

Abstract
Constitutively activated STAT3 plays a critical role in non-small cell lung carcinoma (NSCLC) progression by mediating proliferation and survival. STAT 3 activation in normal cells is transient, making it an attractive target for NSCLC therapy. The therapeutic potential of blocking STAT3 in NSCLC was assessed utilizing a decoy approach by ligating a double-stranded 15-mer oligonucleotide that corresponds to the STAT3 response element of STAT3-target genes, to produce a cyclic STAT3 decoy (CS3D). The decoy was evaluated using NSCLC cells containing either wild-type (WT) EGFR (201T) or mutant EGFR with an additional EGFRi resistance mutation (H1975). These cells are resistant to EGFR inhibitors and require an alternate therapeutic approach. CS3D activity was compared to an inactive cyclic control oligonucleotide (CS3M) that differs by a single base pair, rendering it unable to bind to STAT3 protein. Transfection of 0.3 μM of CS3D caused a 50% inhibition in proliferation in 201T and H1975 cells, relative to CS3M, and a 2-fold increase in apoptotic cells. Toxicity was minimal in normal cells. CS3D treatment caused a significant reduction of mRNA and protein expression of the STAT3 target gene c-Myc, and inhibited colony formation by 70%. The active decoy decreased the nuclear pool of STAT3 compared to the mutant. In a xenograft model, treatments with CS3D (5 mg/kg) caused a potent 96.5% and 81.7% reduction in tumor growth in 201T (P<0.007) and H1975 models (P<.0001), respectively, and reduced c-Myc and p-STAT3 proteins. Targeting STAT3 with the cyclic decoy could be an effective therapeutic strategy for NSCLC.
Keywords: Non-small cell lung cancer, STAT3, Epidermal growth factor receptor, cancer therapeutics, c-Myc, decoy therapy
Introduction
Lung cancer remains the leading cause of cancer-related mortality in the United States, which is largely attributed to the dismal survival of stage IV disease (1–3). In non-small cell lung carcinoma (NSCLC), accounting for 85% of lung cancer cases (4), patients are stratified based on genetic alterations in the tumor to choose targeted therapy. Patients with activating somatic epidermal growth factor receptor (EGFR) mutations initially respond to EGFR tyrosine kinase inhibitors (TKIs), but eventually develop acquired resistance through new mutations such as the EGFR T790M substitution, accounting for about 50% of EGFR mutant patients who develop EGFR TKI resistance. The majority of patients without EGFR mutation, including those who lack other “drivers”, are intrinsically resistant to EGFR TKIs (5). In this study, we tested the ability of a novel therapeutic, a cyclic oligonucleotide, to inhibit the growth of lung tumors with the T790M mutation or with intrinsic resistance to EGFR TKIs, by targeting Signal Transducer and Activator of Transcription 3 (STAT3).
STAT3 is a central node downstream of individual oncogenic tyrosine kinase “drivers,” which could be a therapeutic niche for both intrinsic and acquired resistance to multiple TKIs. STAT3 was often activated during in vitro induction of resistance to erlotinib, as the result of feedback up-regulation of other kinases, suggesting that increased STAT3 activity could commonly occur in response to TKIs (6). During lung cancer progression, STAT3 is often constitutively active, upregulating the expression of important target genes necessary for cellular proliferation and survival (7). As a point of convergence for many dysregulated signaling pathways, STAT3 mediates adaptive mechanisms of resistance to molecular targeted therapy in NSCLC, such as induction of epithelial-to-mesenchymal transition by IL-6 in response to erlotinib (8, 9). Since STAT3 plays a critical role in malignant cell transformation, but is not essential for normal cell growth (10), it is potentially a prime therapeutic target for NSCLC treatment.
Efforts to apply STAT3 inhibition in the clinical setting have been limited due to a paucity of potent and selective inhibitors (11,12). Most currently available STAT3 inhibitors either target upstream kinases, lack specificity, or require exceptionally high concentrations to achieve inhibition. The ability of non-phosphorylated STAT3 to function as an active dimer (13,14) also may limit the ability of kinase inhibitors to completely block STAT3. To address this, we utilized a decoy approach by circularizing (through ligation) a double-stranded oligonucleotide containing a 15 base-pair sequence corresponding to the STAT3 response element (15). The circular molecule was produced through inclusion of two hexaethyleneglycol spacers that provide flexibility, and upon ligation becomes a thermally stable cyclic double-stranded oligonucleotide (16). The cyclic STAT3 decoy (CS3D) was compared to a mutant inactive control (CS3M) molecule that differs from the active compound by a single base-pair and lacks binding to the STAT3 protein (15, 16), allowing an assessment of the specificity of CS3D to block STAT3’s functions. The STAT3 decoy was previously shown to decrease luciferase activity in cells expressing a luciferase reporter gene controlled by the STAT3 consensus sequence; CS3D competitively bound to activated pSTAT3 protein in comparison to CS3M, which showed no affinity for pSTAT3 protein (15).
The decoy approach allows STAT3 dimers to preferentially interact with the oligonucleotide, acting as a molecular sink to competitively inhibit binding of the dimer to the promoters of STAT3 target genes (15,16). This approach demonstrated efficacy in head and neck squamous cell carcinoma models (HNSCC) (17, 18), and the ligated circularized decoy exhibits greater potency when injected in vivo compared to earlier linear versions (16). Here we examined the antitumor effects of CS3D in NSCLC. The STAT3 decoy inhibited proliferation, induced apoptosis, blocked STAT3 target gene expression, and inhibited in vivo tumor growth in NSCLC cells that are EGFR WT (201T cells) and in NSCLC cells that contain both an activating EGFR mutation and the T790M EGFRi resistance mutation (H1975). Targeting STAT3 in NSCLC using a cyclic decoy represents a new potentially effective therapeutic strategy for the treatment of NSCLC with inherent or acquired EGFR TKI resistance.
Materials and Methods
Antibodies and Reagents
Antibodies for STAT3, phospho-STAT3, p-NFKB, cyclin D1, cleaved caspase-3 (CC3), c-Myc, Ki67, GAPDH, Ubiquitin and horseradish peroxidase-conjugated secondary antibodies were purchased from Cell Signaling Technology (Danvers, MA). The CellTiter 96® AQueous One Solution (MTS) Reagent containing 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium was obtained from Promega (Madison, WI).
Cell Culture
The wild-type EGFR (201T) cell line was previously derived from a patient with lung adenocarcinoma (19). H1975 cells with the EGFR intrinsic point mutation L858R and the acquired EGFRi resistance mutation T790M were purchased from American Type Culture Collection (Manassas, VA). 201T and H1975 were cultured in BME and RPMI respectively containing 10% heat-inactivated fetal bovine serum and 1x penicillin/streptomycin (Thermo fisher, Waltham, MA) and 1x GlutaMax (Life Technologies, Carlsbad, CA). A549 and H3255 cells were also purchased from ATCC and cultured in BME and RPMI, respectively. Human bronchial epithelial cells were purchased from ATCC, and primary normal lung fibroblasts were derived from human lung tissue (20). Cell lines were authenticated by short tandem repeat DNA profiling and used within 6 months of testing. Frozen cell stocks were passaged a maximum of 15 times and cells were mycoplasma-free.
Cyclic STAT3 Decoy (CS3D) Transfections
Single-stranded STAT3 decoy and inactive mutant version which differs by a base-pair at position 9 (G to T) as compared to the STAT3 decoy, were generated as previously described (18,19). Both oligonucleotides were synthesized by Integrated DNA Technologies (IDT). The single-stranded oligonucleotide (sense and antisense strands) was further modified with two hexaethyleneglycol linkers attached to both strands, allowing the structure to circularize upon enzymatic ligation of the 3’ and 5’ ends of the annealed oligonucleotide. Both molecules were purified by HPLC according to manufacturer’s reports (IDT). Using T4 DNA ligase (New England Biolabs, Ipswich, MA), we performed overnight ligations to produce the cyclic STAT3 decoy (CS3D) and the inactive circularized mutant (CS3M). Ligation efficiency was analyzed on a 15% TBE gel and it ranged between 85 – 95 %, showing that amount of the cyclic forms predominates over the linear forms. The linear form is also less stable (15,16) and is less likely to contribute to biological effects in the presence of an excess cyclic form.
Fluorescently-labeled CS3M and CS3D (with iFluor) were produced using enzymatic ligation as described above and used to assess uptake efficiency. Transfections were performed using Lipofectamine 2000 (Life technologies) in opti-MEM media (Life Technologies) as follows: Cells were plated at 75–80% confluency, and exposed to transfection media containing the respective circularized oligonucleotides for 24 hours at 37°C. Post-transfection, cell recovery was initiated in media containing 10% heat-inactivated fetal bovine serum.
MTS Assays and Programmed Cell Death Analysis
Dose-response experiments assessing metabolically active viable cells were performed by transfecting cells with increasing concentrations of CS3D or CS3M, followed by performance of MTS assays. After 72 hours, 5mg/ml of MTS reagent was added in the plates and incubated 37°C for 20 minutes. The plates were then read at 490 nm in a Synergy microplate reader (BioTek, Winooski, VT) using Gen5 2.05 software. Data obtained following treatment with C3SD or CS3M were normalized to treatment with lipofectamine alone. IC50s determined from these experiments were used to perform independent assays to confirm the effects of CS3D relative to CS3M. To assess induction of apoptosis, cells were transfected with 100nM of CS3M or CS3D. After 24 hours, cells were resuspended in annexin V-binding buffer, incubated with 5μL of propidium iodide (BD Biosciences) and subjected to flow cytometric analysis, using a BD FACS Canto II Flow Cytometer.
Colony Forming Assays
Following transfection as above, cells transfected with either CS3D or CS3M were detached from the plate using Trypsin-EDTA (Life Technologies) and seeded at a density of 2 × 104 cells/well (in a 6-well plate) in soft agar to determine anchorage-independent growth. Sea Plaque Agarose (Lonza – Rockland, ME) was used to prepare 0.8% base agarose layer and a 0.48% upper agarose layer containing the transfected cells. 1ml of growth media was added into each well and incubated at growth conditions of 37°C with 5% CO2. Media was replaced every 3–4 days and colony formation was monitored. After 15-20 days, media was aspirated and wells were incubated with crystal violet in 10% formalin for 1 h. Using a dissecting scope, colonies were photographed in a 6-well plate (with 4 quadrants/well) using ImageJ software analysis to automatically count colonies, using 35 pixels as a cut-off.
Immunoblotting
Cells were seeded at 0.5 × 106 cells/well in six-well plates in BME/RPMI containing 10% FBS and transfected with 0.1μM CS3D/CS3M for 24 hours. Post-transfection, cell lysates were extracted and protein concentrations quantified using DC assay reagents. Whole cell lysates (20 μg/sample) were electrophoresed on 7.5% SDS-polyacrylamide gels for 1 h and transferred onto Trans-Blot polyvinylidene difluoride (PVDF) membranes (Bio-Rad Laboratories) for 1 h at 100 V. The membranes were blocked using 5% nonfat dry milk, 0.1% Tween 20 in 1x phosphate-buffered saline (TBST) for 1 h. Membranes were incubated in primary antibody diluted at 1:1000 at 4°C overnight, and washed three times with TBST (15 min/wash). Membranes were then incubated with secondary antibody for 1 h (1:2000) at RT, followed by three washes in TBST. Blots were developed using a super-enhanced chemiluminescence substrate according to the manufacturer’s protocol (ThermoScientific). To quantify the results, ImageJ 1.X software assessed changes in protein expression patterns.
Confocal Microscopy
After transfection with fluorescein-tagged CS3D, H1975 cells were fixed in 4% paraformaldehyde and prolong antifade reagent (CST) was applied directly to the slides. Fixed cell imaging was performed using a Nikon Eclipse Ti Confocal Microscope System. Images were captured and analyzed using the imaging software NIS Elements and Image J. Fluorescence was detected at 488 nm. Images were taken and processed at 40X and 60X.
Immunoprecipitation (IP)
Post-transfection with either 100nM CS3D or CS3M, H1975 were lysed in IP lysis buffer (Thermo Scientific, Rockford, IL) supplemented with protease inhibitor cocktail (Roche, Basel, Switzerland) and phosphatase inhibitors (NaF and Na3VO4). Immunoprecipitations were performed using pSTAT3 antibody at 1:500 and Protein A Magnetic Beads (Thermo Scientific) at 4°C. Immunoprecipitated lysates were electrophoresed on 7.5% SDS-polyacrylamide gels followed by exposure to ubiquitin antibody (1:1000) at 4°C overnight and the appropriate secondary antibody while following the immunoblotting procedure as above.
Real-Time qPCR
Post transfection, cells were treated with 10 ng/ml EGF for 1.5 hr to activate STAT3. Trizol (Invitrogen, Carlsbad, CA) was then used to extract total RNA. One microgram of total RNA was reverse-transcribed using a cDNA synthesis kit (Quanta Biosciences) using a T100 Thermal Cycler (BioRad). Real-time qPCR was performed using a SYBR Green Super Mix kit on a CFX connect Real-Time System (BioRad). Gene-specific primers for STAT3 target genes were used to assess mRNA levels normalized to GAPDH mRNA levels as internal control, and the ratio of normalized mRNA to the control conditions was determined using the comparative ∆CT method for analysis. The primers used for real-time qPCR are as follows: Bcl-xL, (F) 5ʹ-ATGCAGGTATTGGTGAGTCG-3ʹ; (R) 5ʹ-CTGCTGCATTGTTCCCATAG-3ʹ; GAPDH, (F) 5ʹ-GGA GCG AGA TCC CTC CAA AAT-3ʹ; (R) 5ʹ-GGC TGT TGT CAT ACT TCT CAT GG-3ʹ; IL-6, (F) 5ʹ-ACT CAC CTC TTC AGA ACG AAT TG-3ʹ; (R) 5ʹ-CCA TCT TTG GAA GGT TCA GGT TG-3ʹ; c-MYC, (F) 5ʹ CCGCATCCACGAAACTTTG-3ʹ; (R) 5ʹ- GGGTGTTGTAAGTTCCAGTGCAA-3ʹ; TIF1 (F) 5′-TTCGGAGAAAGGCATTAGA-3′; (R) 5′ –TCCAGGGCTTCATTCATAT-3′; IRF7 (F) 5′-ATGGGCAAGTGCAAGGTGTA-3’; (R) 5-ACCAGCTCTTGGAAGAAGACTC-3’; IL-8 (F) 5-’TCTGTTAAATCTGGCAACCC-3’; (R) 5-’TAAAGGAGAAACCAAGGCAC- 3; IL-1β (F) 5′- TTCTTCGACACATTGGATAACG-3′ and (R) 5′-TGGAGAACACCACTTGTTGCT-3′.
In Vivo Tumor Xenograft Studies
All studies were approved by the University of Minnesota Institutional Animal Care and Use Committee and were carried out in accordance with institutional guidelines for animal care. Female nude mice (4 – 6 weeks old) were injected with 1×106 cells in the flanks. After 3 weeks, animals with palpable tumors (200mm3) were randomized into two treatment groups with 10 tumors/group. Daily tail vein injections (5 days per week) of the cyclic STAT3 decoy (CS3D) or mutant STAT3 control (CS3M) (100μg in 200μl per injection) was delivered for 2 – 3 weeks. Tumor measurements were made with calipers every 2 days. Volumes were calculated from the formula V= (Length × Width2)/2. Animals were sacrificed according to IACUC guidelines.
Immunohistochemical analysis
Tumors were excised for immunohistochemical staining; 5-μm sections were cut from formalin-fixed paraffin-embedded tissue blocks, deparaffinized and rehydrated using successive washes of xylene followed by ethanol. Antigen retrieval was performed in a microwave oven (for 20 minutes) in an unmasking solution containing sodium citrate buffer followed by peroxidase blocking in 3% hydrogen peroxide. The sections were incubated in ABC blocking buffer (Vector laboratories Inc, Burlingame, CA) for 1 hour at RT. Sections were then stained with H&E or subjected to sequential incubations with different primary antibodies and peroxidase-conjugated goat anti-rabbit secondary antibodies. The following primary antibodies were used (at the indicated dilutions): cleaved caspase-3 (#9664, Cell Signaling, 1:300), p-STAT3 (#9145, Cell Signaling, 1:400), Ki67 (#ab15580, Abcam, 1:1000). Sections were developed with DAB and counterstained with hematoxylin. Bright field microscopy was performed using Leica DM 4000 B LED microscope and images were captured at 20X and 40X magnification using LASv4.7 software. IHC analysis was done as a blinded study with 100 images from each treatment group graded as low, moderate, or high. Staining was graded as low (<30% positive cells per field, scored as 1), moderate (30-60% positive cells per field, scored as 2) or high (> 60% positive cells per field, scored as 3).
Statistical Analyses
For statistical analyses, data were reported as mean ± standard deviation (SD) or standard error of the mean (SEM). To assess significance between different treatment groups, a student’s t test (two tailed) was used to determine significance with p values at least < 0.05 categorized as statistically significant. To analyze IHC scoring data, Chi-Squared test was used to compare the frequency of scores of 1,2, or 3 in treatment groups.
Results
Wild-type and mutant EGFR NSCLC cells exhibit similar sensitivity to the STAT3 decoy (CS3D)
To determine effect of CS3D on cell viability, we transfected increasing concentrations of CS3D, and after 72 hours, MTS assays were used to assess cell viability. Data were normalized to cells treated with lipofectamine alone. The inactive mutant control (CS3M) showed no cytotoxicity (Fig. 1A), and the percent of cytotoxicity induced by CS3D allowed calculation of the IC50s as described in Methods. The IC50 values for CS3D in both the WT (201T) and mutant (H1975) EGFR cell lines were approximately 0.3μM (Fig. 1A). Other cell lines (H3225 with EGFR activating point L858R mutation and A549 which is EGFR WT) were also assessed in the presence of CS3D and CS3M and showed similar sensitivities (IC50s 0.3 μM). Cell lines that are resistant to EGFR TKIs (201T, H1975, and A549) showed the same sensitivity to STAT3 decoy as the EGFR TKI sensitive cell line (H3225). Normal lung fibroblasts and primary bronchial epithelial cells had IC50s that were 100-fold higher (300 μM) than tumor cells.
Figure 1. Effects of the cyclic STAT3 decoy in vitro.
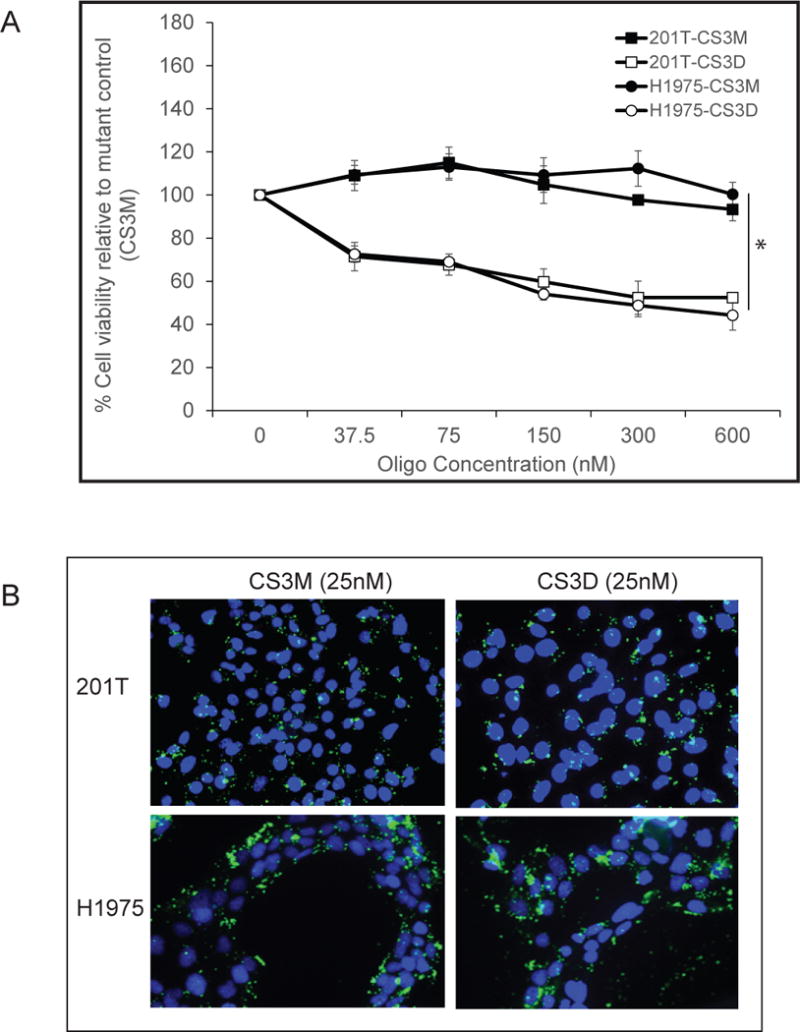
(A) Effect on cell viability. NSCLC cell lines (201T and H1975) were transfected with CS3D or CS3M at concentrations ranging from 0 nM to 600 nM. Using MTS assays, cell viabilities was assessed 72 hours later. Three independent experiments were performed, using 24-well plates and 4 wells/concentration. (B) Uptake of fluorescein-labeled CS3D by NSCLC in vitro. 24 hours post-transfection, confocal imaging shows intracellular localization of CS3M or CS3D in 201T and H1975 cells.
Uptake of CS3D into NSCLC cell lines
To determine whether CS3D and CS3M were equally taken up by NSCLC cells, both molecules were tagged with a fluorescein dye (iFluorT) to assess transfection efficiency. Twenty-four hours post-transfection with the iFluorT-labeled oligonucleotides at a concentration of 25 nM. NSCLC cell lines showed intracellular location of the cyclic molecules (CS3D or CS3M), demonstrating that integrating hexaethyleneglycol linkers into the oligonucleotide, or circularizing the DNA, still allowed intracellular uptake. More importantly, altering the sequence of the CS3D by a single base-pair to generate the inactive STAT3 mutant (CS3M) did not affect its uptake, demonstrating that the cytotoxicity of CS3D was not due to preferential uptake compared to CS3M (Fig 1B). The efficiency of transfection was >90% for both CS3M and CS3D. Further confocal microscopy showed clear fluorescent signal in both the nucleus and the cytoplasm of lung cancer cells, with cytoplasmic signal predominating (Supplemental Figure 1). This suggests that CS3D can interact with its target STAT3 dimers in both compartments.
CS3D inhibits colony-forming ability of NSCLC and induces cell death
We next examined the effect of CS3D on the ability of NSCLC cells to grow in an anchorage-independent manner. 201T and H1975 cells were transfected with either CS3D or CS3M (300 nM), then seeded in soft agar. Compared with CS3M, CS3D produced a significant decrease in colony formation. The number of colonies formed was quantified by using a size cutoff (with greater than 35 pixels counted as a colony). A single transfection with CS3D significantly (P<0.051) disrupted anchorage-independent growth of the NSCLC cells by 70% (at 15 days in H1975 cells) and 50% (at 20 days in 201T cells (P<0.053)) compared to CS3M (Fig. 2A). Flow cytometric analysis of Annexin V/propidium iodide-stained cells also demonstrated that transfection with CS3D significantly increased apoptosis in comparison to CS3M. CS3D treatment (100 nM) caused a 2-fold increase (P<0.05) in the number of cells undergoing apoptosis as compared to CS3M at 24 h (Fig. 2B).
Figure 2. CS3D inhibits anchorage independent growth and promotes apoptosis.
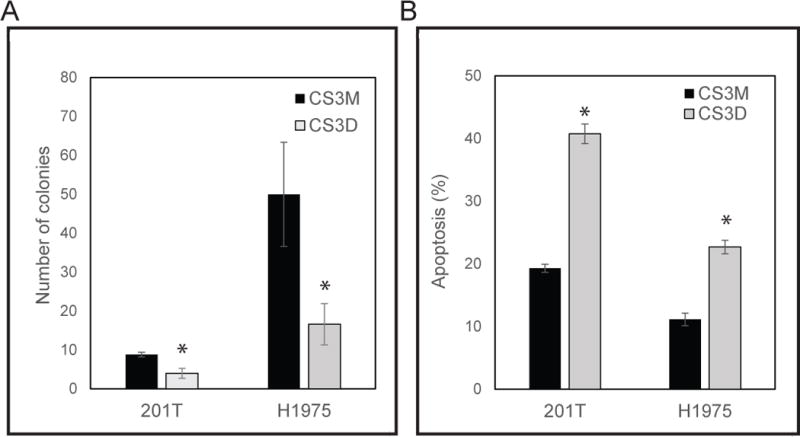
(A) NSCLC cell growth was assessed in colony forming assays in the presence of either 300 nM CS3D or CS3M. CS3D significantly blocks the ability of NSCLC to grow in an anchorage independent manner in soft-agar post-transfection. (B) Detection of annexin V and propidium iodide (PI)-positive cells by flow cytometry: H1975 and 201T cells transfected with 100nM of CS3M or CS3D for 24hr were stained with apoptosis marker (annexin V) and cell viability dye. Statistical significance was determined as *P<0.05, n=3.
CS3D suppresses the expression of c-Myc in response to EGF
c-Myc is a STAT3 target gene that is known to be activated by EGF treatment. To investigate the effects of STAT3 blockade on c-Myc expression, cells were first treated with 10 ng/ml EGF for 1.5 hr (deemed the optimal time to observe increased c-Myc RNA), 24 hr after transfection of the cyclic oligonucleotides. RT-qPCR was used to assess differences in mRNA expression in STAT3 decoy-treated versus mutant decoy-treated cells (Fig 3A & B). A single transfection of CS3D caused a 50% inhibition in mRNA level of c-Myc in 201T cells (Fig 3A) and a 25-30% inhibition in H1975 cells (Fig 3B). Immunoblotting analysis also showed a 31% reduction in c-Myc protein expression post-CS3D transfection compared to either CS3M or control (lipofectamine alone) in 201T cells. Similarly, in H1975 cells, a 40% reduction in c-Myc protein expression was found (Fig 4A). H3225 cells and A549 cells also showed similar decrease in c-Myc expression. Examination of other STAT3 genes (for example Bcl-xl as shown in Fig. 3) also responded with decreased mRNA and/or protein expression across the four cell lines, but not as consistently as c-Myc. Examination of STAT1 target genes (IFIT1, IRF7) and NFκB target genes (IL-1β and IL-8) that are predominantly regulated by these transcription factors showed no significant decrease in mRNA expression level by qRT-PCR when comparing CS3D to CS3M treated cells (Supplemental Fig 2).
Figure 3. Down-regulation of c-Myc and bcl-xL mRNA expression by CS3D.
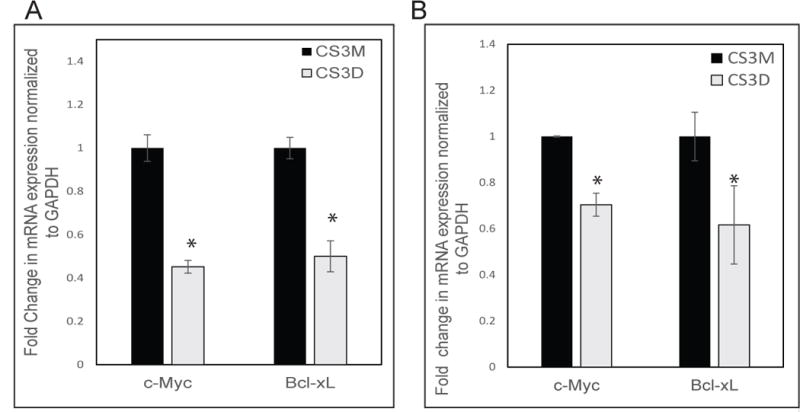
24 hours post-transfection, cells were treated with EGF (10 ng/ml) for 1.5 hr and mRNA was harvested from wild-type (A) and mutant EGFR T790M (B) NSCLC cell lines, and mRNA expression was assessed by RT-qPCR. Relative mRNA expression was normalized to GAPDH mRNA levels as an internal control. Statistical significance was determined as *P<0.05.
Figure 4. Change in c-Myc and STAT3 protein by CS3D.
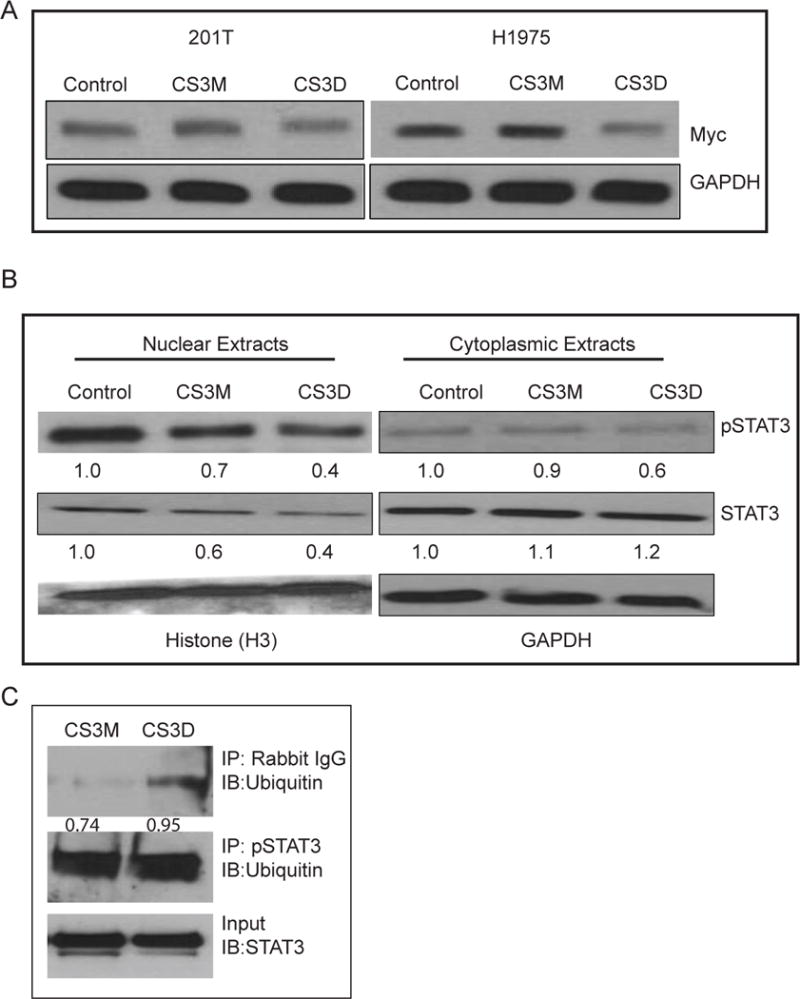
(A) Down-regulation of c-Myc protein by CS3D. Cells were treated as in Fig. 3, and 3 hr after EGF treatment, lysates were collected from wild-type (left) and mutant EGFR T797M (right) NSCLC cell lines. Protein expression was assessed by Western blot. Relative c-Myc expression was normalized to GAPDH protein levels as an internal control. (B) CS3D alters STAT3 levels. 24 hours post-transfection with either CS3M, CS3D, or lipofectamine alone H1975 were stimulated with IL-6 (50ng/mL) for 1 hr to strongly activate STAT3. After cell lysis, nuclear and cytoplasmic extracts were obtained by subcellular fractionation using centrifugation, and Western blotting was used to assessed to protein expression. GAPDH was used as a marker of the cytoplasmic fraction and histone H3 was used as a marker of the nuclear fraction. Densitometric quantification of the c-Myc bands are as indicated. (C) CS3D increases the proportion of pSTAT3-ubiquitin bound complexes. 24 hours post-transfection, P-STAT3 was immunoprecipitated and the presence of ubiquitin was assessed by immunoblotting. Greater ratio of pSTAT3-ubiquitin complexes was detected with CS3D treatment. The increase in pSTAT3-unbiquitin complex correlated with decrease in total STAT3 present. Densitometric analysis revealed a greater ratio of pSTAT3-ubiquitin complex to STAT3 in response to CS3D (0.95) as compared to CS3M (0.74).
Additional immunoblotting analysis of cell extracts showed that after stimulation with IL6, a potent inducer of p-STAT3, there was a 35% reduction in both nuclear pSTAT3 and nuclear total STAT3 after transfection of CS3D compared to CS3M or control (lipofectamine alone) (Fig 4B). In the cytoplasm, the levels of pSTAT3 (but not total STAT3) were also reduced by CS3D, suggesting that the selective binding of pSTAT3 to CS3D might alter its stability and/or its ability to shuttle to the nucleus. (Fig 4B). To test whether CS3D could increase ubiquitination of p-STAT3 protein, an immunoprecipitation was carried out for p-STAT3, followed by immunoblotting for ubiquitin. After 24 hr, CS3D increased the ratio of p-STAT3 that was ubiquitinated relative to amount of STAT3 present (input), compared to CS3M (0.95 vs. 0.74, Fig. 4C). The input of total cell lystate also showed less STAT3 after CS3D treatment, confirming the cell compartment results in Fig. 4B.
Intravenous injection of the cyclic STAT3 decoy (CS3D) inhibits NSCLC tumor growth
Based on the in vitro effects of CS3D, we next evaluated antitumor effects in mice harboring established NSCLC xenografts (10 tumors/group). Mice were given daily intravenous injections (tail vein) of CS3D or CS3M (5mg/kg/d), 5 days per week, and tumor growth was monitored for 14 – 20 days. This method of delivery and dose were previously shown to be efficacious in head and neck cancer models (16, 17). CS3D caused a significant and robust tumor growth inhibition in 201T- and H1975-derived xenografts compared to treatment with CS3M. We observed a 96.5% (P<0.007) in 201T and 81.7% (P<0.0001) reduction in H1975 (Fig 5A). H & E staining of sectioned tumors harvested after treatment showed that the CS3D treated tumors were composed of large areas of debris and infiltration with stroma and lymphocytes, while the CS3M tumors had a high tumor cellularity (Supplemental Fig 3). The animals showed no significant loss of body weight or decreased activity, and histological examination of the lungs, liver, and spleen showed no signs of toxicity with CS3D.
Figure 5. CS3D suppresses NSCLC xenograft tumor growth.
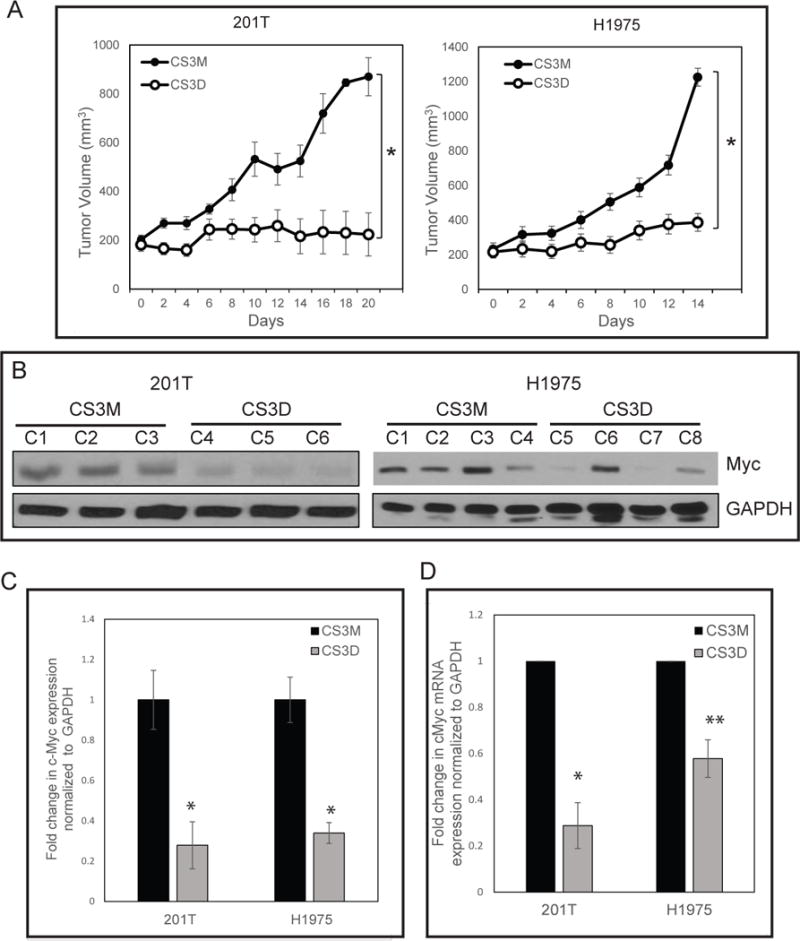
(A) Intravenous delivery of CS3D inhibits tumor growth in both wild-type (201T) and mutant EGFR T790M (H1975) xenografts. Approximately 1x106 cells were inoculated subcutaneously in the flanks of nude mice. Following the development of palpable tumors (about 200mm3), mice were randomized and given daily injections of either CS3D or CS3M (5mg/kg/day; 10 tumors/group). Experiments were done twice as biological replicates. Tumor volumes were recorded every other day while animal weights were also monitored during the course of the treatment. The difference in tumor volume between the CS3D- and CS3M-treated groups was significant for both 201T (P<0.007) and H1975 (P<0.0001). (B) c-Myc is suppressed after CS3D treatment in vivo. At the end of the treatments, tumors were harvested, whole cell lysates were prepared and RNA extracted for target gene analysis by Western blotting and RT-qPCR. GAPDH protein was used as internal loading control for immunoblotting. CS3D treated tumors (C4, C5, and C6) from separate animals show a decrease in c-Myc expression level relative to the CS3M-treated group (C1, C2, and C3) in 201T. Immunoblotting analysis of H1975 residual tumors treated with CS3D (C8-C8) also showed reduction in c-Myc expression levels relative to CS3M (C1-C4). The expression of c-Myc was significantly reduced in response to CS3D, relative to CS3M, in both wild-type (201T, (P<0.042)) and mutant EGFR (H1975, (P<0.05)) derived tumors (*P<0.05). Data are shown as mean +/− SEM. (C) Densitometry quantification of c-Myc protein expression levels in 201T and H1975 from tumor-derived xenografts. The expression of c-Myc was significantly reduced in response to CS3D, relative to CS3M, in both wild-type (201T, (P<0.042)) and mutant EGFR (H1975, (P<0.05)) derived tumors. Data shown are means +/− SEM between two groups (CS3M and CS3D). (D) c-Myc mRNA expression levels in 201T and H1975: CS3D suppresses c-Myc gene expression levels in 201T and H1975-derived tumors. Data are presented as mean +/− SEM. *P<0.05 compared with the mutant control group (CS3M).
STAT3 inhibition In Vivo by CS3D down-regulates c-Myc and promotes cell death
To determine how CS3D blocks NSCLC growth, we performed Western blot, qRT-PCR, and immunohistochemical analysis to determine expression of c-Myc in residual tumors. These analyses identified a substantial downregulation of c-Myc protein (Fig. 5B & C) and mRNA levels (Fig 5D) in individual CS3D-treated tumors relative to CS3M-treated tumors in both 201T and H1975 xenografts. Some variability in protein levels among individual xenografts was observed (Fig 5B & C); the densitometry units for c-Myc protein after normalizing for GAPDH levels ranged from 0.11-0.16 units for CS3D-treated xenografts and 0.25-0.46 units for CS3M-treated tumors in H1975. Similarly, in 201T-derived tumors these measurements ranged from 0.015 - 0.51 units for CS3D and 0.28-0.97 units for CS3M. Even with this variability, the effect across the groups in both cell lines was significant (P<0.05 for H1975 and P<0.042 for 201T xenografts), with a mean decrease of 65% and 70%, respectively.
For mRNA levels in tumor lysates, CS3D produced a 60-70% reduction of c-Myc gene expression relative to GAPDH compared to CS3M (Fig. 5D). Immunohistochemical analysis of tumors harvested after the last day of treatment also revealed a substantial increase in the levels of cleaved caspase-3 (observed predominantly in tumor cells) in response to CS3D. Sections of the H1975 xenografts in response to CS3M showed 10% of fields scoring for high level of cleaved caspase-3 whereas in CS3D-treated xenografts there was an increase to 83% of fields in the high scoring tumor sections (P<0.05). (Fig. 6). Some cleaved caspase 3 staining was localized in nuclei, where caspase-3 is known to be active. Some caspase 3 staining was also evident in fibroblasts.
Figure 6. STAT3 decoy induces caspase-3 cleavage.
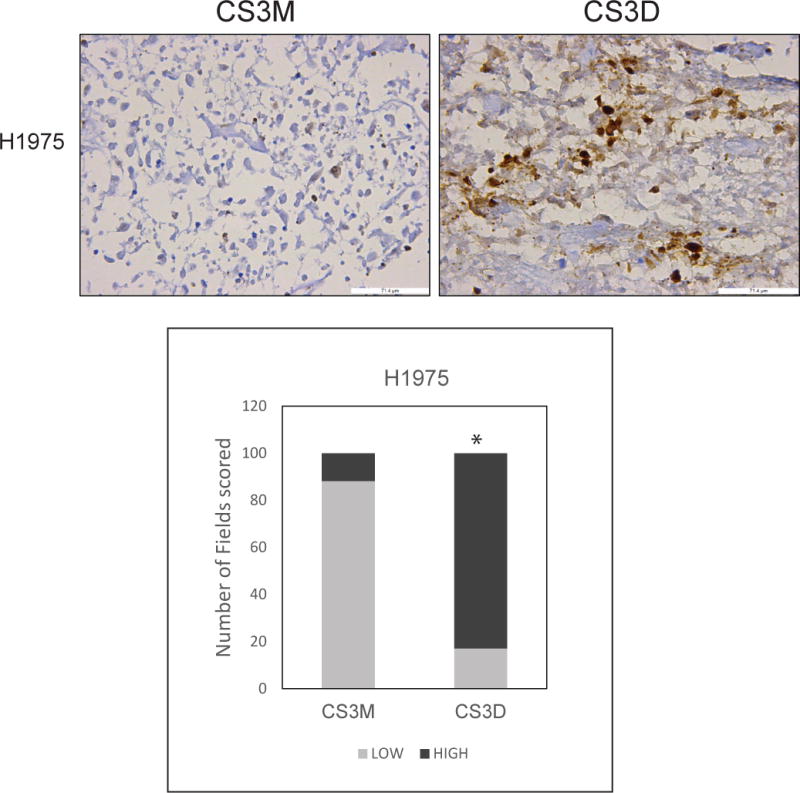
Representative sections of H1975 tumors stained for expression of cleaved caspase-3. H1975-derived xenografts administered daily intravenous injections of either CS3D or CS3M were harvested at the end of treatment (day 14), and expression of cleaved caspase-3 was used as a marker for apoptotic cells. CS3D significantly induces apoptosis relative to CS3M. Similar results was also observed in the 201T derived xenografts.
Short-term CS3D Treatment Shows Anti-Tumor Effects including Reduction in Nuclear p-STAT3
Because tumors treated for several weeks with the STAT3 decoy were largely composed of debris (as illustrated in Supplemental Fig. 3), we examined xenografts after only 5 daily tail vein injections to determine what cellular changes were induced prior to collapse of the intracellular tumor structure. We observed few histological changes in CS3D-treated tumors after only 5 days (Supplemental Fig. 4). Immunohistochemical staining, however, revealed a significant decrease in the proliferative capacity of NSCLC, shown by Ki-67 staining in response to CS3D-treatment (*P<0.001) compared to CS3M-treated tumors after 5 daily injections (Supplemental Fig. 4). We observed a shift from 41% of cells scored as high-grade staining with CS3M treatment to 13% with high-grade staining in the presence of CS3D, with corresponding increases in moderate- and low-grade staining in the STAT3 decoy treated group (Supplemental Fig. 4). Expression levels of p-STAT3 protein (which was mainly observed localized to the nucleus) were likewise significantly suppressed by the STAT3 decoy (*P<0.0001) after 5 days of treatment in vivo compared to mutant decoy (Fig. 7). Frequency of high-grade staining for p-STAT3 differed between CS3D and CS3M treatment groups (23% in CS3D-treated and 51% in CS3M treated, with corresponding increase in low-grade staining in presence of the active decoy (Fig. 7). The p-STAT3 result suggests that multiple doses of the decoy promoted the reduction in p-STAT3 levels in vivo. At this early time point, no cellular debris was observed (Supplemental Fig. 4), suggesting that effects on STAT3 and proliferation occur prior to these histological changes. We also observed no change in NF-κB nuclear protein with the active decoy, suggesting that the decoy does not alter NK-κB movement to the nucleus.
Figure 7. Short-term CS3D-treatment suppresses p-STAT3 levels.
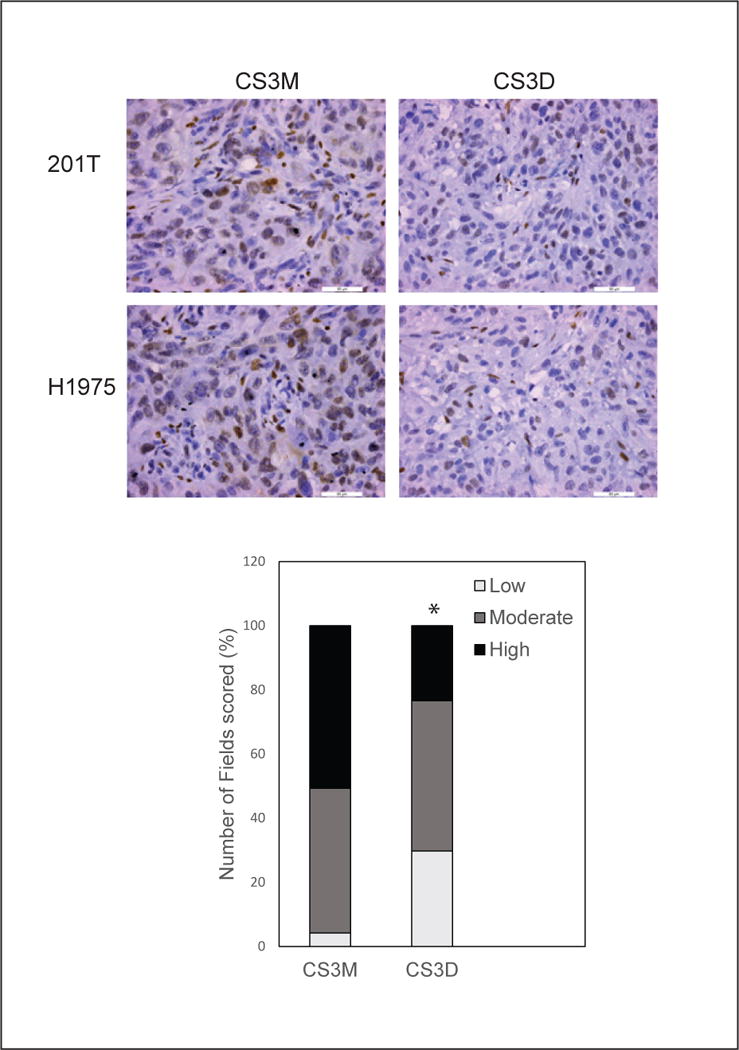
Tumors derived from H1975 xenografts were analyzed by immunohistochemical staining for p-STAT3 after 5 daily iv injections of either STAT3 decoy or mutant decoy. Representative sections of H1975 tumors show a decrease in p-STAT3 levels. Bar graph quantitates the frequency of low-, medium-, and high-grade staining in multiple images; P < 0.001, decoy compared to mutant.Bright field images captured at 40X magnfication
Discussion
STAT3 is a critical mediator of tumor progression in NSCLC. As a transcription factor that regulates the expression of multiple target genes, STAT3 relays signals through multiple tyrosine kinases, including EGFR, IL-6R, and SRC. The key role of STAT3 in mediating proliferation and suppressing apoptosis in NSCLC makes it a prime target for therapeutic intervention (7). Prior STAT3 inhibitors have failed in clinical development due to factors including nonspecificity, low potency, and minimal cell membrane penetrance, limiting their efficacy (11, 12). We examined a novel therapeutic that targets STAT3, a second generation cyclic, double-stranded decoy oligonucleotide that exhibits a long half-life, high potency, and limited toxicity (16). The oligonucleotide decoy was previously found to specifically and competitively bind activated STAT3 protein via its similarity to the STAT3 response element located in the promoter region of the c-fos gene (15), and to reduce expression of STAT3 target genes in head and neck cancer models (15–17). Head and neck cancer cells also responded to the decoy in a xenograft model (16).
Here we demonstrate that the STAT3 decoy produces a robust antitumor effect in NSCLC models known to be resistant to FDA-approved EGFRi therapies. We found that the decoy decreased the viability of NSCLC cells and induced apoptosis, and sensitivity was independent of EGFR mutation status. These effects were specific compared to a mutant version of the double-stranded decoy that is unable to recognize STAT3 protein (15–17), and were demonstrable after a single transfection. In vivo studies illustrated that daily IV injection of the active decoy had strong anti-tumor effects in mouse NSCLC xenografts, accompanied by inhibition of c-Myc protein expression and destruction of the cellular integrity of tumor cells within the xenografts. When tumors were analyzed after only 5 daily treatments, nuclear p-STAT3 protein was greatly reduced and the proliferative state of tumor cells was greatly decreased. Since the phosphorylation step that activates STAT3 is not affected by STAT3 decoy (15), this result suggests one effect of the decoy is to reduce accumulation of p-STAT3 in the nucleus. In cell culture, we observed a decrease in the nuclear pool of STAT3. The cytoplasmic pool of p-STAT3 was reduced as well, with an increase in ubiquitination of p-STAT3 protein following CS3D treatment, which could cause increased degradation of STAT3 dimers when bound to the active decoy. A CS3D-pSTAT3 dimer complex may trigger a ubiquitination process that reduces the amount of active STAT3 available for signaling.
STAT3 is activated by modifications through phosphorylation at tyrosine and serine residues that allow dimerization (11, 12). Small molecule inhibitors of STAT3 that inhibit phosphorylation might be ineffective in blocking the functional activity of STAT3 dimers that do not require phosphorylation to initiate dimerization, and still recognize the STAT3 consensus sequence (13, 14). This strategy also requires a very efficient inhibition of the phosphorylation step, which might limit efficacy. CS3D allows interruption of the function of STAT3 dimers, without requiring the interruption of phosphorylation. Potentially this strategy could be more effective, because it focuses on the active dimeric moieties. We showed that CS3D downregulates the expression of the STAT3 target gene c-Myc in NSCLC cell lines in culture and in residual tumors after systemic administration. The transcriptional activity of STAT3 overlaps with other transcription factors such as STAT1 and NF-κB, but genes controlled predominantly by STAT1 or NF-κB such as IRF7 and IL-8 were unaffected by CS3D. Although c-Myc is a direct STAT3 target gene, it is possible that indirect effects on c-Myc mRNA or protein expression could also be occurring in response to the STAT3 decoy, which we were unable to detect.
Previously we demonstrated that the cyclic double-stranded oligonucleotide showed enhanced stability compared to linear versions (16), and was efficacious after iv injection in HNSCC (16). In addition to lack of toxicity seen with the STAT3 decoy in normal lung cells and in mice bearing NSCLC xenografts, the decoy effects were also specific since the mutant control oligonucleotide was unable to induce anti-tumor effects. The cellular uptake of fluorescently labeled CS3D and CS3M also did not differ, supporting the specificity of the decoy that is designed to bind with selectivity to dimeric STAT3 protein. Imaging analysis also showed that the localization of CS3D mostly stained positively in the cytoplasm, although some was present in the nucleus, suggesting the main effect is to bind to pSTAT3 dimers in the cytoplasm.
The anti-tumor effects of CS3D were comparable in both EGFR wild-type and mutant NSCLC. It has been reported that the constitutively active mutant form of STAT3, STAT3-C, which is dimerized by cysteine-cysteine residues, mediates epithelial cell transformation by promoting anchorage-independent growth (21), suggesting that this phenotype is especially dependent on STAT3 signaling. We found that after a single transfection, CS3D was very effective in suppressing anchorage-independent growth. The inhibition of c-Myc expression in cultured cells and in xenografts also was a strong indicator of CS3D activity, suggesting that the ability to suppress this STAT3 target gene may be critical for its anti-tumor mechanism in NSCLC. c-Myc regulates genes encoding lung cancer stem cell factors such as SOX2 and nanog and cooperates with STAT3 in this regulation (22), further suggesting that c-Myc down-regulation caused by reduction in STAT3 action is important in the anti-tumor action of CS3D in NSCLC, especially in reducing stem cell self-renewal.
As a transcription factor, STAT3 has been generally considered an “undruggable” target. Previous STAT3 oligonucleotide decoys were unstable (23, 24) and small molecule STAT3 inhibitors proved relatively ineffective. The cyclic decoy approach exhibited strong therapeutic activity in NSCLC. Previously published observations with the non-cyclic version of the STAT3 decoy injected intratumorally into HNSCC patients in a Phase 0 trial produced remarkable effects by downregulating STAT3 target genes (16). Our findings support the ability of a systemically administered, double-stranded, cyclic oligonucleotide decoy to attenuate STAT3 signaling in a lung cancer model, leading to tumor growth arrest. We observed no increase in nuclear NF-κB protein in NSCLC xenografts after treatment, suggesting that the STAT3 decoy does not increase NF-κB nuclear translocation, as has been reported in a STAT3 knockout KRAS lung cancer model (25). The effects in NSCLC support the idea that a circularized double-stranded oligonucleotide targeting STAT3 has promise as a therapeutic agent, and may be effective in lung tumors that lack sensitivity to EGFR TKIs. The decoy lacked toxicity in immunocompetent mice (17), and we observed no toxic effects in immunosuppressed animals. Efforts are underway to complete the IND-directed pharmacology and toxicology studies needed to initiate a Phase I clinical trial of cyclic STAT3 decoy.
Supplementary Material
Acknowledgments
This research was funded in part by a philanthropic gift from the 5th District Eagles of Minnesota to the Masonic Cancer Center (to JMS) and by P50 CA097190 (to JRG) from National Cancer Institute and an American Cancer Society Professorship (to JRG). CN was supported by a fellowship from a T32 Cancer Biology Training Grant (T32 CA009138) from National Cancer Institute and by an F31 individual fellowship (F31 CA213982) from National Cancer Institute. JMS was supported by the Frederick & Alice Stark Chair in Pharmacology. AK was supported by an undergraduate summer fellowship from grant R25 CA200508 (to JMS) from the National Cancer Institute.
Abbreviations
- STAT3
signal transducer and activator of transcription 3
- NSCLC
non-small cell lung cancer
- EGFR
epidermal growth factor receptor
- TK
tyrosine kinase
- TKI
tyrosine kinase inhibitor
- CS3D
cyclic STAT3 decoy
- CS3M
cyclic STAT3 mutant decoy
Footnotes
The authors declare no potential conflicts of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics: 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Ward EM, Johnson CJ, Cronin KA, Ma J, Ryerson B, Mariotto A, Lake AJ, et al. Annual Report to the Nation on the Status of Cancer: 1975-2014. Featuring Survival J Natl Cancer Inst. 2017;109(9) doi: 10.1093/jnci/djx030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Torre LA, Siegel RL, Ward EM, Jemal A. Global Cancer Incidence and Mortality Rates and Trends: An Update. Cancer Epidemiol Biomarkers Prev. 2016;25:16–27. doi: 10.1158/1055-9965.EPI-15-0578. [DOI] [PubMed] [Google Scholar]
- 4.Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14:535–46. doi: 10.1038/nrc3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gainor JF, Shaw AT. Emerging paradigms in the development of resistance to tyrosine kinase inhibitors in lung cancer. J Clin Oncol. 2013;31:3987–96. doi: 10.1200/JCO.2012.45.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee H-J, Zhuang G, Cao Y, Du P, Kim H-J, Settleman J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell. 2014;26:207–221. doi: 10.1016/j.ccr.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 7.Dutta P, Sabri N, Li J, Li WX. Role of STAT3 in lung cancer. JAKSTAT. 2015;3 doi: 10.1080/21623996.2014.999503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yao Z, Fenoglio S, Gao DC, Camiolo M, Stiles B, Lindsted T, et al. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc Natl Acad Sci USA. 2010;107:15535–40. doi: 10.1073/pnas.1009472107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao SP, Mark KG, Leslie K, Pao W, Motoi N, Gerald WL, et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest. 2007;117:3846–56. doi: 10.1172/JCI31871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schlessinger K, Levy DE. Malignant transformation but not normal cell growth depend on STAT3. Cancer research. 2005;65:5828–5834. doi: 10.1158/0008-5472.CAN-05-0317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fagard R, Metelev V, Souissi I, Baran-Marszak F. STAT3 inhibitors for cancer therapy: Have all roads been explored? JAK-STAT. 2013;2 doi: 10.4161/jkst.22882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furtek SL, Backos DS, Matheson CJ, Reigan P. Strategies and Approaches of Targeting STAT3 for Cancer Treatment. ACS Chem Biol. 2016;11:308–18. doi: 10.1021/acschembio.5b00945. [DOI] [PubMed] [Google Scholar]
- 13.Timofeeva OA, Chasovskikh S, Lonskaya I, Tarasova NI, Khavrutskii L, Tarasov SG, et al. Mechanisms of unphosphorylated STAT3 transcription factor binding to DNA. J Biol Chem. 2012;287:14192–200. doi: 10.1074/jbc.M111.323899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, et al. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005;65:939–47. [PubMed] [Google Scholar]
- 15.Leong PL, Andrews GA, Johnson DE, Dyer KF, Xi S, Mai JC, et al. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proc Natl Acad Sci USA. 2003;100:4138–43. doi: 10.1073/pnas.0534764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sen M, Thomas SM, Kim S, Yeh JI, Ferris RL, Johnson JT, et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: implications for cancer therapy. Cancer Discov. 2012;8:694–705. doi: 10.1158/2159-8290.CD-12-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sen M, Paul K, Freilino ML, Li H, Li C, Johnson DE, et al. Systemic administration of a cyclic signal transducer and activator of transcription 3 (STAT3) decoy oligonucleotide inhibits tumor growth without inducing toxicological effects. Mol Med. 2014;20:46–56. doi: 10.2119/molmed.2013.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klein JD, Sano D, Sen M, Myers JN, Grandis JR, Kim S. STAT3 oligonucleotide inhibits tumor angiogenesis in preclinical models of squamous cell carcinoma. PLoS One. 2014;9 doi: 10.1371/journal.pone.0081819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siegfried JM, Krishnamachary N, Gaither Davis A, Gubish C, Hunt JD, Shriver SP. Evideince for autocrine actions of neuromedin B and gastrin-releasing peptide in non-small cell lung cancer. Pulm Pharmacol Ther. 1999;12:291–302. doi: 10.1006/pupt.1999.0210. [DOI] [PubMed] [Google Scholar]
- 20.Stabile LP, Gaither AL, Gubish CT, Hopkins TM, Luketich JS, Christie NC, et al. Human non-small lung tumors and cells derived from normal lung express both estrogen receptor α and β and show biological responses to estrogen. Cancer Res. 2002;62:2141–50. [PubMed] [Google Scholar]
- 21.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an Oncogene. Cell. 1999;99:238–239. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 22.Kidder BL, Yang J, Palmer S. Stat3 and cMyc genome-wide promoter occupany in embroyonic stem cells. PLOS one. 2008;3 doi: 10.1371/journal.pone.0003932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castanotto D, Stein CA. Antisense oligonucleotides in cancer. Curr Opin Oncol. 2014 Nov;26:584–589. doi: 10.1097/CCO.0000000000000127. [DOI] [PubMed] [Google Scholar]
- 24.Dean NM, Bennett CF. Antisense oligonucleotide-based therapeutics for cancer. Oncogene. 2003;22:9087–96. doi: 10.1038/sj.onc.1207231. [DOI] [PubMed] [Google Scholar]
- 25.Grabner B, Schramek D, Mueller KM, Moll HP, Svinka J, Hofmann T, et al. Disruption of STAT3 signaling promotes KRAS-induced lung tumorigenesis. Nature Communications. 2015;6:6285. doi: 10.1038/ncomms7285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.