

Abstract
Recent thymic emigrants (RTEs) are peripheral T cells that have most recently completed selection and thymic egress and comprise a population that is phenotypically and functionally distinct from its more mature counterpart. Antigen-activated RTEs are less potent effectors than are activated mature T cells, due in part to reduced aerobic glycolysis (correctable by exogenous IL-2), which in turn impacts IFN-γ production. Mitochondria serve as nodal regulators of cell function but their contribution to the unique biology of RTEs is unknown. Here, we show that activated mouse RTEs have impaired oxidative phosphorylation, even in the presence of exogenous IL-2. This altered respiratory phenotype is the result of decreased CD28 signaling, reduced glutaminase induction, and diminished mitochondrial mass in RTEs relative to mature T cells. These results suggest an uncoupling, whereby IL-2 tunes the rate of RTE glycolytic metabolism while the unique profile of RTE mitochondrial metabolism is ‘hard wired’.
Introduction
T cells were once thought to be fully mature following thymic selection and egress (1). This notion has been progressively revised as improved models have allowed for the study of intrinsic peripheral T cell age (reviewed in 2). The Rag2p-GFP transgenic (Tg) mouse (3) has been used to isolate live, untouched RTEs, facilitating previously unfeasible experiments (4). Using this model, it has become clear that for the first 3 weeks in the lymphoid periphery, T cells occupy a distinct phenotypic and functional niche. Relative to mature T cells, CD8+ RTEs are poor proliferators and producers of effector cytokines (IFN-γ, TNF-α, and IL-2) following activation in the absence of inflammation (4–7). Critically, Ag exposure during the period when T cells occupy the RTE compartment imparts lasting functional differences that distinguish RTE- from mature T cell-derived memory cells (8). Importantly, the unique physiology of RTEs is not just a feature of the mouse but also of humans (9–12).
RTE maturation is independent of many of the signals required for mature but naïve (MN) T cell homeostasis, including IL-7, self-MHC molecules, and CD80/CD86 (13, 14). However, thymic egress and access to secondary lymphoid organs are strictly required for RTE maturation (15). Recently, the lymphocyte migration factor sphingosine 1-phosphate, which is present at high concentrations in the blood, has been shown to regulate MN T cell homeostasis by maintaining mitochondrial content (16). These data underscore the possibility that RTE maturation has a metabolic component.
MN T cells undergo profound metabolic reprogramming following activation (17) that is associated with an increase in the rate of aerobic glycolysis (18). However, even in the presence of this large glycolytic flux, mitochondrial metabolism remains critically important both for T cell activation and effector function (19, 20). The requirement for ongoing respiration in activated T cells extends beyond the requisite ATP generation (reviewed in 21). In CD4+ T cells, production of mitochondrial reactive oxygen species (mROS) is required for NFAT-mediated IL-2 production (20). Additionally, mitochondria-derived acetyl-CoA is critical for histone acetylation at the ifng locus in the context of ongoing aerobic glycolysis (22). Mitochondrial metabolism also regulates memory T cell formation (23–25). Specifically, mitochondrial morphology distinguishes CD8+ effector from memory T cells, the former having round mitochondria with loose cristae and the latter, elongated mitochondria with tight cristae, in which key membrane components are brought into close spatial proximity, thereby enhancing the efficacy of electron transport (26–28). Thus, mitochondria represent a central hub through which T cell fate decisions are orchestrated.
In addition to signals triggered through the TCR, costimulatory signals delivered through CD28 play a well established role in regulating the induction of glycolytic metabolism in activated T cells (29) and an emerging role in modulating mitochondrial function (30). CD28 signals are required for the establishment of tight cristae and carnitine palmitoyltransferase 1a (Cpt1a)-mediated spare respiratory capacity (SRC). 4-1BB signaling has also been implicated in the regulation of mitochondrial metabolism (31), being critical for optimal oxidative phosphorylation (OxPhos) via peroxisome proliferator-activator receptor gamma coactivator 1-α (PGC-1α)-mediated accumulation of mitochondrial mass.
We recently found that activated CD8+ RTEs have a reduced rate of glycolysis relative to mature T cells, a defect that underlies their diminished IFN-γ production (32). Activation in the presence of exogenous IL-2 corrects this deficit. As mitochondria are central drivers of effector function, we now assess mitochondrial metabolism in RTEs to better understand their unique biology. Here, we show that activated CD8+ RTEs have reduced OxPhos and SRC relative to their more mature counterparts. RTEs fail to increase glutaminase (GLS) expression and mitochondrial mass following activation, defects that persist even in the presence of exogenous IL-2. These alterations are driven by reduced CD28 signaling in RTEs. Collectively, these results suggest that, while glycolytic metabolism in activated CD8+ RTEs is subject to modulation by IL-2, mitochondrial metabolism appears to be ‘hard wired’ differently in RTEs than in mature T cells. These results have profound implications for T cell responses that are dominated by RTEs, such as those in neonates and in adults recovering from therapeutic or accidental lymphodepletion.
Materials and Methods
Mice
C57BL/6J, B6.129P2-Tcrbtm1MomTcrdtm1Mom/J (TCRβδ−/−), B6.129S4-Cd80tm1ShrCD86tm2Shr/J (CD80/86−/−), and OT-I [B6-Tg (TcraTcrb) 1100Mjb/J] mice expressing a transgene-encoded TCR that recognizes the chicken ovalbumin-derived peptide SIINFEKL in the context of H-2Kb were purchased from The Jackson Laboratory and maintained in-house. CD80/86−/− mice were co-housed with wild type (WT) controls for >14 days prior to experimentation. Rag2p-GFP Tg mice (3) were backcrossed onto the C57BL/6J background in our laboratory for at least 12 generations. Mice of both sexes were used at 8 - 10 weeks of age. All mice were housed under specific pathogen-free conditions and used in accordance with the University of Washington Institutional Animal Care and Use Committee guidelines.
Cell isolation and T cell stimulations
RTE and MN OT-I Tg T cells were isolated from Rag2p-GFP Tg mice (7) and stimulated (32) as previously described. In brief, sort-purified CD45.1 RTEs and MN T cells were plated at 1 × 105 per well + 5 × 105 irradiated CD45.2 TCRβδ−/− or T cell depleted CD80/86−/− splenocytes as APCs in complete RPMI 1640 (Life Technologies) containing 10% heat-inactivated FBS. Cells were stimulated with 10 nM SIINFEKL (Genmed Synthesis) ± 20 U/mL rIL-2 (Cetus) and maintained for 3 days at 37 °C in 7% CO2.
Abs, cell surface staining, and flow cytometry
Fc receptors were blocked using anti-16/32 (2.4G2; BD Biosciences) and cells were stained with fluorescent Abs to detect cell surface expression of 4-1BB (17B5), CD4 (RM4-5), CD11b (M1/70), CD25 (PC61.5), CD44 (IM7), CD45.1 (A20), B220 (RA3-6B2), ICOS (7E.17G9) NK1.1 (PK136), and Ter119 (Ly-76), all from eBioscience, in addition to CD8α (53-6.7), CD28 (37.51), and CD62L (MEL-14) from BD Biosciences. Data were acquired using a FACSCanto II (BD Biosciences) and analyzed with FlowJo software (TreeStar).
Intracellular staining
Surface stained cells were fixed and permeabilized using the Cytofix/Cytoperm Kit (BD Biosciences) and stained with Abs against GLS (polyclonal), ATPB (3D3), Cpt1a (EPR12740(B)), and voltage-dependent anion channel 1 (VDAC1; 20B12AF2), all from Abcam, and PGC-1α (D-5; Santa Cruz Biotechnology), p-S6 S240/S244 (D68F8), p-NF-κB (931H1) and p-p38 (D3F9), all from Cell Signaling Technology. For IL-2 staining, cells were plated and activated as above for 6 h; GolgiPlug (BD Biosciences) was added after the first hour. Cells were stained with a labeled Ab against IL-2 (JES6-5H4; eBioscience).
Metabolic bioassays
Oxygen consumption rate (OCR) measurements (in pmol/min) were generated using an XF24 bioanalyzer (Seahorse Bioscience). T cells were plated (1 × 106/well) into a XF24 microplate coated with Cel-Tak (Sigma). Cells were allowed to equilibrate in non-buffered medium (Seahorse Bioscience) containing 10 mM glucose for 30 min in a non-CO2 incubator. Four baseline OCR readings were acquired before sequential injection of 2.5 μM Oligomycin A (Oligo), 500 nM Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), and 2 μM Rotenone + 2 μM Antimycin A (Rot/AA), all from Sigma. To determine the contribution of glutamine to OCR, basal OCR was measured in cells plated in non-buffered medium containing 10 mM glucose and glutamine at either 0.2 mM (- Gln) or 2 mM (+ Gln).
Widefield microscopy
CD8+ RTEs and MN T cells were stimulated as above and mitochondria and nuclear DNA were visualized using Mitotracker Red CMXRos (Thermo Fisher) and NucBlue Live ReadyProbes Reagent (Molecular Probes), respectively. Cells were allowed to settle onto poly-D-lysine (Sigma) coated glass-bottom petri dishes (MatTek) for 30 minutes before imaging. A Z-series with a 0.3 μm step size was collected at 37°C and 7% CO2 using a Ti-E widefield microscope with a 100X NA 1.45 oil objective (Nikon), a solid-state light source (Spectra X, Lumencor), and a sCMOS camera (Zyla 5.5 Megapixel). Mitochondrial morphology was scored in a blinded fashion as “fragmented”: spherical mitochondria < 1.5 μm in diameter; “intermediate”: oblong and < 2.5 μm in length; and “reticular”: elongated and highly connected.
Electron microscopy
RTEs and mature T cells were activated as above and 70 nm sections imaged using a JEM-1230 transmission electron microscope (Joel USA) in the University of Washington Vision Science Center. Image analysis was performed blinded on 35 – 40 measurements of maximum crista width per condition, across 20 image fields.
Statistics
P-values were calculated in Prism (Graphpad) using a paired two-tailed Student t-test except where indicated. P-values < 0.05 were considered significant.
Results and Discussion
Defective glutamine-fueled OxPhos in activated RTEs
Our previous work indicated that activated RTEs display glycolytic defects that can be corrected by exogenous IL-2 (32). However, mitochondrial and glycolytic metabolism play non-redundant roles in T cell activation and effector function (19). Indeed, exogenous IL-2 failed to restore defective IL-2 production by activated RTEs (Fig. 1A). As IL-2 production requires mitochondrial metabolism (20), we assessed the rate of OxPhos, which generates ATP and is coupled to NAD+/NADH homeostasis and the generation of mROS in activated T cells (21). We found that RTEs and MN T cells measured directly ex vivo displayed equivalent OCRs. However, the robust, activation-dependent induction of OCR seen in mature T cells was significantly attenuated in RTEs (Fig. 1B, left). Additionally, activated RTEs displayed less SRC (a measure of mitochondrial reserve). Importantly, exogenous IL-2 had no effect on RTE basal OCR or SRC (Fig. 1B), despite restoring maximum glycolytic rate (32). The TCA-cycle is critical for generating NADH and FADH2. In activated CD8+ T cells, glutaminolysis and fatty acid oxidation (FAO) are critical for replenishing TCA-cycle intermediates depleted by cataplurotic processes (33). As RTEs display reduced OCR and SRC, we next assessed the expression of rate-determining enzymes for glutaminolysis (GLS) and FAO (Cpt1a). To this end, we assessed enzyme expression in RTEs and mature T cells left resting or activated ± exogenous IL-2. While resting RTEs and mature T cells express similar levels of GLS (Fig. 1C) and Cpt1a (Supplemental Fig. 1A), enzyme induction is significantly diminished in activated RTEs. Critically, IL-2 fails to restore GLS expression in RTEs (Fig. 1C). To confirm that altered glutamine flux drives the differences in OCR seen in RTEs and mature T cells, OCR was measured in cells activated in medium containing glutamine and then plated in the presence of low (0.2 mM; - Gln) or high (2 mM; + Gln) glutamine. Activated mature T cells had a significantly reduced OCR in the presence of low glutamine (Fig. 1D). By contrast, glutamine access did not impact the OCR of activated RTEs. These results suggest that the defective OCR observed in RTEs primed in the presence of IL-2 is associated with a reduced rate of glutaminolysis. Interestingly, GLS is a Myc target gene (17) and IL-2 promotes Myc expression (34) and restores Myc protein levels in RTEs (32). This discrepancy suggests that the GLS locus may be in an altered epigenetic state in RTEs, as has been observed for certain cytokine loci (35). Alternatively, Myc may regulate a subtly different set of genes in RTEs and mature T cells.
Figure 1. Activated RTEs display defective OxPhos and GLS induction in the presence of exogenous IL-2.
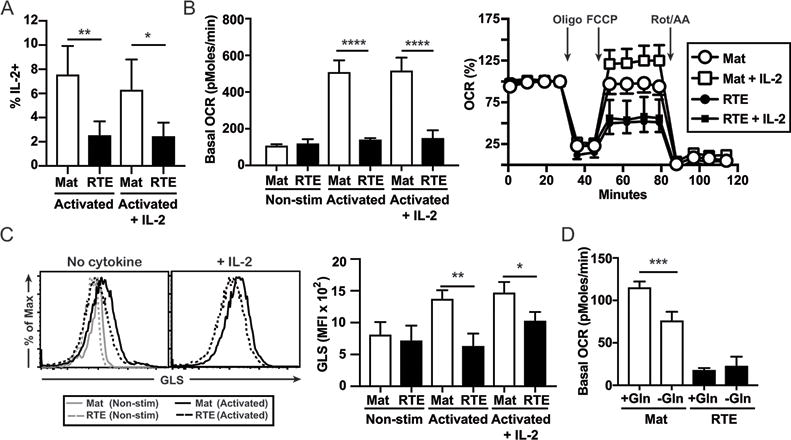
For each experiment, sort-purified CD8+ RTEs and MN T cells (Mat) were left unstimulated (non-stim) or stimulated in vitro for 3 days with peptide Ag and APCs ± IL-2. (A) The percent of IL-2+ cells after restimulation was determined. (B) Basal (left) and maximal (right) OCR were determined on cells freshly isolated (non-stim) or stimulated with Ag ± IL-2; arrows denote injection of the indicated compounds. A single timepoint of these data was previously published as Supplemental Fig. 1C in reference 32. Intracellular expression of GLS (C) was determined on cells freshly isolated (non-stim) or stimulated with Ag ± IL-2; representative histograms are shown on the left. The bar graphs in the right panels depict the average mean fluorescence intensity (MFI) ± SD from >3 independent experiments. (D) Basal OCR was determined on activated RTEs and mature T cells plated in the presence of either 0.2 mM (- Gln) or 2 mM (+ Gln) glutamine. *p<0.05, **p<0.01, *** p<0.001, ****p<0.0001 by unpaired (B,D) or paired (A,C) Student t-test.
Altered mitochondrial mass and morphology in activated RTEs
Respiratory capacity in activated CD8+ T cells is regulated by mitochondrial content (23) and morphology (26, 30). To determine mitochondrial mass, RTEs and mature T cells were left resting or activated ± exogenous IL-2 and the level of VDAC1 determined. Resting CD8+ RTEs and mature T cells had equivalent VDAC1 levels, similar to recent observations in resting CD4+ RTEs (36). However, upon activation, RTEs expressed significantly less VDAC1 than their mature T cell counterparts (Fig. 2A). This defect was also reflected in the expression of the ATPB subunit of Complex V (Fig. 2B), Tom20 and staining with the mitochondrial membrane potential-sensitive dye Mitotracker Red (data not shown). Critically, activation of RTEs in the presence of IL-2 failed to correct this defect in mitochondrial mass (Fig. 2A,B). By mitotracker staining and live-cell microscopy, the majority of activated mature T cells contained mitochondria that were fragmented, with few cells having mitochondria of intermediate length or reticular appearance (Fig. 2C). In contrast, fewer activated RTEs had fragmented mitochondria and a large proportion contained mitochondria of intermediate length or reticular appearance. Crista structure is generally coupled to mitochondrial morphology (26), and both resting RTEs and mature T cells had tight cristae (Fig. 2D). Upon activation, mature T cells displayed greater crista width than RTEs and this difference was unaffected by activation in the presence of IL-2. Thus, activated RTEs have more elongated mitochondria with tighter crista structure (parameters associated with greater respiratory capacity); however, this apparent respiratory advantage is offset by a profound reduction in mitochondrial mass.
Figure 2. RTE mitochondria have reduced mass and altered morphology.
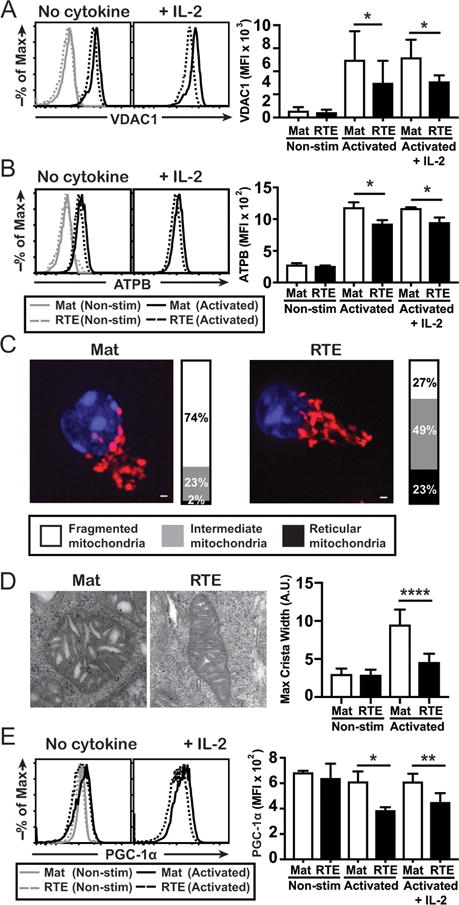
Sort-purified CD8+ RTEs and MN T cells were freshly isolated (non-stim) or stimulated with Ag ± IL-2 in vitro for 3 days and the intracellular level of VDAC1 (A) and ATPB (B) determined; representative histograms are shown on the left. The bar graphs in the right panels depict the average MFI ± SD from 4 independent experiments. CD8+ RTEs and MN T cells were stimulated with Ag for 3 days and imaged by widefield microscopy (C). Mitochondria (red) and nuclear DNA (blue) are shown in representative maximum intensity projections (scale bar = 1 μm). Bar graphs represent the fraction of cells containing mitochondria of the indicated morphology. RTEs and mature T cells (> 100/experiment) were imaged across 2 independent experiments. (D) Cells were left resting or activated as in (A) and mitochondrial morphology determined by electron microscopy. The bar graph on the right depicts the average maximum crista width. Expression level of PGC-1α (E) was determined on cells left resting or activated as in (A); representative histograms are shown on the left. The bar graph depicts the average MFI ± SD from 3 independent experiments. *p < 0.05, **p < 0.01, ****p<0.0001 by paired (A,B,E) or unpaired (D) Student t test.
Reduced mitochondrial mass even in the presence of IL-2 implies a defect in mitochondrial biogenesis following activation, a process that is regulated by PGC-1α (37–39). Resting RTEs and mature T cells had equivalent levels of PGC-1α; however, activated RTEs expressed significantly less PGC-1α than their more mature counterparts (Fig. 2E). Importantly, the addition of IL-2 failed to restore PGC-1α levels in RTEs, suggesting that the reduced mitochondrial mass in RTEs is the result of lower PGC-1α expression.
Reduced CD28 expression and defective 4-1BB induction in activated RTEs
Seminal work has shown that the costimulatory receptor CD28 regulates glycolytic metabolism in CD8+ T cells (29), a role that has been expanded to include respiratory metabolism (30, 31). To determine if costimulatory receptors contribute to the distinct mitochondrial metabolism in RTEs, we assessed expression levels of 4-1BB, inducible costimulator (ICOS), and CD28. Compared to mature T cells, resting RTEs expressed equivalent levels of 4-1BB (Fig. 3A) and ICOS (Fig. 3B) but had a subtle, reproducible reduction in CD28 expression (Fig. 3C; 4). Upon activation, RTEs expressed significantly less 4-1BB, ICOS, and CD28 than their more mature T cell counterparts. However, in the presence of IL-2 (which fails to restore OCR and SRC), RTEs expressed less 4-1BB but equivalent ICOS and CD28; hence, reduced CD28 expression on naïve cells and reduced 4-1BB expression on activated cells correlates with the diminished respiratory capacity in activated RTEs.
Figure 3. RTEs display reduced basal CD28 expression and defective 4-1BB induction in the presence of exogenous IL-2.
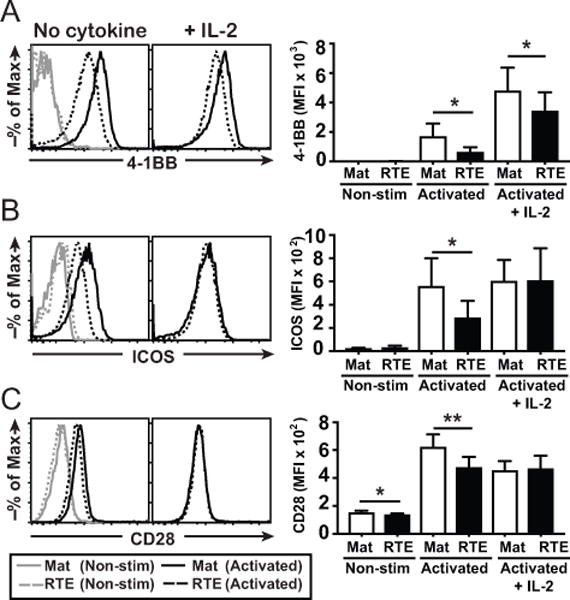
Cells were left resting or activated as in Fig 2A and the surface expression levels of 4-1BB (A), ICOS (B), and CD28 (C) determined; representative histograms are shown on the left. The bar graphs on the right depict the average MFI ± SD from 4 independent experiments. * p < 0.05, **p < 0.01 by paired Student t test.
Figure 4. Altered CD28 signals account for the reduced mitochondrial mass in RTEs.
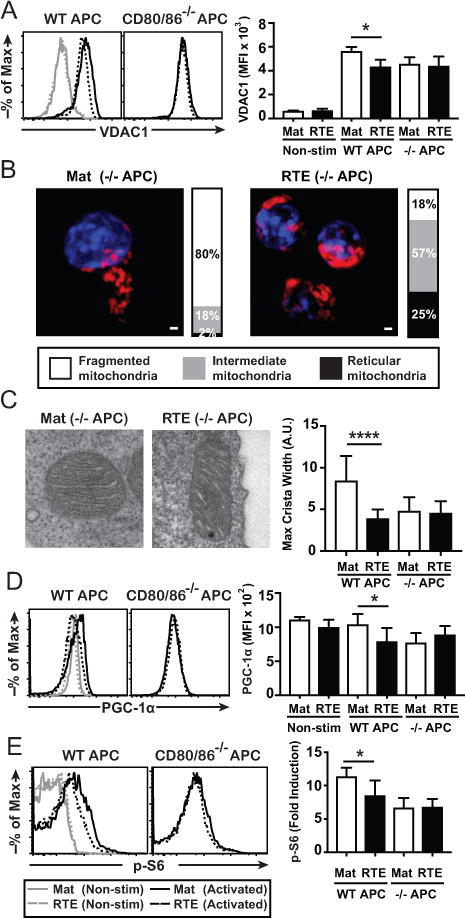
Sort-purified CD8+ RTEs and MN T cells were freshly isolated (non-stim) or stimulated with WT or CD80/86−/− APCs + Ag in vitro for 3 days and the intracellular expression levels of VDAC1 (A) determined; representative histograms are shown on the left. The bar graphs on the right depict the average MFI ± SD from 3 independent experiments. (B) RTEs and MN T cells stimulated with WT or CD80/86−/− APCs were imaged as in Fig. 2C. Bar graphs represent the fraction of cells containing mitochondria of the indicated morphology. RTEs and mature T cells (> 100/experiment) were imaged across 2 independent experiments. (C) Cells were activated with WT or CD80/86−/− APC and mitochondrial morphology determined by electron microscopy. The bar graph on the right depicts the average maximum crista width. Cells were stimulated as in (A) and PGC-1α (D), and p-S6 (E) levels determined; representative histograms are shown on the left. The bar graphs on the right depict the average MFI ± SD (D) or Fold Induction ± SD (E) from 3 independent experiments. * p < 0.05, **** p<0.0001 by paired (A, D, E) or unpaired (C) Student t test.
CD28 signals drive alterations in mitochondrial mass
Compared to mature T cells, resting RTEs express less CD28 and have more elongated mitochondria with tighter crista structure, alterations seen in CD8+ T cells primed without costimulation (30). To determine if altered CD28 signaling accounts for the diminished mitochondrial mass in RTEs, cells were left resting or activated with Ag presented by WT or CD80/86−/− APCs. As expected, RTEs have lower mitochondrial mass than mature T cells when activated with WT APCs. By contrast, RTEs and mature T cells had equivalent mitochondrial mass when activated with CD80/86−/− APCs (Fig. 4A). Mitochondrial morphology in both activated RTEs and mature T was unaffected by the loss of CD28 signaling at this timepoint (Fig. 4B, 2C). By contrast, crista width remained tight in both RTEs and mature T cells activated with CD80/86−/− APCs (Fig. 4C). In support of these findings, RTEs and mature T cells expressed equivalent PGC-1α levels when activated with CD80/86−/− APCs (Fig. 4D). These data suggest that alterations in CD28 signaling account for the diminished mitochondrial mass observed in activated RTEs. To determine whether the reduced CD28 expression by RTEs translates to quantitatively different signaling, we assessed the level of p-S6 as a readout of CD28 signaling intensity. As observed previously (32), RTEs activated with WT APCs have reduced p-S6 levels compared to their mature T cell counterparts (Fig. 4E). However, RTEs and mature T cells had similarly reduced levels of p-S6 when activated with CD80/86−/− APCs. These data argue that the reduced surface expression of CD28 on RTEs results in a quantitatively reduced downstream signal. 4-1BB can also support mitochondrial biogenesis in activated T cells (31). However, 4-1BB transduces a similar downstream signal in RTEs and mature T cells as measured by p-p38 (Supplemental Fig. 1B) and p-NF-κB (Supplemental Fig. 1C). Blocking 4-1BBL/4-1BB interactions did not affect VDAC1 levels in either RTEs or mature T cells (Supplemental Fig. 1D), further suggesting that diminished signaling via CD28, but not 4-1BB, accounts for the reduced mitochondrial mass observed in RTEs.
In sum, our data show that activated RTEs have defective OxPhos, even in the presence of exogenous IL-2. This reduced respiratory rate is due to defective GLS induction and reduced accumulation of mitochondrial mass following activation. Altered CD28 signals account for the reduced PGC-1α expression and diminished accumulation of mitochondrial mass in RTEs. As shown schematically in Supplemental Fig. 2, our study highlights that, while defective glycolytic metabolism in RTEs is correctable by the addition of IL-2, the altered mitochondrial metabolism in these transitional T cells appears to be ‘hard wired’. The reduced mitochondrial content in activated RTEs may also confer RTEs with a survival advantage, as mitochondria serve as central hubs in the regulation of apoptosis (40). Lastly, this work has important implications in the context of anti-tumor immune responses in which mitochondrial metabolism in CD8+ T cells appears to be the central driver of disease outcome (31).
Supplementary Material
Acknowledgments
We thank Edward Parker, Daciana Margineantu, and David Hockenbery for technical assistance and Ramon Klein-Geltink and Erika Pearce for helpful discussions.
Grant Support: This work was supported by NIH Grants R01 AI 064318 and R21 AI 132960 (to P.J.F.), R01 GM 118509 (to S.H.), and T32 DK 007247 (to C.A.C.)
Abbreviations
- Cpt1a
carnitine palmitoyltransferase 1a
- FAO
fatty acid oxidation
- FCCP
carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone
- GLS
glutaminase
- ICOS
inducible T cell costimulator
- MFI
mean fluorescence intensity
- Mat
mature
- MN
mature but naïve
- mROS
mitochondrial reactive oxygen species
- non-stim
non-stimulated
- OCR
oxygen consumption rate
- Oligo
Oligomycin A
- OxPhos
oxidative phosphorylation
- PGC-1α
peroxisome proliferator-activator receptor gamma coactivator-1α
- Rot/AA
rotenone/antimycin A
- RTE
recent thymic emigrant
- SRC
spare respiratory capacity
- Tg
transgenic
- VDAC1
voltage-dependent anion channel 1
- WT
wild type
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Starr TK, Jameson SC, Hogquist KA. Positive and negative selection of T cells. Annu Rev Immunol. 2003;21:139–176. doi: 10.1146/annurev.immunol.21.120601.141107. [DOI] [PubMed] [Google Scholar]
- 2.Fink PJ, Hendricks DW. Post-thymic maturation: young T cells assert their individuality. Nat Rev Immunol. 2011;11:544–548. doi: 10.1038/nri3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu W, Nagaoka H, Jankovic M, Misulovin Z, Suh H, Rolink A, Melchers F, Meffre E, Nussenzweig MC. Continued RAG expression in late stages of B cell development and no apparent re-induction after immunization. Nature. 1999;400:682–687. doi: 10.1038/23287. [DOI] [PubMed] [Google Scholar]
- 4.Boursalian TE, Golub J, Soper DM, Cooper CJ, Fink PJ. Continued maturation of thymic emigrants in the periphery. Nat Immunol. 2004;5:418–425. doi: 10.1038/ni1049. [DOI] [PubMed] [Google Scholar]
- 5.Makaroff LE, Hendricks DW, Niec RE, Fink PJ. Postthymic maturation influences the CD8 T cell response to antigen. Proc Natl Acad Sci USA. 2009;106:4799–4804. doi: 10.1073/pnas.0812354106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Priyadharshini B, Welsh RM, Greiner DL, Gerstein RM, Brehm MA. Maturation-dependent licensing of naive T cells for rapid TNF production. PLoS One. 2010;5:e15038. doi: 10.1371/journal.pone.0015038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friesen TJ, Ji Q, Fink PJ. Recent thymic emigrants are tolerized in the absence of inflammation. J Exp Med. 2016;213:913–920. doi: 10.1084/jem.20151990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deets KA, Berkley AM, Bergsbaken T, Fink PJ. Cutting Edge: Enhanced clonal burst size corrects an otherwise defective memory response by CD8+ recent thymic emigrants. J Immunol. 2016;196:2450–2455. doi: 10.4049/jimmunol.1502525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haines CJ, Giffon TD, Lu LS, Lu X, Tessier-Lavigne M, Ross DT, Lewis DB. Human CD4+ T cell recent thymic emigrants are identified by protein tyrosine kinase 7 and have reduced immune function. J Exp Med. 2009;206:275–285. doi: 10.1084/jem.20080996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pekalski ML, Garcia AR, Ferreira RC, Rainbow DB, Smyth DJ, Mashar M, Brady J, Savinykh N, Dopico XC, Mahmood S, Duley S, Stevens HE, Walker NM, Cutler AJ, Waldron-Lynch F, Dunger DB, Shannon-Lowe C, Coles AJ, Jones JL, Wallace C, Todd JA, Wicker LS. Neonatal and adult recent thymic emigrants produce IL-8 and express complement receptors CR1 and CR2. JCI Insight 2. 2017 doi: 10.1172/jci.insight.93739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van den Broek T, Borghans JAM, van Wijk F. The full spectrum of human naive T cells. Nat Rev Immunol. 2018;18:423–437. doi: 10.1038/s41577-018-0001-y. [DOI] [PubMed] [Google Scholar]
- 12.Cunningham CA, Helm EY, Fink PJ. Reinterpreting recent thymic emigrant function: defective or adaptive? Curr Opin Immunol. 2017;51:1–6. doi: 10.1016/j.coi.2017.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Houston EG, Jr, Fink PJ. MHC drives TCR repertoire shaping, but not maturation, in recent thymic emigrants. J Immunol. 2009;183:7244–7249. doi: 10.4049/jimmunol.0902313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Houston EGJ, Boursalian TE, Fink PJ. Homeostatic signals do not drive post-thymic T cell maturation. Cell Immunol. 2012;274:39–45. doi: 10.1016/j.cellimm.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Houston EG, Jr, Nechanitzky R, Fink PJ. Cutting Edge: contact with secondary lymphoid organs drives postthymic T cell maturation. J Immunol. 2008;181:5213–5217. doi: 10.4049/jimmunol.181.8.5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mendoza A, Fang V, Chen C, Serasinghe M, Verma A, Muller J, Chaluvadi VS, Dustin ML, Hla T, Elemento O, Chipuk JE, Schwab SR. Lymphatic endothelial S1P promotes mitochondrial function and survival in naive T cells. Nature. 2017;546:158–161. doi: 10.1038/nature22352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, Green DR. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hedeskov CJ. Early effects of phytohaemagglutinin on glucose metabolism of normal human lymphocytes. Biochem J. 1968;110:373–380. doi: 10.1042/bj1100373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang CH, Curtis JD, Maggi LB, Jr, Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, Weber JD, Pearce EJ, Jones RG, Pearce EL. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, Wang CR, Schumacker PT, Licht JD, Perlman H, Bryce PJ, Chandel NS. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. 2013;38:225–236. doi: 10.1016/j.immuni.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mehta MM, Weinberg SE, Chandel NS. Mitochondrial control of immunity: beyond ATP. Nat Rev Immunol. 2017;17:608–620. doi: 10.1038/nri.2017.66. [DOI] [PubMed] [Google Scholar]
- 22.Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science. 2016;354:481–484. doi: 10.1126/science.aaf6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, Pearce EL. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van der Windt GJ, O’Sullivan D, Everts B, Huang SC, Buck MD, Curtis JD, Chang CH, Smith AM, Ai T, Faubert B, Jones RG, Pearce EJ, Pearce EL. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci USA. 2013;110:14336–14341. doi: 10.1073/pnas.1221740110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phan AT, Doedens AL, Palazon A, Tyrakis PA, Cheung KP, Johnson RS, Goldrath AW. Constitutive glycolytic metabolism supports CD8+ T cell effector memory differentiation during viral infection. Immunity. 2016;45:1024–1037. doi: 10.1016/j.immuni.2016.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, Qiu J, Kretz O, Braas D, van der Windt GJ, Chen Q, Huang SC, O’Neill CM, Edelson BT, Pearce EJ, Sesaki H, Huber TB, Rambold AS, Pearce EL. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell. 2016;166:63–76. doi: 10.1016/j.cell.2016.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patten DA, Wong J, Khacho M, Soubannier V, Mailloux RJ, Pilon-Larose K, MacLaurin JG, Park DS, McBride HM, Trinkle-Mulcahy L, Harper ME, Germain M, Slack RS. OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 2014;33:2676–2691. doi: 10.15252/embj.201488349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, Perales-Clemente E, Salviati L, Fernandez-Silva P, Enriquez JA, Scorrano L. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–171. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH, Thompson CB. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769–777. doi: 10.1016/s1074-7613(02)00323-0. [DOI] [PubMed] [Google Scholar]
- 30.Klein Geltink RI, O’Sullivan D, Corrado M, Bremser A, Buck MD, Buescher JM, Firat E, Zhu X, Niedermann G, Caputa G, Kelly B, Warthorst U, Rensing-Ehl A, Kyle RL, Vandersarren L, Curtis JD, Patterson AE, Lawless S, Grzes K, Qiu J, Sanin DE, Kretz O, Huber TB, Janssens S, Lambrecht BN, Rambold AS, Pearce EJ, Pearce EL. Mitochondrial priming by CD28. Cell. 2017;171:385–397. doi: 10.1016/j.cell.2017.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Menk AV, Scharping NE, Rivadeneira DB, Calderon MJ, Watson MJ, Dunstane D, Watkins SC, Delgoffe GM. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J Exp Med. 2018;215:1091–1100. doi: 10.1084/jem.20171068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cunningham CA, Bergsbaken T, Fink PJ. Cutting Edge: Defective aerobic glycolysis defines the distinct effector function in antigen-activated CD8+ recent thymic emigrants. J Immunol. 2017;198:4575–4580. doi: 10.4049/jimmunol.1700465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buck MD, O’Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015;212:1345–1360. doi: 10.1084/jem.20151159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Preston GC, Sinclair LV, Kaskar A, Hukelmann JL, Navarro MN, Ferrero I, MacDonald HR, Cowling VH, Cantrell DA. Single cell tuning of Myc expression by antigen receptor signal strength and interleukin-2 in T lymphocytes. EMBO J. 2015;34:2008–2024. doi: 10.15252/embj.201490252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berkley AM, Hendricks DW, Simmons KB, Fink PJ. Recent thymic emigrants and mature naïve T cells exhibit differential DNA methylation at key cytokine loci. J Immunol. 2013;190:6180–6186. doi: 10.4049/jimmunol.1300181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang S, Zhang X, Wang K, Xu X, Li M, Zhang J, Zhang Y, Hao J, Sun X, Chen Y, Liu X, Chang Y, Jin R, Wu H, Ge Q. Newly generated CD4(+) T cells acquire metabolic quiescence after thymic egress. J Immunol. 2018;200:1064–1077. doi: 10.4049/jimmunol.1700721. [DOI] [PubMed] [Google Scholar]
- 37.Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr. 2011;93:884–890. doi: 10.3945/ajcn.110.001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, Stelekati E, McLane LM, Paley MA, Delgoffe GM, Wherry EJ. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8+ T cell exhaustion. Immunity. 2016;45:358–373. doi: 10.1016/j.immuni.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, Ferris RL, Delgoffe GM. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity. 2016;45:374–388. doi: 10.1016/j.immuni.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang C, Youle R. The role of mitochondria in apoptosis. Annu Rev Genet. 2009;43:95–118. doi: 10.1146/annurev-genet-102108-134850. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.