further suggests that the other six classes of monomeric OMBBs which show sequence alignments of E-value >1 to the prototypical barrels may indeed be related to the prototypical barrels as it would be more difficult to create a monomeric β-barrel with no precursor than it would be to start with a multi-chain barrel. However, we see no low E-value connector proteins between these groups and the prototypical barrels to support that they have divergently evolved.
We find three classes of homo-oligomeric multi-chain barrels, the lysins, the OMFs of efflux pumps, and the autotransporters. We have suggested that two of the three classes, the lysins and the OMFs of efflux pumps, have convergently evolved. The third class, the autotransporters, show an alignment with E-value 2.2 between the multi-chain adhesin YadA and the single-chain barrel adhesin Ail. The similarity in function between these two proteins further supports the likelihood of this being a true relationship. Moreover, that YadA has four strands per chain and Ail has 8 strands per chain is suggestive of a genetic duplication which is the most common form of diversification (Katju and Bergthorsson, 2013). Finally, we find a low E-value connector protein between the prototypical barrels and the autotranporters (Table S1). Therefore, if the prototypical, single-chain barrels did evolve from a multi-chain barrel it would most likely be from the class of autotransporters. These autotransporters may be the progenitors of our prototypical barrels.
Reasons for evolutionary convergence on a barrel
Our theory of having at least two groups of membrane beta barrel proteins (OMFs of efflux pumps and lysins) convergently evolve to such similar folds suggests that there is evolutionary pressure for proteins to find and utilize this fold in the membrane or that this fold is easily ‘designable’ by nature (Koonin et al., 2002).
If the efflux pump β-barrel fold were to be a case of convergent evolution, the question is: what are the pressures that have led to convergence onto this fold? The membrane barrel topology is fostered by a variety of overlapping factors. The first factor is chemical. Being hydrophobic, the outer membrane disfavors long stretches of protein without secondary structure because the backbone’s hydrogen bonding requirements cannot be satisfied. Secondary structure is the primary means of proteins satisfying backbone hydrogen bonds, which limits the fold repertoire to all-alpha or all-beta architectures.
The second factor is biological. Most alpha helical proteins are sorted into the inner membrane. The insertion mechanism of the translocon in the inner membrane causes more uniformly hydrophobic segments to be partitioned into the inner membrane (Osborne et al., 2005). More consistently hydrophobic segments have preference for alpha-helical formation wherein more amino acid side chains have contact with the membrane. Only less hydrophobic segments make it through to the periplasm and allow for folding into the outer membrane. β-barrels fold with two faces, one that can face the membrane and one that can face a proteinaceous or aqueous interior. This favors more hydrophilic membrane proteins. The Bam machinery, especially BamA—itself an OMBB—then assist in insertion of the barrels into the outer membrane in an unknown manner that likely takes advantage of the POTRA domain (Kim et al., 2007) and the weak connection between the first and last strand of BamA (Doerner and Sousa, 2017). Chemically, barrels are preferred over β-sandwiches as barrels allow for complete hydrogen bonding whereas in the β-sandwich architecture the metaphorical crust would have an edge of unsatisfied bonds.
Overall, the sequence and structural relationships among OMBBs demonstrate the possibility that the OMBB fold may have been converged upon from different origins. This convergence on a fold would be facilitated by the outer membrane environment requiring β-barrel formation. More work will still need to be done to understand and disentangle the evolutionary pathway of the large group of more clearly inter-related, prototypical OMBBs.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Joanna Slusky (slusky@ku.edu).
METHOD DETAILS
Protein Structure Set
We assembled our dataset by compiling native structures from those present in any of the following membrane-focused databases: mpstruc (http://blanco.biomol.uci.edu/mpstruc/, Stephen White lab at UC Irvine), Orientation of Proteins in Membranes or OPM (Lomize et al., 2006), and MemProtMD (Stansfeld et al., 2015). We then used PISCES (Dunbrack and Wang, 2003) to eliminate sequences with greater than 85% sequence identity. This resulted in a set of 110 single-chain β-barrels (including 20 single-chain multi-barrels), and 20 multi-chain barrels: 130 barrels in total. Single-chain barrels are defined as having a single chain contributing to a single complete barrel. In contrast, multi-chain β-barrels have multiple identical chains contributing to a single barrel; only two barrels are heterooligomers (PDB IDs 3b07 and 4tw1) while the rest are homomeric assemblies.
Strand Definition and Barrel Characteristics
Barrel characteristics were determined using in house software, Polar Bearal, as previously described (Slusky and Dunbrack, 2013). Some updates were included as described below.
Each residue of a chain was classified as belonging to a helix, sheet or other secondary structure using the ϕ and φ angles. Strands were then defined using a combination of backbone hydrogen bonding and a pattern of sheet-like residues. Each barrel was visually checked for appropriate strand definition.
The coordinates of the Cα atoms of the first or last residues of each strand were used to create the top and bottom of the β-barrel into ellipses (Pauly et al., 2002). The angle between the upper and lower ellipses is the angle between the surface normal of the top and bottom ellipsis. The eigenvectors of the two highest eigenvalues were normalized to form the semi-major and semi-minor of the ellipse, which were then used to calculate the eccentricity. The barrel axis is the vector connecting the centroid of each ellipse while the barrel height is the distance between the two centroids. The radius is the average distance to the barrel axis from the Cα atom of each residue. Barrel tilt is the average angle between the barrel axis and the vector formed between the previous Cαatom and next Cα atom for all residues except the first and last residue in each strand.
Barrel Network Creation
The barrel network is the relevant sub-network of protein structure space. In it, each node represents a protein, and edges connect proteins that are evolutionary related. We identify evolutionary relationships using the sensitive HMM sequence aligner HHSearch (Soding, 2005), keeping only alignments with a significant score, and sufficient length (Nepomnyachiy et al., 2014, 2017)
HHSearch (Soding, 2005) was used with default parameters to find significant alignments of the set of 130 β-barrels with proteins from the database of 39,386 70% non-redundant PDB chains (April 2017) available with the HHsuite. The profiles were precomputed by the Soding group and downloaded from http://wwwuser.gwdg.de/~compbiol/data/hhsuite/databases/hhsearch dbs/. HMM profiles for the 57 β-barrels in our list but not in this database were generated using the webserver for HHblits (Remmert et al., 2012) and the database uniclust30_2017_04. The search yielded 2974 alignments at E-value < 1. Alignments with no structural component were removed. We then applied a 20-residue cutoff which is a length that includes 99.7% of the hairpins in our database. All alignments to a non-OMBB structure were visually inspected.
Although it has been shown that using membrane-specific alignment functions yields more accurate alignments for membrane proteins (Stamm et al., 2013), such methods have not yet been applied to β-barrels.
To identify sequence homologues shared between the OMFs of efflux pumps and prototypical barrels and shared between the efflux pump OMFs and PAPs, the webserver for pHMMER was used. The sequence database used was “Reference Proteomes”. We reported all homologoues of two groups with E-values < 10−2.
Protein visualization
Images were generated using PyMOL (DeLano, 2002).
QUANTIFICATION AND STATISTICAL ANALYSIS
All details of statistical tests can be found in the Results in the text. P-values < 0.05 were determined to be significant.
ADDITIONAL RESOURCES
We organized the set of proteins and their alignments as a network, using CyToScape (Saito et al., 2012), and CyToStruct (Nepomnyachiy et al., 2015)). Multiple alignments between nodes frequently exist, but only the edge with the lowest E-value is shown. To easily view the alignments in a molecular viewer. The resulting network is available online at: http://cytostruct.info/rachel/barrels/. The network including soluble alignments is available online at: http://cytostruct.info/rachel/barrels/pumps.html. In both networks, users can change the E-value cutoff and see the resulting networks: using a more stringent cutoff removes edges, resulting in a more fragmented network. The structure of a protein can be viewed in a molecular viewer by clicking its node. The structural and sequence alignment can be viewed by clicking the edge.
Supplementary Material
Highlights:
Most outer membrane β-barrels (OMBBs) are clearly related to each other.
Efflux OMBBs have no known homology to other OMBBs and have distinctive structure.
Efflux OMBBs and soluble pump components may have evolved from a common ancestor.
Autotransporters may be best known models for primordial OMBB structure.
Acknowledgements
Professors James Walters, Mark Holder, Vikas Nanda, Christian Ray and Eric Deeds for helpful discussions. NIH awards DP2GM128201, P20GM103418, P20GM113117, T32-GM008359, NSF MCB160205, The Gordon and Betty Moore Inventor Fellowship, KU-startup, and Israel Science Foundation grant number 450/16.
Author contributions
Conceptualization, J.S.G.S.; Methodology, J.S.G.S., R.K., M.W.F., and S.N.; Software, M.W.F., S.N., and R.K.; Investigation, M.W.F., R.F.; Writing—original draft preparation, J.S.G.S., M.W.F.; Writing—Review & Editing Preparation, N.B., R.K., J.S.G.S, M.W.F.
Declaration of Interest
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersen C, Koronakis E, Bokma E, Eswaran J, Humphreys D, Hughes C, and Koronakis V (2002). Transition to the open state of the TolC periplasmic tunnel entrance. Proceedings of the National Academy of Sciences 99, 11103–11108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball C, Blake J, Botstein D, Butler H, Cherry J, Davis A, Dolinski K, Dwight S, Eppig J, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Genetics 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagos PG, Liakopoulos TD, and Hamodrakas SJ (2005). Evaluation of methods for predicting the topology of beta-barrel outer membrane proteins and a consensus prediction method. BMC Bioinformatics 6, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battistuzzi FU, Feijao A, and Hedges SB (2004). A genomic timescale of prokaryote evolution: insights into the origin of methanogenesis, phototrophy, and the colonization of land. BMC Evolutionary Biology 4, 44–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Schaeffer RD, Liao Y, Kinch LN, Pei J, Shi S, Kim B-H, and Grishin NV (2014). ECOD: An Evolutionary Classification of Protein Domains. PLOS Computational Biology 10, e1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeds EJ, Shakhnovich B, and Shakhnovich EI (2004). Proteomic Traces of Speciation. Journal of Molecular Biology 336, 695–706. [DOI] [PubMed] [Google Scholar]
- DeLano WL (2002). The PyMOL Molecular Graphics System (San Carlos, CA, USA: DeLano Scientific; ). [Google Scholar]
- Doerner PA, and Sousa MC (2017). Extreme Dynamics in the BamA β-Barrel Seam. Biochemistry 56, 3142–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Beis K, Nesper J, Brunkan-LaMontagne AL, Clarke BR, Whitfield C, and Naismith JH (2006). Wza the translocon for E. coli capsular polysaccharides defines a new class of membrane protein. Nature 444, 226–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbrack J, R. L and Wang G (2003). PISCES: a protein sequence culling server. Bioinformatics 19, 1589–1591. [DOI] [PubMed] [Google Scholar]
- Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, Bateman A, and Eddy SR (2015). HMMER web server: 2015 update. Nucleic Acids Research Web Server Issue 43, W30–W38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox NK, Brenner SE, and Chandonia J-M (2014). SCOPe: Structural Classification of Proteins—extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Research 42, D304–D309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene-Ontology-Consortium (2017). Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Research 45, D331–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Morales D, and Liang J (2011). Pattern of Amino Acid Substitutions in Transmembrane Domains of β-Barrel Membrane Proteins for Detecting Remote Homologs in Bacteria and Mitochondria. PLoS ONE 6, e26400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S, and Altschul SF (1990). Methods for assessing the statistical significance of molecular sequence features by using general scoring schemes. Proc Natl Acad Sci USA 87, 2264–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katju V, and Bergthorsson U (2013). Copy-number changes in evolution: rates, fitness effects and adaptive significance. Frontiers in Genetics 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Malinverni JC, Sliz P, Silhavy TJ, Harrison SC, and Kahne D (2007). Structure and Function of an Essential Component of the Outer Membrane Protein Assembly Machine. Science 317, 961–964. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Wolf YI, and Karev GP (2002). The structure of the protein universe and genome evolution. Nature 420, 218–223. [DOI] [PubMed] [Google Scholar]
- Liu A, Tran L, Becket E, Lee K, Chinn L, Park E, Tran K, and Miller JH (2010). Antibiotic Sensitivity Profiles Determined with an Escherichia coli Gene Knockout Collection: Generating an Antibiotic Bar Code. Antimicrobial Agents and Chemotherapy 54, 1393–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomize MA, Lomize AL, Pogozheva ID, and Mosberg HI (2006). OPM: Orientations of Proteins in Membranes database. Bioinformatics 22, 623–625. [DOI] [PubMed] [Google Scholar]
- McLachlan AD (1979). Gene duplications in the structural evolution of chymotrypsin. Journal of Molecular Biology 128, 49–79. [DOI] [PubMed] [Google Scholar]
- Menestrina G, Serra MD, Pederzolli C, Bregante M, and Gambale F (1995). Bacterial Hemolysins and Leukotoxins Affect Target Cells By Forming Large Exogenous Pores Into Their Plasma Membrane. Escherichia coli Hemolysin A as a Case Example. Biosci Rep 15, 543–551. [DOI] [PubMed] [Google Scholar]
- Murzin AG, Lesk AM, and Chothia C (1994). Principles determining the structure of beta-sheet barrels in proteins I. A theoretical analysis. Journal of Molecular Biology 236, 1369–1381. [DOI] [PubMed] [Google Scholar]
- Nepomnyachiy S, Ben-Tal N, and Kolodny R (2014). Global view of the protein universe. Proceedings of the National Academy of Sciences 111, 11691–11696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepomnyachiy S, Ben-Tal N, and Kolodny R (2015). CyToStruct: Augmenting the Network Visualization of Cytoscape with the Power of Molecular Viewers. Structure 23, 941–948. [DOI] [PubMed] [Google Scholar]
- Nepomnyachiy S, Ben-Tal N, and Kolodny R (2017). Complex evolutionary footprints revealed in an analysis of reused protein segments of diverse lengths. Proceedings of the National Academy of Sciences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuwald AF, Liu JS, and Lawrence CE (1995). Gibbs motif sampling: Detection of bacterial outer membrane protein repeats. Protein Science 4, 1618–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne AR, Rapoport TA, and Berg B.v.d.(2005). Protein Translocation by the Sec61/SecY Channel. Annual Review of Cell and Developmental Biology 21, 529–550. [DOI] [PubMed] [Google Scholar]
- Pauly M, Gross M, and Kobbelt LP (2002). Efficient simplification of point-sampled surfaces In Proceedings of the conference on Visualization ‘02 (Boston, Massachusetts: IEEE Computer Society; ), pp. 163–170. [Google Scholar]
- Pearson WR (2013). An Introduction to Sequence Similarity (“Homology”) Searching. Current protocols in bioinformatics / editoral board, Andreas D Baxevanis [et al] 0 3, 10.1002/0471250953.bi0471250301s0471250942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting CP, and Russell RB (2000). Identification of distant homologues of fibroblast growth factors suggests a common ancestor for all β-trefoil proteins11Edited by J. Thornton. Journal of Molecular Biology 302, 1041–1047. [DOI] [PubMed] [Google Scholar]
- Poole K (2005). Efflux-mediated antimicrobial resistance. Journal of Antimicrobial Chemotherapy 56, 20–51. [DOI] [PubMed] [Google Scholar]
- Pruitt K, Brown G, Tatusova T, and Maglott D (2002). The Reference Sequence (RefSeq) Database In The NCBI Handbook [Internet], McEntyre J, and Ostell J, eds. (Bethesda, MD: National Center for Biotechnology Information (US)). [Google Scholar]
- Reddy BL, and Saier MH Jr. (2016). Properties and Phylogeny of 76 Families of Bacterial and Eukaryotic Organellar Outer Membrane Pore-Forming Proteins. PLOS ONE 11, e0152733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmert M, Biegert A, Hauser A, and Soding J (2012). HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nature Methods 9, 173–175. [DOI] [PubMed] [Google Scholar]
- Remmert M, Biegert A, Linke D, Lupas AN, and Soding J (2010). Evolution of Outer Membrane Beta-Barrels from an Ancestral Beta Beta Hairpin. Molecular Biology and Evolution 27, 1348–1358. [DOI] [PubMed] [Google Scholar]
- Saito R, Smoot ME, Ono K, Ruscheinski J, Wang P-L, Lotia S, Pico AR, Bader GD, and Ideker T (2012). A travel guide to Cytoscape plugins. Nature methods 9, 1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz GE (2002). The structure of bacterial outer membrane proteins. Biochimica et Biophysica Acta (BBA) - Biomembranes 1565, 308–317. [DOI] [PubMed] [Google Scholar]
- Sillitoe I, Lewis TE, Cuff A, Das S, Ashford P, Dawson NL, Furnham N, Laskowski RA, Lee D, Lees JG, et al. (2015). CATH: comprehensive structural and functional annotations for genome sequences. Nucleic Acids Research 43, D376–D381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slusky JSG, and Dunbrack RL (2013). Charge Asymmetry in the Proteins of the Outer Membrane. Bioinformatics 29, 2122–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soding J (2005). Protein homology detection by HMM-HMM comparison. Bioinformatics 21, 951–960. [DOI] [PubMed] [Google Scholar]
- Sojo V, Dessimoz C, Pomiankowski A, and Lane N (2016). Membrane Proteins Are Dramatically Less Conserved than Water-Soluble Proteins across the Tree of Life. Molecular Biology and Evolution 33, 2874–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamm M, Staritzbichler R, Khafizov K, and Forrest LR (2013). Alignment of Helical Membrane Protein Sequences Using AlignMe. PLOS ONE 8, e57731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfeld PJ, Goose JE, Caffrey M, Carpenter EP, Parker JL, Newstead S, and Sansom MSP (2015). MemProtMD: Automated Insertion of Membrane Protein Structures into Explicit Lipid Membranes. Structure 23, 1350–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swick MC, Morgan-Linnell SK, Carlson KM, and Zechiedrich L (2011). Expression of Multidrug Efflux Pump Genes acrAB-tolC, mdfA, and norE in Escherichia coli Clinical Isolates as a Function of Fluoroquinolone and Multidrug Resistance. Antimicrobial Agents and Chemotherapy 55, 921–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsirigos KD, Govindarajan S, Bassot C, Vastermark A, Lamb J, Shu N, and Elofsson A (2018). Topology of membrane proteins — predictions, limitations and variations. Current Opinion in Structural Biology 50, 9–17. [DOI] [PubMed] [Google Scholar]
- Vaccaro L, Scott KA, and Sansom MSP (2008). Gating at Both Ends and Breathing in the Middle: Conformational Dynamics of TolC. Biophysical Journal 95, 5681–5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgurskaya HI, and Nikaido H (1999). Bypassing the periplasm: Reconstitution of the AcrAB multidrug efflux pump of Escherichia coli. Proceedings of the National Academy of Sciences 96, 7190–7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.