

Abstract
Asparagine (N)-linked glycosylation is a post-translational modification essential for the function of complex transmembrane proteins. However, targeting glycosylation for cancer therapy has not been feasible due to generalized effects on all glycoproteins. Here we perform sensitivity screening of 94 lung cancer cell lines using NGI-1, a small molecule inhibitor of the oligosaccharyltransferase (OST) that partially disrupts N-linked glycosylation, and demonstrate a selective loss of tumor cell viability. This screen revealed NGI-1 sensitivity in just 11/94 (12%) cell lines, with a significant correlation between OST and EGFR inhibitors. In EGFR mutant NSCLC with EGFR TKI resistance (PC9-GR, HCC827-GR, and H1975-OR), OST inhibition maintained its ability to induce cell cycle arrest and a proliferative block. Addition of NGI-1 to EGFR TKI treatment was synthetic lethal in cells resistant to gefitinib, erlotinib, or osimertinib. OST inhibition invariably disrupted EGFR N-linked glycosylation and reduced activation of receptors either with or without the T790M TKI resistance mutation. OST inhibition also dissociated EGFR signaling from other co-expressed receptors like MET via altered receptor compartmentalization. Translation of this approach to preclinical models was accomplished through synthesis and delivery of NGI-1 nanoparticles, confirmation of in vivo activity through molecular imaging, and demonstration of significant tumor growth delay in TKI resistant HCC827 and H1975 xenografts. This therapeutic strategy breaks from kinase-targeted approaches and validates N-linked glycosylation as an effective target in tumors driven by glycoprotein signaling.
INTRODUCTION:
The epidermal growth factor receptor (EGFR) is a transmembrane glycoprotein and receptor tyrosine kinase (RTK) that is over-expressed in diverse cancer subtypes. In NSCLC, a subset of adenocarcinomas harbor EGFR activating kinase domain mutations that drive both the initiation and maintenance of oncogenic signaling (1,2). These tumors are sensitive to EGFR specific tyrosine kinase inhibitors (TKIs), which block EGFR signaling, induce cell death, and lead to dramatic clinical responses (3).
Although TKIs have revolutionized treatment for EGFR mutant NSCLC, resistance to therapy inevitably develops and progression typically occurs within a year of treatment (4,5). Mechanisms of therapeutic resistance include secondary (T790M) and tertiary kinase domain mutations (C797S) that prevent TKI access to the kinase active site (6–8). The discovery of these mutations has led to the design and synthesis of next generation EGFR TKIs that target these mechanisms of resistance and block EGFR kinase activity. However, despite significant initial clinical responses, therapeutic resistance to these EGR TKIs also occurs and leads to progressive disease.
EGFR TKI therapeutic resistance also develops through parallel, or bypass, mechanisms. These include amplification and enhanced signaling through co-expressed MET and ERBB2 RTKs, as well as in association with less well understood phenotypic changes such as acquisition of epithelial to mesenchymal transition (EMT) or small cell differentiation (9–11). At the genetic level co-occurring mutations to pathways that regulate membrane signaling, transcription, or control of cell cycle progression have been implicated (12). Because EGFR bypass resistance mechanisms can occur after initial TKI treatment, emerge later in the disease course after treatment with second or third generation inhibitors, and are difficult to treat with standard therapeutic options, they now represent a category with the greatest need for development of novel treatment strategies.
RTKs and other highly complex cell surface signaling molecules require post-translational modification by N-linked glycans to achieve appropriate cell compartment distribution, conformations, and function. N-linked glycan assembly and transfer to nascent proteins is completed in the endoplasmic reticulum by a multi-subunit protein complex called the oligosaccharyltransferase (OST). Although N-linked glycosylation is an essential process, partial inhibition with a recently discovered small molecule inhibitor of the OST catalytic subunit suggests a selective effect on tumor cells with RTK dependent signaling (13). In this work, we therefore examined the effects of this inhibitor (NGI-1) on proliferation and apoptosis in EGFR mutant NSCLC with therapeutic resistance. Our results indicate that targeting the OST is a novel approach for treating diverse mechanisms of resistance to EGFR TKI therapy.
MATERIALS AND METHODS:
Cell Culture and Cell Line Derivation:
The H1975 and A549 cell lines were purchased from ATCC (Manassas, VA), the PC9 cell line was a gift from Katie Politi, and the HCC-827 and HCC-827-GR lines were gifts from Jeff Engelman (MGH, Boston Mass). Cell lines were cultured in RPMI 1640 + 10% FBS supplemented with penicillin and streptomycin (Gibco, Life Technologies, Grand Island, NY, US) in a humidified incubator with 5% CO2, and they were kept in culture no more than 4 months after resuscitation from the original stocks. No additional authentication was performed. Mycoplasma cell culture contamination was routinely checked and ruled out using the MycoAlert Mycoplasma Detection Kit (Lonza, Rockland, ME USA). To generate a TKI resistant cell lines, either PC9 or H1975 cells were exposed to increasing concentrations of gefitinib or osimertinib, respectively. Gefitinib or Osimertinib concentrations were increased stepwise when cells resumed growth kinetics similar to the untreated parental cells over a dose range from 10 to 500 nM. Resistant cell cultures were obtained ~8–12 weeks after initiation of drug exposure. To confirm the emergence of a therapeutic resistant, MTT assays were performed after allowing the cells to grow in drug-free conditions for at least 4 days.
Cell Line Screening:
NGI-1 activity was screened in 94 lung cancer cell lines at the Center for Molecular Therapeutics at the Massachusetts General Hospital Center for Cancer Research with previously described methods (14). Briefly, cells were treated in 384-well microplates using 9 serial NGI-1 dilutions, returned to an incubator for 96 hours, then stained with 55 μg/ml resazurin (Sigma) prepared in Glutathione-free media for 4 hours. Fluorescent signal intensity was quantified with a plate reader at excitation and emission wavelengths of 535/595 nm to determine viability. Viability ratio across the 9 doses were fitted to determine the half maximal inhibitory concentration (IC50). Cell lines with an IC50 less than 10uM NGI-1 were considered sensitive to the drug. For data base comparisons, cell lines with EGFR inhibitor IC50s less than 750 nM were considered EGFR TKI sensitive.
Proliferation Assays:
Growth rates were determined by CellTiter 96 NonRadioactive Cell Proliferation Assay (Promega; Madison, WI, USA) according to the manufacturer’s directions. Briefly, NSCLC cells (2×103) untreated or treated with 10 μM NGI-1, 100nM Gefitinib, or 1μM Osimertinib (Selleck Chemicals), were seeded in triplicate in 96-wells plates and grown in culture medium containing 10% serum. The media was changed with or without new inhibitor every 48h. Cell numbers were estimated after 0, 3, and 5 days by adding MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reagent to the wells 4h before taking the spectrophotometric reading (absorbance at 570 nm).
Cell Cycle Distribution:
For the assessment of cell cycle distribution, 1 × 106 cells were collected, washed once with ice-cold PBS and fixed in ice-cold 70% ethanol overnight at −20°C. Thereafter, cells were washed twice with PBS and incubated for 30 min at room temperature in 200 μL of Guava Cell Cycle Reagent (Guava Technologies). Cytofluorometric acquisitions were performed on a LSRII cytometer (BD Biosciences). First-line analysis was performed with Flow Jo software, upon gating of the events characterized by normal forward and side scatter parameters and discrimination of doublets in a FSC-A vs. FSC-H bivariate plot. Approximately 30,000 cells were analyzed per experiment, and the mean value was obtained from 3 independent assays.
Assessment of Apoptosis:
Redistribution of plasma membrane phosphatidylserine (PS) is a marker of apoptosis and was assessed by annexin V phycoerythrin (BD Biosciences) according to the manufacturer’s protocol. Briefly, 1 × 106 cells/sample were collected, washed in PBS, pelleted, and resuspended in incubation buffer (10 mm HEPES/NaOH, pH 7.4, 140 mm NaCl, 2.5 mm CaCl2) containing 1% annexin V and 1% 7-Amino-actinomycin D or propidium iodide, to identify dead cells. The samples were kept in the dark and incubated for 15 min prior to analysis by flow cytometry on a LSRII cytometer (BD Biosciences) using BD FACSDiva software (BD Biosciences). First-line analysis was performed with Flow Jo software. Approximately 50,000 cells were analyzed per experiment, and the mean value was obtained from 3 independent assays.
Western Blot Analysis:
Western blot analyses were performed as previously described (13). Cell fractionation and recovery of cell surface proteins was performed with HCC827 and HCC827-GR cells using Qproteome Plasma Membrane Protein kit (Qiagen, Gaitherberg MD) according the manufacturer’s protocol. Immunoprecipitation of EGFR was performed on whole cell lysate of intact HCC827 and HCC827-GR cell monolayers using Protein A magnetic beads (Cell Signaling) to isolate EGFR coupled to rabbit anti–EGFR (1:100) antibody from Cell Signaling (Danvers, MA, USA). Equilibrated Protein A magnetic beads equilibrated with a whole cell lysate were used as negative controls for non-specific protein binding. We used the following primary antibodies, rabbit anti–EGFR (1:1,000) antibody from Santa Cruz biotechnology, Inc. (Santa Cruz, CA); and rabbit anti–pEGFR-Y1068 (1:1,000), rabbit anti–MET (1:1,000), rabbit anti–pMET-Y1234/1235 (1:500) from Cell Signaling (Danvers, MA, USA), anti-GADPH (HRP-60004 Proteintech; 1:10,000). The nitrocellulose-bound primary antibodies, were detected with anti–rabbit IgG horseradish peroxidase–linked antibody or anti–mouse IgG horseradish peroxidase– linked antibody (EMD Millipore; Temecula, CA USA), and were detected by the enhanced chemoluminescence staining ECL (GE Healthcare–Amersham Pharmacia, Buckinghamshire, U.K.). For phospho-protein array analysis, H1975 and H1975-OR cells were cultured in 6-well plates in serum-containing medium. The Human Phospho-Protein Array kit (ARY001B; R&D Systems) was used to simultaneously detect the relative level of tyrosine phosphorylation of human receptor tyrosine kinases (RTKs) according to the manufacturer’s protocol.
Microscopy:
For immunofluorescence, HCC827-GR cell lines were grown on glass coverslips to 80% confluence. Cell cultures were washed with PBS and fixed with 4% (w/v) formaldehyde in PBS for 30 min at 37°C. After washing with PBS, cells were permeabilized with 0.1% v/v Triton X-100 in PBS for 10 min, rinsed three times in PBS and treated with 5% w/v bovine serum albumin for 1 h. Cells were then incubated overnight at 4°C with mouse anti-EGFR pAb (sc-03; Santa Cruz Biotechnology; 1:800) and rabbit anti-MET mAb (Cell Signaling; 1:1,000) primary antibodies and for 1 h at room temperature with either Alexa Fluor 543-conjugated goat anti-rabbit IgG or Alexa Fluor 488-conjugated goat anti-mouse IgG (ThermoFisher Scientific; 1:1,000) secondary antibodies. All antibodies were diluted in PBS containing 5% w/v bovine serum albumin. Nuclei were stained using ToPro3 (Invitrogen). Confocal cellular images were captured with an inverted Zeiss LSM 510 Pascal laser confocal microscope (Carl Zeiss, Jenna, Germany), using a 63/1.4 Plan-Apochromat objective.
Clonogenic Survival Assays:
Clonogenic survival assays were performed with cells treated in the presence of TKI inhibitors or NGI-1. Cultures were grown for 14 days, washed once with 1x PBS, and stained with 0.25% crystal violet in 80% methanol. Colonies with >50 cells were counted and clonogenic cell survival differences for each treatment were compared using survival curves generated from the linear quadratic equation using GraphPad Prism7 (GraphPad Software Inc.).
Deep Sequencing of EGFR Mutations:
Somatic mutations were identified and quantified from cell-line DNA using an enhanced version of the previously published Error-Suppressed Deep Sequencing method (15,16) Genomic DNA was purified from cell lines and fragmented to an average size of ~200 bp on the Covaris E210 system. The following parameters were used for fragmentation: Duty cycle =20%, Intensity = 5, Cycle per burst = 200, Time = 255 seconds. 10 ng of fragmented genomic DNA was used for next generation sequencing library preparation. High-throughput DNA sequencing was performed in 75 base-pair, paired-end mode on an Illumina HiSeq2500 instrument. Allelic fraction of mutant DNA was calculated based on mutant and wild-type sequence counts obtained from next-generation sequencing data.
NGI-1 nanoparticle (NP) preparation:
Polyethylene glycol (PEG)-b-Polylactic acid (PLA) diblock polymer (Mw PEG = 5kDa, Mw PLA = 10kDa) was purchased from Polysciences, Inc. (Warrington, PA, USA). Dimethylsulfoxide were obtained from J.T. Baker (Avantor Performance Materials, Central Valley, PA, USA). Polyethyeleneimine (PEI; branched – average Mw ~800, average Mn ~600) was purchased from Sigma-Aldrich (USA). PLA-PEG NPs were synthesized using a nanoprecipitation technique, similar to one previously reported (17). Briefly, to create control NPs, 100 mg of polymer was dissolved in 5 ml DMSO at RT for 2 h. The polymer solution was then divided into 200 μL aliquots. Each 200 μL aliquot was added drop-wise to 1mL deionized (DI) water under strong vortex to create a NP suspension. These suspensions were immediately pooled and diluted 5x with DI water. This diluted suspension was then transferred to an Amicon Ultracell 100k centrifugal filter unit, and centrifuged at 4000 g, 4°C for 30 min. The NPs were washed twice with DI water and centrifuged for another 30 min each time. After a final wash with DI water, the NPs were then centrifuged for 1 h to achieve a final concentration of 100 mg NP/mL DI water. The final NP suspension was then either immediately used for in vivo or in vitro experiments, or snap-frozen at − 80°C until use.
For drug-loaded NPs, NGI-1 was dissolved in DMSO at a concentration of 50 mg/ml, and PEI was dissolved in DMSO at 50mg/ml. The NGI-1 and PEI solutions were then mixed at a 6:1 ratio (by weight) of PEI:drug. The solution was vortexed for ~10 s and then incubated at room temperature for 15 min. After the 15 min incubation period, the PEI/NG-1 solution was then added to the PLA-PEG solution at a 10% ratio of NGI-1:PLA-PEG by weight. This combined solution was briefly vortexed and then water-bath sonicated to ensure uniform mixing. The solution was then added dropwise to diH2O under vortex at a final ratio of 1:5 organic:aqueous phase. These suspensions were then pooled, diluted, and filtered, washed, and frozen and stored or used as above. For PEI-only loaded control NPs, PEI was dissolved in DMSO at a concentration of 50 mg/ml, the PEI solution was then added to the PLA-PEG solution at a 60% ratio of PEI:PLA-PEG by weight. This combined solution was briefly vortexed and then water-bath sonicated to ensure uniform mixing. The solution was then added dropwise to diH2O under vortex at a final ratio of 1:5 organic:aqueous phase. These suspensions were then pooled, diluted, and filtered, washed, and frozen and stored or used as above. All NP preparations were tested for particle size distribution by dynamic light scattering (DLS) using a Malvern Nano-ZS (Malvern Instruments).
To assess particle loading a 100 μL solution of a nanoparticle sample was lyophilized in a pre-weighed eppendorf tube to measure particle yield. Drug loading was determined by dissolving 10mg of NPs in 1ml of acetonitrile (ACN) for 24 h at RT. The dissolved NP solution was then size-filtered at 0.22 μM and the filtrate was analyzed using a Shimadzu HPLC System (SpectraLab Scientific, Markham, ON, Canada) and compared against a previously established standard curve for NGI-1. Drug loading was repeated with three technical replicates per batch of NPs.
NGI-1 Imaging in Vivo:
Bioluminescent imaging of mice bearing PC9 ERlucT flank tumors were generated as previously described (18). Tumors were grown in six week old female athymic Swiss nu/nu mice (Envigo) by subcutaneous flank implantation of ~1X107 cells into the hind limb. Ten days following injection, mice bearing palpable tumors were anesthetized with a 1% isoflurane/air mixture and given a single I.P. dose of 150 mg/kg luciferin in normal saline. Bioluminescent imaging was performed from 5 to 30 minutes after luciferin administration, and mice were anesthetized and kept warm with a temperature controlled bed during image acquisition. Signal intensity was quantified for a region of interest (ROI) for each tumor over the imaging time period to determine the peak of bioluminescent activity. Tumor bioluminescence prior to drug treatment was used to establish a baseline of activity and to calculate induction of Luc activity. After obtaining baseline images, mice were treated i.v. with blank nanoparticles or 20 mg/Kg of NGI-1 nanoparticles. Tumor bioluminescence was assessed at day 1 and day 2 after treatment. All experimental procedures were approved in accordance with IACUC and Yale university institutional guidelines for animal care and ethics and guidelines for the welfare and use of animals in cancer research.
NGI-1 Therapeutic Studies in Xenograft Tumors:
The effects of NGI-1 were evaluated in Six-to-eight-week old female athymic Swiss nu/nu mice bearing bilateral flank xenograft tumors. Five days after inoculation, mice were randomized to receive i.v. NGI-1 NPs (20 mg/kg) or blank NPs three times per week for a total of 8 doses. TKI inhibitors (erlotinib or osimertinib) were orally administered every day at a dose of 25 mg/kg and 5 mg/kg, respectively, over the same time period as NGI-1 treatment. Tumor size was measured three times per week and volume was calculated according to the formula π/6 × (length) × (width)2.
Statistics:
Data points are reported as experimental averages and error bars represent standard deviations or standard errors, as indicated, from at least three independent experiments. No samples were excluded from the analysis. The Chi-square test was used for cell line sensitivity comparisons. Otherwise statistical significance was determined using a two-sided Student’s t-test with Graph-Pad Prism 6 (GraphPad Software Inc.). A P value < 0.05 or less was considered to be statistically significant.
RESULTS:
Lung Cancer Cell Line Screen for OST Inhibitor Sensitivity.
To determine cellular characteristics of OST inhibitor sensitivity we screened a panel of 94 lung cancer cell lines and measured viability following four days of NGI-1 treatment. Fifty percent inhibitory concentration (IC50) values were determined from a nine point drug dilution series that used a maximum concentration of 10μM, which is 10 fold higher than the IC50 of NGI-1 in cell culture. The screening results showed that only 11/94 cell lines demonstrated an IC50 less than 10μM for NGI-1 (Suppl. Table 1). We compared these responses to those from EGFR inhibitor treatments (afatinib, erlotinib, and gefitinib) performed in the same cell line screening platform (http://www.cancerrxgene.org), and these results indicated a strong correlation between sensitivity to EGFR inhibition, defined as an IC50 < 0.75μM, and NGI-1 sensitivity (Fig. 1). These results support the hypothesis that NGI-1 preferentially affects tumor cells that are dependent on glycoprotein driven proliferation. Using Fisher’s exact test, a correlation between NGI-1 and afatinib sensitivity (73 cell lines; p<.0001) or NGI-1 and gefitinib sensitivity (75 cell lines; p=.0015) were significant. Only four EGFR TKI sensitive cell lines were not NGI-1 sensitive, including EGFR kinase domain mutant H3255 and LOU-NH91. Because we previously demonstrated significant inhibition of H3255 proliferation by NGI-1 (13), these screening results are most consistent with false negative values that could be due to the growth characteristics or reduced proliferation time of the screening assay. The two other cell lines, LU-139 and EPLC-272H, were afatinib sensitive but not as responsive to other EGFR TKIs or NGI-1, which may reflect borderline TKI sensitivity. Of the seven lung cancer cell lines with sensitivity to both NGI-1 and EGFR TKIs, four harbor known genetic aberrations in the ErbB RTK family; EGFR 747–749 deletion, EGFR amplification, ErbB2 amplification, and ErbB3 Y523C mutation. In summary, the data from this screen shows that OST inhibition has a largely selective effect on lung cancer cell lines with genetic or phenotypic characteristics of EGFR and ErbB RTK driven proliferation.
Figure 1: Lung Cancer Cell Line drug sensitivity.
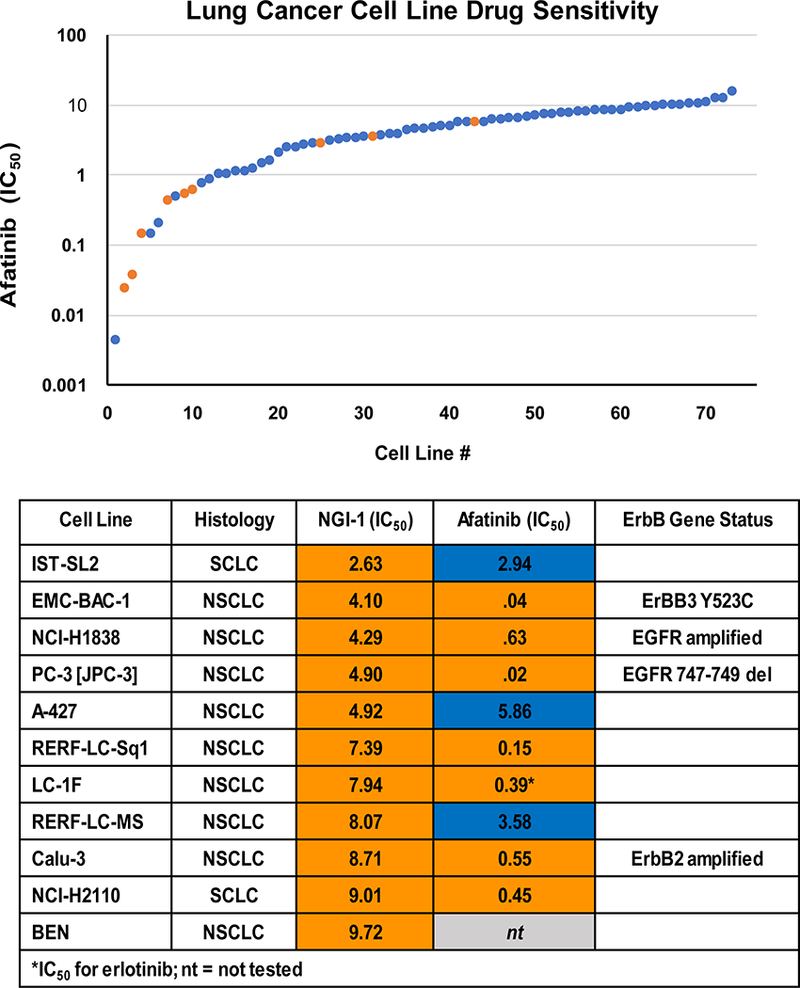
Comparisons of cell viability in 73 lung cancer cell lines after dose response treatment with afatinib or NGI-1. Afatinib IC50 is plotted and NGI-1 sensitive (orange) or insensitive (blue) cell lines are marked to show a correlation of sensitivity between the two inhibitors (upper panel). The lower panel details NGI-1 sensitive lung cancer cell line identities, and drug IC50 with notable genetic abnormalities of EGFR family receptors.
OST inhibition blocks proliferation of EGFR mutant NSCLC with T790M mutation.
The selective effect of NGI-1 on EGFR dependent lung cancer proliferation in the cell line screen led us to further investigate its effects in the setting of NSCLC TKI resistance. Gefitinib resistant cultures of the EGFR exon 19 deletion containing PC-9 cell line were generated through serial passaging in the presence of gefitinib as described in Materials and Methods. These cell lines, referred to as PC9-GR1 and PC9-GR2, were analyzed by EGFR sequencing and demonstrated to have the T790M resistance mutation. Because EGFR membrane localization and activation can be reduced through OST inhibition (13), a mechanism that is independent of the intracellular kinase domain, the effects of NGI-1 were examined on proliferation of these resistant PC9 cell lines. NGI-1 reduced the proliferation of both parental and T790M expressing PC9 cell lines by ~90% (p <.001; Fig. 2A). NGI-1 also caused a G1 arrest in both GR cell lines (60% vs 40% p<.05 in PC9-GR1 and 80% vs 60% p<.05 in PC9-GR2), consistent with the block in proliferation (Fig. 2B). This data suggests that despite the EGFR-T790M mutation, the EGFR remains sensitive to inhibition of N-linked glycosylation.
Figure 2: NGI-1 blocks proliferation in NSCLC TKI-resistant cells.
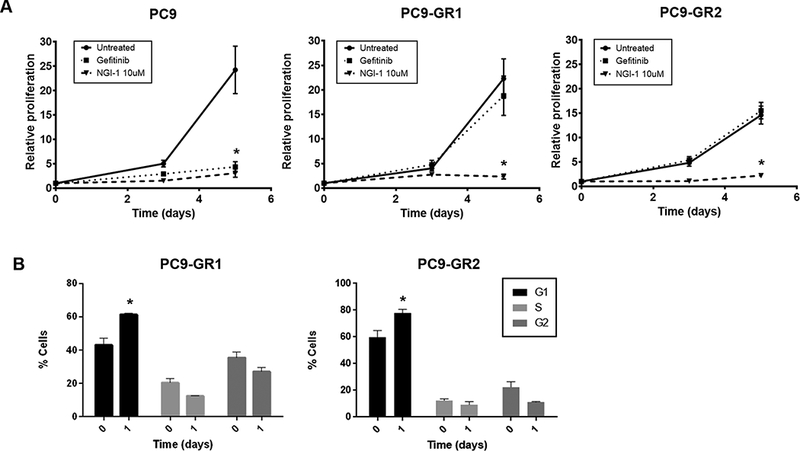
a. Fold proliferation measured by MTT over 5 days with 10μM NGI-1 or 100nM Gefitinib treatment in PC9 and PC9-GR cells. b. Flow cytometry and cell cycle distribution of PC9-GR cells after NGI-1 treatment for 24 h. Data are represented as mean ± s.d., n = 3. P values were determined using two-tailed t-tests. *P < 0.01.
OST inhibition re-sensitizes EGFR T790M NSCLC to EGFR TKIs.
Unlike gefitinib or erlotinib, NGI-1 does not induce apoptosis in parental PC9 cells (Fig.3A; left panel). NGI-1 treatment alone also has no pro-apoptotic effect in the PC9-GR cells. However, the combination of NGI-1 and erlotinib treatment for 48 h was sufficient to induce apoptosis in PC9-GR cell lines as measured by Annexin V flow cytometry (10% vs 40% p<.05 Fig. 3A; right panel). The combined treatment, however, could not induce apoptosis in EGFR independent lung cancer such as the KRAS mutant A549 cells (Suppl Fig. 1). Treatment with NGI-1 and erlotinib also produced a greater reduction of EGFR phosphorylation measured by phosphoblot analysis (Fig. 3B) and reduced clonogenic survival by more than 30% compared to treatment with either inhibitor alone (p<.05, Fig. 3C, D).
Figure 3: NGI-1 overcomes EGFR T790M resistance.
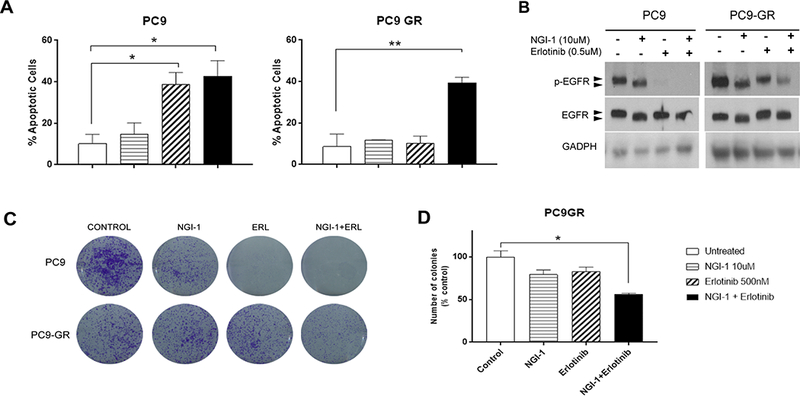
a. Apoptosis susceptibility of PC9 and PC9-GR NSCLC cell lines following 48 h treatment with NGI-1 (10μM), Erlotinib (0.5μM) or a combination of both measured with Annexin-V and 7-AAD flow cytometry. Representative fluorescence data for each condition are displayed as bar graphs using the Flojo software. 100% values correspond to 50,000 cells and the data is represented as mean ± s.d., n = 3. P values were determined using two-tailed t-tests. *P < 0.01. b. EGFR phosphorylation (Y1173) and increased gel mobility after 48 h treatment with NGI-1 (10μM), Erlotinib (0.5μM) or a combination of both in PC9 and PC9-GR NSCLC cell lines. GADPH was used as a loading control. Representative data from two independent experiments are shown. c. Clonogenic survival of PC9 and PC9-GR NSCLC cell lines treated with vehicle, NGI-1 (10μM), Erlotinib (0.5μM), or a combination of both. d. Clonogenic survival from three independent experiments for PC9-GR cells shown as the mean ± s.d., n = 3. *P < 0.01.
The effects of NGI-1 on PC9 and PC9-GR cell proliferation can be accounted for by inhibition of EGFR glycosylation, which is invariant between the cell lines. However, a mechanism to account for NGI-1’s re-sensitization of PC9 cells with EGFR T790M to erlotinib was not readily explained. The data suggest that an erlotinib sensitive target is present in the PC9 GR cell lines. To further investigate this phenomenon, we performed deep sequencing of the PC9-GR cell lines in order to determine proportions of the wild type, exon 19 deletion mutation, and T790M EGFR alleles (Suppl. Table 2). Comparison of the number of reads for each mutation versus the number of wild type reads showed that the exon 19 deletion is found in a ratio of approximately 7:2. This ratio is consistent with amplification of the allele encoding the exon 19 deletion. In comparison, the ratio of the T790M methionine mutation to the wild type threonine sequence is approximately 1:7, showing that although it effectively confers resistance to erlotinib, this mutation is only present in a small fraction of the EGFR alleles. Sequencing data for both PC9-GR cell lines was similar suggesting the outgrowth of a common resistant clone that has an allele ratio of 2:5:1 for wild type, Exon19 deletion, and exon 19 deletion/T790M EGFR. This persistence of amplified EGFR alleles with the activating exon 19 deletion mutation thus provides the explanation for a combined effect of OST inhibition with erlotinib and suggests that NGI-1 disrupts protein from the allele with T790M while erlotinib targets the other five alleles with Exon 19 deletion and no T790M resistance mutation. Together the data suggests that inhibition of EGFR-T790M glycosylation reduces EGFR phosphorylation and survival signaling and re-sensitizes EGFR mutant NSCLC to EGFR TKIs.
OST inhibition re-sensitizes EGFR mutant NSCLC with MET amplification to EGFR TKIs.
We have previously demonstrated that lung cancer cells driven by RTKs other than EGFR, such as the FGFR1 dependent H1580, are sensitive to OST inhibition with NGI-1 (13). Because therapeutic resistance to EGFR TKIs can also be mediated by other co-expressed RTKs, we next examined the effects of NGI-1 on MET amplified HCC827-GR cells (19). We found that the proliferation of both parental and HCC827-GR cells were sensitive to NGI-1 treatment with a reduction of proliferation by 80% (p<.005; Fig. 4A). In HCC-827-GR cells a significant G1 arrest was again observed (75% vs 45% p<.05; Fig 4B) similar to effects in PC9-GR cells. NGI-1 treatment was not sufficient to induce apoptosis in HCC827-GR cells, however, NGI-1 was able to re-sensitize these cells to erlotinib with significant induction of apoptosis (10% vs 50% p<.005; Fig 4C). In agreement with these findings clonogenic survival was also decreased by more than 60% in HCC827-GR cells using both inhibitors (p<.005 Fig 4D). These results suggest that OST inhibitors may be able to overcome MET-mediated resistance to EGFR kinase inhibitors.
Figure 4: NGI-1 overcomes MET mediated therapeutic resistance.
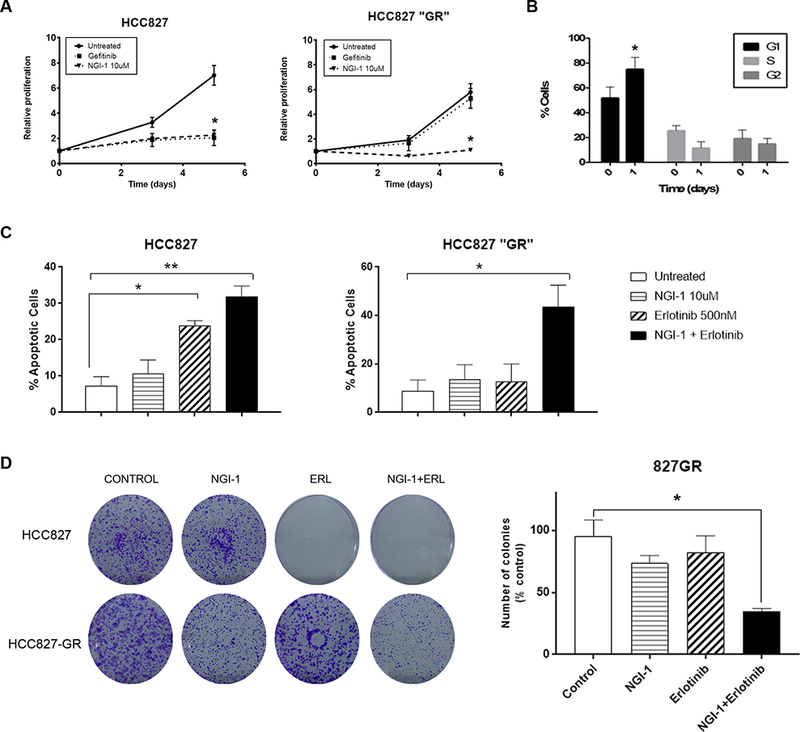
a. Fold proliferation measured by MTT over 5 days with NGI-1 (10μM) or Gefitinib (0.1nM) treatment in HCC827 and HCC827-GR cells. b. Flow cytometry and cell cycle distribution of HCC827-GR cells after NGI-1 treatment for 24 h. Data are represented as mean ± s.d., n = 3. *P < 0.01. c. Apoptosis susceptibility of HCC827 and HCC827-GR NSCLC cell lines following 48 h treatment with NGI-1 (10μM), Erlotinib (0.5μM) or a combination of both measured with Annexin-V and 7-AAD flow cytometry as in Fig. 1. **P < 0.01. d. Clonogenic survival of HCC827 and HCC827-GR NSCLC cell lines treated with vehicle, NGI-1 (10 μM), Erlotinib (0.5μM) or a combination of both. The results represent data from three independent experiments for each cell line. Data for three independent experiments are shown as the mean ± s.d. *P < 0.01.
NGI-1 blocks EGFR and Met co-localization.
To elucidate a mechanism for the combined effect of NGI-1 and erlotinib in HCC827-GR cells we evaluated changes in RTK phosphorylation (Fig. 5A). MET phosphorylation was sensitive to NGI-1 in parental cells but was not affected in MET amplified HCC827-GR cells. EGFR phosphorylation, however, was significantly reduced by both NGI-1 and erlotinib in HCC827-GR cells, and eliminated by a combination of both inhibitors. The complete inhibition of EGFR phosphorylation, similar to treatment of HCC827 parental cells with erlotinib alone, provides a mechanistic explanation for increased apoptosis and reduced clonogenic survival with combined erlotinib and NGI-1 treatment. However, because MET phosphorylation was not affected our observations could not provide a clear rationale for why EGFR phosphorylation was reduced. We therefore investigated EGFR and MET localization after NGI-1 treatment using a cell fractionation approach and found that MET was only detected at the plasma membrane in the setting of MET amplification, showing the potential for increased EGFR and MET interactions in the HCC827-GR cells (Fig. 5B). NGI-1 treatment reduced EGFR and MET in the plasma membrane fraction demonstrating an effect on trafficking for both receptors. Analysis of the non-membrane fraction showed a second NGI-1 effect with inhibition of pro-MET processing to the mature receptor and elimination of mature receptor phosphorylation (Fig. 5C). In the setting of MET amplification, however, pro-MET processing and phosphorylation was increased at baseline and not blocked by NGI-1. The production of mature MET in HCC827-GR in the presence of NGI-1 provides a reasonable explanation for why NGI-1 does not reduce global MET phosphorylation. In contrast, NGI-1 reduced EGFR phosphorylation in the non-membrane fraction in both HCC827 and HCC827-GR cells.
Figure 5: NGI-1 disrupts MET and EGFR interactions.
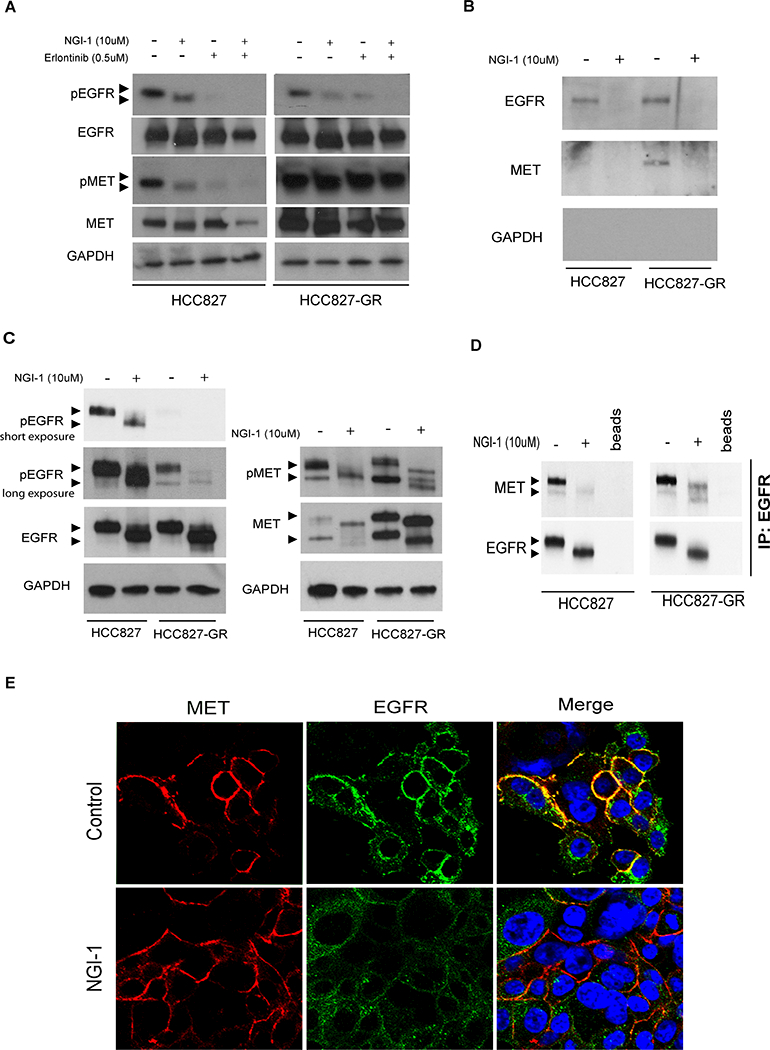
a. Western blots demonstrating changes in EGFR and MET molecular weight and phosphorylation after 24 h treatment with NGI-1 (10μM), Erlotinib (0.5μM), or a combination of both in HCC827 and HCC827-GR NSCLC cell lines. GAPDH was used as a loading control. b. Western blots of cell fractionation studies demonstrating changes of EGFR and MET localization in the plasma membrane fraction of HCC827 and HCC827-GR NSCLC cell lines. c. Western blots of cell fractionation studies demonstrating changes of EGFR and MET in the non-plasma membrane fraction of HCC827 and HCC827-GR NSCLC cell lines. Arrows for MET blots identify the uncleaved Pro-Met (upper arrow) and cleaved Met isoforms (lower arrow) d. Western blots of EGFR immunoprecipitation studies with or without NGI-1 treatment HCC827 and HCC827-GR NSCLC cell lines. e. Confocal microscopy and immunofluorescence of MET protein (red) and EGFR (green) with nuclear ToPro3 counterstain (cyan). Image overlays (yellow) demonstrate the presence or absence of c-localization. Results for each panel are representative of at least three independent experiments unless otherwise noted. Scale bars, 10 μm.
Based on these results we hypothesized that decreased EGFR phosphorylation in HCC827-GR could be caused an effect of NGI-1 on EGFR and MET receptor interactions. In support of this, immune precipitation experiments of EGFR showed reduced association with MET in both parental and GR cell lines (Fig. 5D). We therefore examined MET localization in HCC827-GR cells using confocal microscopy with a phospho-specific MET antibody (Fig. 5E). Phosphorylated MET was detected near the cell surface in both control and NGI-1 treated samples (red). However, EGFR (green) was detected near the cell surface in control HCC827-GR cells samples but not in NGI-1 treated samples, again demonstrating a dissociation of interactions between phosphorylated MET and EGFR. Together the data show that although NGI-1 does not reduce MET processing when it is over-expressed it does alter MET trafficking and also reduces EGFR and MET interactions. These results indicate that NGI-1 reduces MET dependent activation of EGFR phosphorylation by altering RTK localization in the cell.
OST inhibition re-sensitizes osimertinib resistant H1975 cells to osimertinib.
Third generation irreversible TKI inhibitors have been developed to successfully treat EGFR mutant NSCLC with T790M mutations. However, similar to first and second generation inhibitors, therapeutic resistance also develops. We therefore sought to generate an osimertinib resistant NSCLC model using the H1975 cell line, known to have both the L858R and T790M EGFR mutations. Deep sequencing of EGFR in this cell line revealed a ratio of ~4:1 for both L858R and T790M mutations compared to wild type EGFR (Suppl. Table 2), indicating a T790M mutation in cis with L858R (1:1 ratio) as well as amplification of this allele. The T790M cells were then exposed to increasing concentrations of osimertinib as described in materials and methods to produce the H1975-OR cell line. This resistant cell line was also sequenced and re-demonstrated the identical EGFR mutations as well as the absence of the C797S mutation, a known osimertinib resistance mutation. This data suggests that osimertinib resistance in H1975 cells is not mediated by genetic alterations of the EGFR.
We next compared the effects of osimertinib and NGI-1 on parental H1975 and H1975-OR cells (Fig. 6A). As expected the results demonstrated that H1975 cells were sensitive and H1975-OR cells were resistant to osimertinib. However, like the previously described EGFR TKI resistant cell lines (PC9-GR, HCC827-GR), NGI-1 reduced proliferation of both parental and H1975-OR cell lines by more than 70% (p<.001). Again, the block in proliferation was accompanied by a G1 arrest for both H1975 (40% vs 60% p<.05) and H1975-OR cells (45% vs 75% p<.05; Fig. 6B). NGI-1 blocked proliferation of H1975 expressing EGFR T790M cells, similar to effects in PC9-GR cells with EGFR T790M, and consistent with the dependency of EGFR on N-linked glycosylation for cell surface expression. Combination experiments of NGI-1 plus osimertinib in H1975 cells also demonstrated re-sensitization to the TKI as measured by a significant enhancement of apoptosis (20% vs 50%, p<.05, Fig. 6C) and a significant reduction of clonogenic survival by greater than 80% (p<.0001, Fig. 6D). The effects of NGI-1 on H1975-OR cells suggest that just as the PC9-GR and HCC827-GR cell lines achieved TKI resistance through parallel glycoprotein signaling, H1975-OR also circumvents EGFR signaling by stimulating signaling cascades that are glycoprotein dependent. Taken together this in vitro data suggests that NGI-1 treatment in combination with osimertinib can overcome therapeutic resistance to 3rd generation TKIs in EGFR-T790M expressing cells.
Figure 6: NGI-1 overcomes osimertinib resistance.
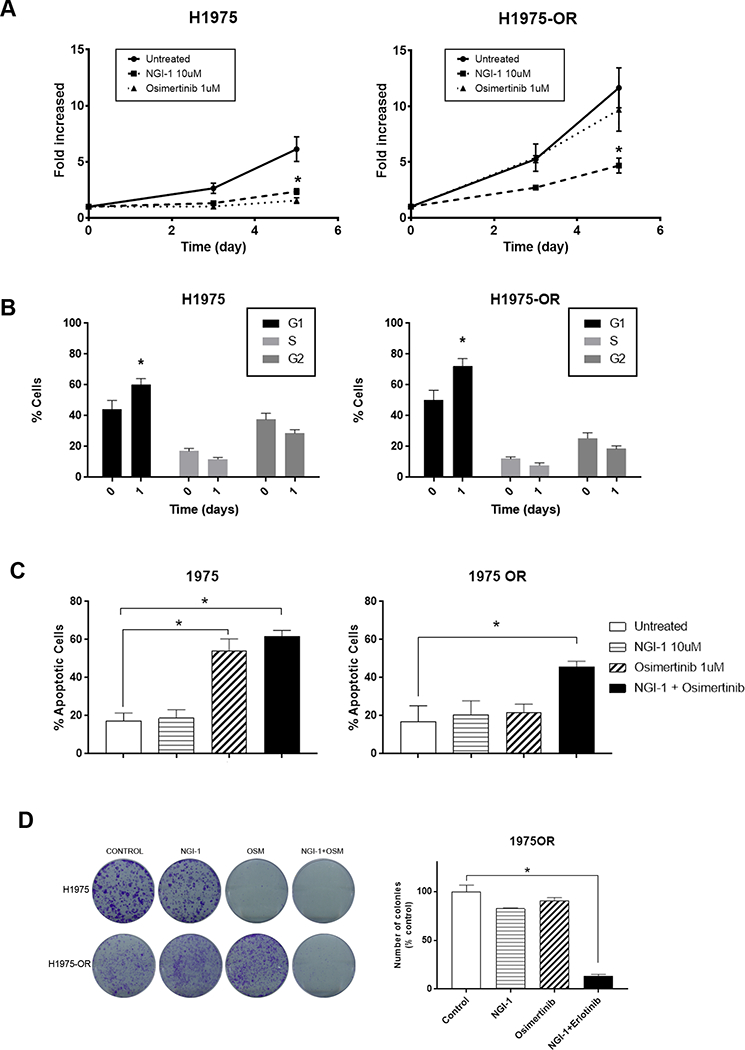
a. Fold proliferation measured by MTT over 5 days with NGI-1 (10μM), or Osimertinib (1μM) treatment in H1975 and H1975-OR cells. b. Flow cytometry and cell cycle distribution of H1975 and H1975-OR cells after NGI-1 treatment for 24 h. Data are represented as mean ± s.d., n =3. P values were determined using two-tailed t-tests. *P < 0.01. c. Apoptosis susceptibility of H1975 and H1975-OR NSCLC cell lines following treatment with NGI-1 (10μM), Osimertinib (1μM) or a combination of both for 48 h measured with Annexin-V and 7-AAD flow cytometry as in Fig. 1. The data represents the mean ± s.d., n = 3. *P < 0.01. d. Clonogenic survival of H1975 and H1975-OR NSCLC cell lines treated with vehicle, NGI-1 (10 μM), Osimertinib (1μM) or a combination of both. The bar graph shows data from three independent experiments and data represented as the mean ± s.d., n = 3. *P < 0.01.
During the course of these studies, work detailing H1975-OR cells was published by another group (20). This work suggested that H1975-OR cells have reduced EGFR activation and increased ERK and AKT signaling, two major downstream proteins of the EGFR pathway. This finding is similar to our H1975 resistant cell line (Suppl Fig. 2A), suggesting we have isolated a similar H1975 resistant clone. Notably Tang et. al. could not identify a discrete resistance mechanism and our RTK phospho-array screen did not show up-regulation of receptor activation (Suppl Fig. 2B), and thus we suggest that osimertinib therapeutic resistance could be regulated by individual or groups of glycoproteins in this cell line.
Formulation and imaging of NGI-1 nanoparticle activity in vivo:
NGI-1 is a drug-like compound with favorable physico-chemical properties; however, solubility remains a significant challenge for in vivo translation of this inhibitor. To test the hypothesis that NGI-1 could enhance TKI therapy in EGFR mutant and TKI resistant NSCLC, we developed a NGI-1 nanoparticle (NGI-NP) formulation by nanoprecipitation of a NGI-1 PEI complex. All nanoparticle preparations were confirmed by DLS and the average particle size was 120 ± 10nm. To assess the activity of the NGI-1 NPs we established a PC9 cell line with stable expression of the ER-LucT glycosylation reporter (18), a luciferase engineered to be expressed in the endoplasmic reticulum with N-linked glycosylation sites. Under normal cell conditions glycosylation of the luciferase blocks activity and luminescence. However, under conditions of decreased glycosylation luciferase activity is restored with a measurable increase in luciferase activity. We found that equivalent concentrations of NGI-1, either dissolved in DMSO or via NP, activated luminescence ~6 fold after 24 h in these PC9-ERLucT cells in vitro (Fig. 7A).
Figure 7: NGI-1 nanoparticle activity and tumor growth delay in vivo.
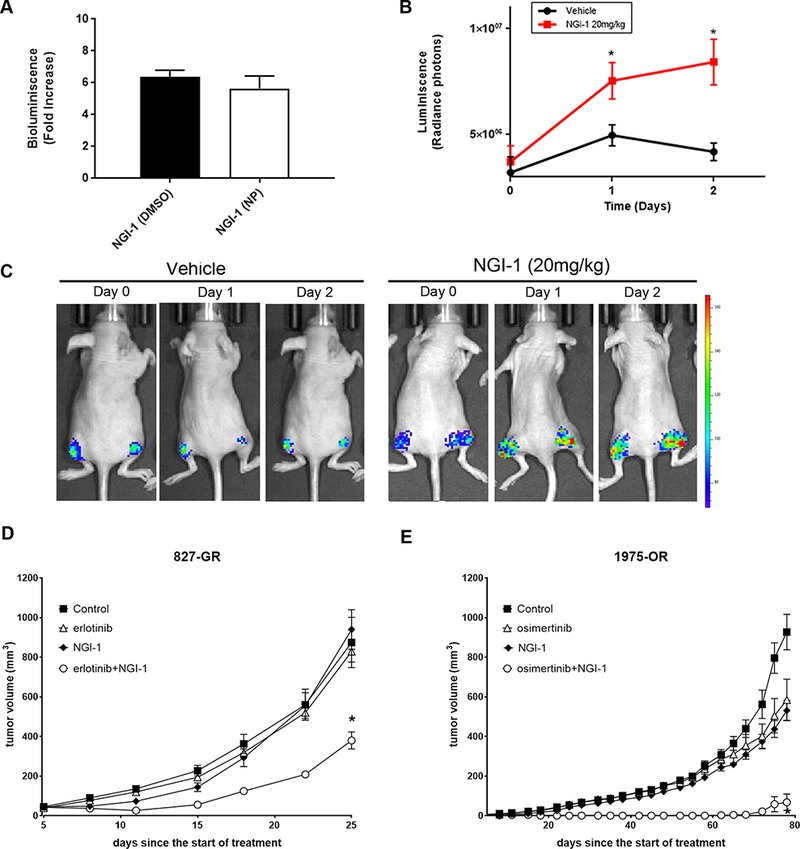
a. Fold increase of luciferase activity by NGI-1 dissolved in DMSO compared to the NGI-1 nanoparticle (NP) formulation. b. In vivo imaging over 48 h to detect inhibition of N-linked glycosylation in PC9 ER-LucT xenograft tumors following i.v. administration of blank (control) or NGI-1 NPs at a dose of 20 mg/Kg. c. Relative luminescence changes after i.v. blank NP (black) or NGI-1 NP (red) administration for the entire cohort. Data represents the average and SE for four animals (eight tumors) for each group. d., e. Tumor growth experiments in mice bearing HCC827-GR and H1975-OR xenografts, respectively. Tumors were randomized to four treatment groups: control; TKI, NGI-1, 20 mg/kg, and TKI+NGI-1. Mice were treated with a daily dose of TKI and every other day (3 times per week) with NGI-1 NPs. The data shows mean tumor volume for eight tumors in each group and error bars represent the SE. *P < 0.01.
To test whether intravenous delivery of NGI-1-NP could alter N-linked glycosylation in vivo, we performed serial bioluminescent imaging of PC9-ERLucT xenograft tumors. Luminescence was quantified to estimate the magnitude and duration of NLG inhibition after a single i.v. injection of either NGI-1-NP (20mg/Kg) or blank NPs. The results demonstrated a significant induction of bioluminescence at 24 hours in mice that received NGI-1 NPs (1.5 fold, p =.04; Fig. 7B), and 2.0 fold enhancement at 48 h (p =.01; Fig. 7C). This data confirmed the efficacy of the NGI-1 when administered as a NP formulation and suggested that daily treatment was not necessary to achieve sustained OST inhibition.
NGI-1 reduces growth of EGFR mutant, TKI-resistant NSCLC in vivo.
To evaluate the potential therapeutic effects of NGI-1 in vivo, we tested NGI-1 both alone and in combination with EGFR TKIs on tumor growth using both HCC827-GR and H1975-OR cell lines. These TKI resistance models were chosen because EGFR mutant, TKI resistant NSCLC that is not driven by an EGFR T790M allele remains the major clinical challenge in this patient population. For both HCC827-GR and H1975-OR xenografts, growing tumors were randomly assigned to receive NGI-NP or EGFR-TKI as either a monotherapy or in combination. HCC827-GR-xenografts were treated with control NPs, 25mg/kg erlotinib, 5mg/kg NGI-NP, or erlotinib + NGI-1-NP. We found that similar to in vitro findings, the combination of erlotinib and NGI-1 had the greatest impact on tumor growth. The mean tumor volume of erlotinib + NGI-NP at day 25 was 380 ± 121 mm3, significantly less than mice that received erlotinib (829 ± 149 mm3; p = .001) or NGI-1-NP (940 ± 283 mm3; p = .001) alone (Fig. 7D). Similar results favoring the combined treatment were observed in the H1975-OR xenografts. In this resistant line, the combination of osimertinib and NGI-NP caused a comparatively larger reduction in tumor growth. The mean tumor volume at day 78 for the osimertinib + NGI-1-NP group was 68 ± 117 mm3. In comparison tumor volumes for blank NPs (926 ± 253 mm3; p = .001), 5mg/kg osimertinib (584 ± 296 mm3; p = .001) and 5mg/kg NGI-1-NP (531 ± 142 mm3; p = .001) were significantly greater (Fig. 7E). In this xenograft model, the combined therapy group caused an ~50 day delay in tumor growth. For both in vivo experiments, there was no evidence of toxicity or significant weight loss in animals treated with NGI-NP. Taken together, these results indicate that the combination of an EGFR TKI with NGI-1 provides a potential therapeutic approach for NSCLC with resistance to EGFR inhibitors.
DISCUSSION:
The emergence of therapeutic resistance to EGFR TKIs prevents long term disease control and a cure for patients with EGFR mutant NSCLC. Tumor cell adaptation to EGFR targeted therapy is a familiar pattern that has been observed for 1st, 2nd , and 3rd generation inhibitors (21,22) and arises from both selection of existing tumor cell clones or from spontaneous acquisition of mechanisms for therapeutic resistance (23). Similar to the clinical observations from other disease sites employing TKIs, recurrent kinase domain mutations (ie T790M or C797S) that eliminate EGFR TKI action are a major form of therapeutic resistance that can be further targeted through structure based modeling and the ingenuity of medicinal chemistry. However, a significant portion of the EGFR TKI resistant population initially develops bypass mechanisms, which are not responsive to EGFR targeting, and ultimately even tumors with kinase domain resistance mutations will also undergo progression through an EGFR kinase insensitive mechanism. In this work, we demonstrate that a novel small molecule inhibitor of the OST can overcome EGFR TKI therapeutic resistance, both in settings of EGFR dependent and independent resistance, and identify inhibition of N-linked glycosylation as a strategy for treating EGFR mutant NSCLC.
The EGFR is a highly glycosylated protein with 11 canonical N-linked glycosylation sequons (NXS/T) and one non-canonical NXC site (24). Either complete or partial inhibition of this post-translational modification alters the receptor’s stability, localization, and function (13,25,26) suggesting that blocking N-linked glycosylation is an approach for reducing EGFR driven signaling. Recently we described the first OST inhibitor which targets the catalytic subunit and causes partial inhibition of N-linked glycosylation, an effect that is consistent with an allosteric mechanism of action. Herein we demonstrate that kinase domain mutant EGFR, with or without the T790M mutation, is sensitive to partial inhibition of N-linked glycosylation by pharmacologic inhibition of the OST. The data confirms our hypothesis that glycosylation of the extracellular domain is a key regulator of receptor function that remains crucial in the setting of kinase domain resistance mutations. Inhibition of glycosylation thus has advantages of providing a kinase domain independent mechanism for EGFR inhibition, which couples favorably with kinase domain targeting, as well as the potential for targeting other RTKs and glycoproteins that contribute to tumor cell survival signaling.
Investigation into the synergistic effects of NGI-1 and EGFR TKIs also provided new insights into the cellular mechanism of EGFR TKI resistance. In the PC9-GR cell line the combined effect of NGI-1 plus erlotinib led to the observation that a minority of alleles harboring the T790M mutation can drive TKI resistance. This data has implications for TKI therapy where the affinity of TKIs may vary for Exon 19 deletions, L858R point mutations, and T790M resistance mutations. For example, the acquisition of a C797S mutation on a single allele could drive 3rd generation TKI resistance and be sensitive to 4th generation EGFR TKIs, yet EGFR alleles with only an exon 19 deletion would be insensitive to this new class of allosteric inhibitors (27). Our results investigating TKI resistance in the setting of MET amplification also provided novel insights into bypass mechanism of resistance. Surprisingly, although reduction of MET glycosylation reduces MET phosphorylation in the HCC827 cell line, a significant effect in the MET amplified HCC827-GR cell line was not seen. In this model of TKI resistance we found that interference of EGFR and MET interactions was associated with reduced EGFR phosphorylation. This suggests changes in RTK trafficking and localization may also be important for establishing a TKI resistant phenotype. The inability of NGI-1 to block MET activation or processing of the pro-receptor to the mature form in the setting of MET amplification is itself of interest as EGFR overexpression and amplification does not cause insensitivity to NGI-1. This difference between MET and EGFGR reflects distinct requirements for receptor processing and maturation in the secretory pathway and indicates that EGFR may be more sensitive to inhibition of N-linked glycosylation by NGI-1 than other RTKs.
EGFR TKI therapeutic resistance (including the EGFR T790M mutation) is largely characterized by bypass mechanisms initiated by plasma membrane receptors. Although downstream mutations of the Ras/MAPK pathway have been described (28,29), whether tumor cells become absolutely dependent on these mutations for proliferation and survival remains unclear. On the genetic level, in addition to MET, mutations or amplification of ErbB2 have also been observed after EGFR TKI resistance develops (30). AXL and IGF-1R are additional glycoproteins implicated in TKI resistance through initiation of parallel and compensatory survival signaling (31,32), and it has been demonstrated that RTK stimulation by growth factors rescues EGFR dependent NSCLC cell lines from TKI therapy (33,34). The analysis of clinical and cell culture samples has also implicated epithelial to mesenchymal transition as a cellular maneuver to evade drug toxicity. CRIPTO1, NRP2, and TGFB have all been identified as factors that induce EMT and cause EGFR TKI resistance (35–37). Even more recently ß-adrenergic signaling and Il-6 signaling have been demonstrated to mediate therapeutic resistance in EGFR mutant NSCLC (38). Together these mechanisms of resistance can be practically classified with respect to the proteins involved, and thus the majority of EGFR TKI resistance can be ascribed to preservation of and dependence on cell surface glycoprotein signaling. In this context, the discovery of NGI-1 and demonstration of therapeutic effect is noteworthy as it suggests that OST inhibition may be effective against diverse resistance mechanisms.
On the cellular level, EGFR TKI resistance manifests itself with cell cycle progression, proliferation, and tumor growth. Although EGFR inhibition typically induces a cell cycle arrest in the G1 phase followed by apoptosis (39–41), bypass mechanisms either uncouple or replace EGFR dependent progression through cell cycle checkpoints. The importance of dysregulated cell cycle check points as a mechanism of EGFR TKI therapeutic resistance has recently been emphasized by a large genomic analysis of serial patient samples where co-occurring mutations or copy number variations of cell cycle regulatory genes (eg CDK4 and CDK6), were a significant predictor of progression free survival (12). The ability of NGI-1 to induce G1 arrest in EGFR TKI resistant cells is therefore likely to be a fundamental consequence of the inhibitor, and when coupled with EGFR kinase inhibition, leads to restoration of an apoptotic response and cell death.
Targeting the OST and disrupting N-linked glycosylation in eukaryotic cells is anticipated to affect multiple downstream glycoprotein targets. Until now, and with the discovery a partial inhibitor of N-linked glycosylation, blockade of this post-translational modification has not been investigated due to anticipated toxicity. Indeed tunicamycin, which blocks the first committed step in glycan precursor biosynthesis and all N-linked glycosylation has significant toxicity both in vitro and in vivo (18). NGI-1 on the other hand was identified in a cell based phenotypic HTS that inherently selected against small molecules which induce cell death. Herein we demonstrate a relatively selective effect on proliferation for EGFR and ErbB family receptor driven lung cancer through the profiling of 94 lung cancer cell lines. Specificity for RTK (and other glycoprotein) driven proliferation is also consistent with observations of no significant toxicity in non-transformed cell lines, primary human dermal fibroblasts, and neural progenitor cells (13,42,43). In this work, we overcame the in vivo pharmacokinetic challenges of NGI-1 using a novel nanoparticle formulation to effectively deliver the drug to the tumor by intravenous injection. The re-sensitization of TKI resistant HCC827-GR and H1975-OR to EGFR TKIs without significant side effects thus shows for the first time that targeting N-linked glycosylation is feasible in vivo. However, because multiple weekly i.v. infusions are not likely to be practical in the clinical setting, future directions include optimization of nanoparticle properties to produce a sustained release over time as well as medicinal chemistry approaches to identify improved NGI-1 analogs for in vivo use.
In summary, we have identified a small molecule inhibitor of the OST that partially inhibits N-linked glycosylation and re-sensitizes EGFR mutant NSCLC to EGFR TKIs. Targeting N-linked glycosylation has the advantage of disrupting the dependence of NSCLC tumor cells on cell surface receptor signaling and couples well with EGFR TKIs to produce blockade of both primary and redundant mechanism of cell growth. Our demonstration of significant tumor growth delay with little toxicity provides a strong rationale to further develop this targeted approach for reducing oncogenic signaling in NSCLC.
Supplementary Material
{kind=link}
{kind=link}
Significance:
EGFR mutant NSCLC is incurable despite the marked sensitivity of these tumors to EGFR TKIs. Our findings identify N-linked glycosylation, a post-translational modification common to EGFR and other oncogenic signaling proteins, as an effective therapeutic target that enhances tumor responses for EGFR mutant NSCLC.
Acknowledgements:
This work is supported by US National Institutes of Health (NIH) R01CA172391, a Research Scholar Grant from the American Cancer Society, and in part by a Developmental Research Program Grant from the Yale SPORE in Lung Cancer (J.N. Contessa). Additional support is from NIH RO1 CA149128 (W.M. Saltzman), NIH F30 CA206386 (A.Quijano), and NIH RO1 CA197486 (A.A. Patel). Cell line screening was supported by the Wellcome Trust (102696) (C.H. Benes).
Footnotes
With respect to potential conflicts of interest; Dr. Saltzman, Dr. Quijano, and Dr. Contessa are listed as inventors in a preliminary patent application for the nanoparticle formulation of NGI-1 that is described in this work.
REFERENCES:
- 1.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004;305(5687):1163–7. [DOI] [PubMed] [Google Scholar]
- 2.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 2004;101(36):13306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med 2004;350(21):2129–39. [DOI] [PubMed] [Google Scholar]
- 4.Sequist LV, Martins RG, Spigel D, Grunberg SM, Spira A, Janne PA, et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol 2008;26(15):2442–9. [DOI] [PubMed] [Google Scholar]
- 5.Mok TS, Wu YL, Ahn MJ, Garassino MC, Kim HR, Ramalingam SS, et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N Engl J Med 2017;376(7):629–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A 2005;102(21):7665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005;2(3):e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat Med 2015;21(6):560–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 2011;3(75):75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res 2013;19(8):2240–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun 2015;6:6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blakely CM, Watkins TBK, Wu W, Gini B, Chabon JJ, McCoach CE, et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat Genet 2017;49(12):1693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopez-Sambrooks C, Shrimal S, Khodier C, Flaherty DP, Rinis N, Charest JC, et al. Oligosaccharyltransferase inhibition induces senescence in RTK-driven tumor cells. Nat Chem Biol 2016;12(12):1023–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012;483(7391):570–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Narayan A, Carriero NJ, Gettinger SN, Kluytenaar J, Kozak KR, Yock TI, et al. Ultrasensitive measurement of hotspot mutations in tumor DNA in blood using error-suppressed multiplexed deep sequencing. Cancer Res 2012;72(14):3492–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldberg SB, Narayan A, Kole AJ, Decker RH, Teysir J, Carriero NJ, et al. Early Assessment of Lung Cancer Immunotherapy Response via Circulating Tumor DNA. Clin Cancer Res 2018;24(8):1872–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.King AR, Corso CD, Chen EM, Song E, Bongiorni P, Chen Z, et al. Local DNA Repair Inhibition for Sustained Radiosensitization of High-Grade Gliomas. Mol Cancer Ther 2017;16(8):1456–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Contessa JN, Bhojani MS, Freeze HH, Ross BD, Rehemtulla A, Lawrence TS. Molecular imaging of N-linked glycosylation suggests glycan biosynthesis is a novel target for cancer therapy. Clin Cancer Res 2010;16(12):3205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316(5827):1039–43. [DOI] [PubMed] [Google Scholar]
- 20.Tang ZH, Jiang XM, Guo X, Fong CM, Chen X, Lu JJ. Characterization of osimertinib (AZD9291)-resistant non-small cell lung cancer NCI-H1975/OSIR cell line. Oncotarget 2016;7(49):81598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JK, Shin JY, Kim S, Lee S, Park C, Kim JY, et al. Primary resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) in patients with non-small-cell lung cancer harboring TKI-sensitive EGFR mutations: an exploratory study. Ann Oncol 2013;24(8):2080–7. [DOI] [PubMed] [Google Scholar]
- 22.Chong CR, Janne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med 2013;19(11):1389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med 2016;22(3):262–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ullrich A, Coussens L, Hayflick JS, Dull TJ, Gray A, Tam AW, et al. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 1984;309(5967):418–25. [DOI] [PubMed] [Google Scholar]
- 25.Contessa JN, Bhojani MS, Freeze HH, Rehemtulla A, Lawrence TS. Inhibition of N-linked glycosylation disrupts receptor tyrosine kinase signaling in tumor cells. Cancer Res 2008;68(10):3803–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsuda T, Ikeda Y, Taniguchi N. The Asn-420-linked sugar chain in human epidermal growth factor receptor suppresses ligand-independent spontaneous oligomerization. Possible role of a specific sugar chain in controllable receptor activation. J Biol Chem 2000;275(29):21988–94. [DOI] [PubMed] [Google Scholar]
- 27.Jia Y, Yun CH, Park E, Ercan D, Manuia M, Juarez J, et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016;534(7605):129–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Mello RA, Marques DS, Medeiros R, Araujo AM. Epidermal growth factor receptor and K-Ras in non-small cell lung cancer-molecular pathways involved and targeted therapies. World J Clin Oncol 2011;2(11):367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2005;2(1):e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Landi L, Cappuzzo F. HER2 and lung cancer. Expert Rev Anticancer Ther 2013;13(10):1219–28. [DOI] [PubMed] [Google Scholar]
- 31.Vazquez-Martin A, Cufi S, Oliveras-Ferraros C, Torres-Garcia VZ, Corominas-Faja B, Cuyas E, et al. IGF-1R/epithelial-to-mesenchymal transition (EMT) crosstalk suppresses the erlotinib-sensitizing effect of EGFR exon 19 deletion mutations. Sci Rep 2013;3:2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 2013;19(1):279–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshida T, Zhang G, Smith MA, Lopez AS, Bai Y, Li J, et al. Tyrosine phosphoproteomics identifies both codrivers and cotargeting strategies for T790M-related EGFR-TKI resistance in non-small cell lung cancer. Clin Cancer Res 2014;20(15):4059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012;487(7408):505–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park KS, Raffeld M, Moon YW, Xi L, Bianco C, Pham T, et al. CRIPTO1 expression in EGFR-mutant NSCLC elicits intrinsic EGFR-inhibitor resistance. J Clin Invest 2014;124(7):3003–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gemmill RM, Nasarre P, Nair-Menon J, Cappuzzo F, Landi L, D’Incecco A, et al. The neuropilin 2 isoform NRP2b uniquely supports TGFbeta-mediated progression in lung cancer. Sci Signal 2017;10(462). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yao Z, Fenoglio S, Gao DC, Camiolo M, Stiles B, Lindsted T, et al. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc Natl Acad Sci U S A 2010;107(35):15535–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nilsson MB, Sun H, Diao L, Tong P, Liu D, Li L, et al. Stress hormones promote EGFR inhibitor resistance in NSCLC: Implications for combinations with beta-blockers. Sci Transl Med 2017;9(415). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song L, Morris M, Bagui T, Lee FY, Jove R, Haura EB. Dasatinib (BMS-354825) selectively induces apoptosis in lung cancer cells dependent on epidermal growth factor receptor signaling for survival. Cancer Res 2006;66(11):5542–8. [DOI] [PubMed] [Google Scholar]
- 40.Helfrich BA, Raben D, Varella-Garcia M, Gustafson D, Chan DC, Bemis L, et al. Antitumor activity of the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor gefitinib (ZD1839, Iressa) in non-small cell lung cancer cell lines correlates with gene copy number and EGFR mutations but not EGFR protein levels. Clin Cancer Res 2006;12(23):7117–25. [DOI] [PubMed] [Google Scholar]
- 41.de La Motte Rouge T, Galluzzi L, Olaussen KA, Zermati Y, Tasdemir E, Robert T, et al. A novel epidermal growth factor receptor inhibitor promotes apoptosis in non-small cell lung cancer cells resistant to erlotinib. Cancer Res 2007;67(13):6253–62. [DOI] [PubMed] [Google Scholar]
- 42.Hafirassou ML, Meertens L, Umana-Diaz C, Labeau A, Dejarnac O, Bonnet-Madin L, et al. A Global Interactome Map of the Dengue Virus NS1 Identifies Virus Restriction and Dependency Host Factors. Cell Rep 2017;21(13):3900–13. [DOI] [PubMed] [Google Scholar]
- 43.Puschnik AS, Marceau CD, Ooi YS, Majzoub K, Rinis N, Contessa JN, et al. A Small-Molecule Oligosaccharyltransferase Inhibitor with Pan-flaviviral Activity. Cell Rep 2017;21(11):3032–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.