

Abstract
Although successes in cancer immunotherapy have generated considerable excitement, this form of treatment has been largely ineffective in patients with pancreatic ductal adenocarcinoma (PDA). Mechanisms that contribute to the poor antitumor immune response in PDA are not well understood. Here, we demonstrated that cytokine IL35 is a major immunosuppressive driver in PDA and potentiates tumor growth via the suppression of endogenous antitumor T-cell responses. The growth of pancreatic tumors in mice deficient for IL35 was significantly reduced. An analysis of tumor-infiltrating immune cells revealed a role for IL35 in the expansion of regulatory T cells and the suppression of CD4+ effector T cells. We also detected a robust increase in both the infiltration and activation of cytotoxic CD8+ T cells, suggesting that targeting IL35 may be an effective strategy to convert PDA from an immunologically ‘cold’ to ‘hot’ tumor. Although PDA is typically resistant to anti–PD-1 immunotherapy, we demonstrated robust synergistic reduction in tumor growth when IL35 deficiency was combined with anti–PD-1 treatment. These findings provide new insight into the function of IL35 in the pathogenesis of pancreatic cancer and underscore the potential significance of IL35 as a therapeutic target for use in combination immunotherapy approaches in this deadly malignancy.
Keywords: pancreatic cancer, IL35, Kras, immunotherapy
Introduction
Pancreatic ductal adenocarcinoma (PDA) carries one of the worst prognoses of human cancers. It is a disease characterized by late diagnosis and a poor response to therapy (1–3). The formation of PDA is accompanied by changes in stromal composition and dampened immune surveillance, which are now recognized as some of the major drivers in PDA tumor evolution and resistance to therapy (4–8). Components of the PDA tumor microenvironment (TME) have been described in human patient samples and mouse models of PDA and include infiltrating leukocytes from both the lymphoid and myeloid lineages (9, 10). Tumor-infiltrating immune cells with known immunosuppressive capabilities, such as tumor-associated macrophages and myeloid-derived suppressor cells, as well as regulatory T cells (Tregs) and B cells, play important roles in pancreatic tumor growth (11–16). These cells are thought to contribute to an ineffective initial antitumor response and create barriers for immunotherapeutic approaches in established tumors, although the mechanisms by which immune cells in the PDA TME operate in successful establishment of tumor growth remain to be fully understood.
We previously demonstrated that interleukin-35 (IL35)-producing regulatory B cells (Bregs) constitute part of the PDA TME (13). Although B cells can support tumor growth in an IL35-dependent manner, the mechanism of IL35 function in pancreatic cancer remains unclear. IL35 belongs to the IL12 family of cytokines and is formed by the heterodimerization of the subunits p35 and Ebi3 (17). IL35 expression is induced under conditions of elevated inflammatory signaling, and studies in lung cancer, leukemia, lymphoma, colorectal, and pancreatic cancers have documented elevated IL35 and predicted poor patient outcome (18–23). To date, the major stromal sources of IL35 were shown to be T cells (predominantly Tregs) and B cells (13, 24–28). The mechanisms that IL35 acts upon in cancer have not been well established. In a model of melanoma, IL35 promoted tumor growth via enhanced accumulation of myeloid-derived suppressor cells and increased angiogenesis (29), whereas in a study in melanoma and colon cancer models, IL35 expression in Tregs was linked to the exhaustion of CD8+ effector T cells (30). It is likely that depending on disease setting, IL35 may impact functionality of diverse immune cell populations (17, 31).
In this study, we demonstrated that IL35 promotes pancreatic tumor growth by antagonizing endogenous antitumor T-cell immune responses. Using established mouse models of PDA combined with deficiency for IL35, we demonstrated acute dependency of PDA growth on IL35. IL35 enhanced the Treg response and downregulated endogenous effector T-cell responses. We also found that deficiency in IL35 renders PDA sensitive to anti–PD-1 immunotherapy, suggesting that IL35 expression in PDA confers resistance not only to endogenous antitumor responses but also to current immunotherapeutic approaches.
Materials and Methods
Animal models
All mouse protocols were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the University of North Carolina at Chapel Hill. Animals were maintained in a specific pathogen-free facility. Six to eight-week old wild-type C57Bl/6J mice were purchased form the Charles River Laboratories (stock #027). Six to eight-week old p40−/− (stock #002693), Il10−/− (stock #002251), p35−/− (stock #002692), Il27ra−/− (stock #018078), and Ebi3−/− (stock #008691) mouse strains were purchased from the Jackson Laboratory. All of these strains were on C57Bl/6J background. Both male and female mice were used for orthotopic injections of PDA cells. Briefly, for injection of cancer cells into the pancreas, mice were anesthetized using a ketamine (100 mg/kg)/ Xylazine (10 mg/kg) cocktail administered via intraperitoneal injection. The depth of anesthesia was confirmed by verifying an absence of response to toe pinch. An incision in the left flank was made, and 75,000 KPC cells in ice-cold PBS mixed at 1:1 dilution with Matrigel (#354234, Corning) in a volume of 50 μl were injected using a 28-gauge needle into a tail of the pancreas. The wound was closed in two layers, with running 5–0 Vicryl RAPIDE sutures (Ethicon) for the body wall, and 5–0 PROLENE sutures (Ethicon) for the skin. All animals were given the pain reliever buprenorphine (0.1 mg/kg) subcutaneously once, directly after the conclusion of surgical procedure. Mice were euthanized by carbon dioxide-induced narcosis at three weeks post injection of cancer cells. Tumors were weighed and measured using digital caliper (VWR) at the endpoint. Part of the tumor tissue was fixed in 10% buffered formalin phosphate (Fisher) and reserved for histology, and the rest was processed for flow cytometry as described below in the ‘Lymphocyte isolation’ section. Spleens were harvested for flow cytometry analysis as also described in ‘Lymphocyte isolation’ section below. The LSL-KrasG12D/+, LSL-Trp53R172H/+, and p48Cre/+ strains have been described previously (32–35). These mouse strains were bred at the Charles River Laboratories for Dr. Bar-Sagi, prior to being transferred to the University of North Carolina at Chapel Hill.
Cell lines
Murine PDA cell lines 4662 or 2173 were derived from primary pancreatic tumors of LSL-KrasG12D/+; LSL-Trp53R172H/+; p48Cre/+ (KPC) mice, as reported in Bayne et al. (12). KPC cells were a kind gift from Dr. Vonderheide and were derived from C57Bl6/J background strain (12). GFP-labeled 4662 and 2173 KPC lines were generated as previously described (11). Cell lines used for implantation studies were tested and confirmed to be mycoplasma free (MycoAlert Plus Mycoplasma detection kit, Lonza), and were used at <10 passages. All cell lines were maintained at 37°C and 5% CO2 in complete DMEM, which contained Dulbecco’s Modified Eagle medium (Gibco) supplemented with 10% FBS (Sigma), 1% L-glutamine, and gentamicin (50 μg/mL; Thermo Scientific).
Immunohistochemistry
Mouse splenic and tumor tissues were fixed in 10% buffered formalin (Fisher Scientific) for 48 hours and embedded in paraffin at UNC’s Animal Histopathology and Laboratory Medicine Core. Six micron sections were deparaffinized and rehydrated. A solution of 1% hydrogen peroxide (stock of 30% hydrogen peroxide, Sigma) in methanol at room temperature for 10 minutes was used to quench endogenous peroxidase activity. Antigen retrieval was done in a 10 mM sodium citrate plus 0.05% Tween-20 solution (pH 6.1) for 15 minutes in a microwave oven. Blocking was performed for 1 hour at room temperature in a solution of 10% goat serum, 10 mM Tris-HCl, 0.1M magnesium chloride, 1% BSA, and 0.5% Tween-20. Sections were incubated with primary rat anti-CD8α (clone 53–6.7, BD Pharmingen) diluted in 2% BSA/PBS (final concentration of 2.5 μg/mL) overnight at 4°C. Secondary biotinylated goat anti-rat (Vector Laboratories) was diluted in 2% BSA/PBS (final concentration of 3.75 μg/mL) and incubated for 1 hour at room temperature. Tertiary ABC solution was prepared according to manufacturer’s instructions (Vectastain ABC kit, Vector Laboratories) and incubated for 45 minutes at room temperature. Sections were developed using a 3,3’-diaminobenzidine tetrahydrochloride kit (DAB peroxidase substrate kit, Vector Laboratories). Slides were then counterstained with Harris hematoxylin (Sigma), dehydrated, and mounted with DPX mounting media (Sigma). Slides were scanned using the Aperio ScanScope XT at UNC’s Translational Pathology Lab, and slides were examined and images were generated using ImageScope Viewer (Leica Biosystems).
Immunofluorescence (IF) on human samples
For the purposes of analyzing CD8+ T-cell and IL35+ immune cell infiltration, we examined 11 samples containing PDA lesions. Samples consisted of 5 μm sections that were cut from FFPE blocks provided by the Tissue Pathology Laboratory (TPL) of the UNC School of Medicine and were obtained after informed written consent. This study was conducted in accordance with the Declaration of Helsinki. All samples were anonymized prior to being transferred to the investigator’s laboratory and, therefore, met exempt human subject research criteria. Samples were selected on the basis of patient diagnosis with PDAC. Triple IF (3-plex IF) stains were carried out in the Leica Bond-Rx fully automated staining platform (Leica Biosystems Inc., Norwell, MA). All steps were carried out at room temperature, except for the epitope retrieval, which was done at 100°C. Slides were dewaxed in Bond™ Dewax solution (#AR9222) and hydrated in Bond Wash solution (#AR9590). Epitope retrieval for all targets was done for 20 minutes in Bond-epitope retrieval solution 1, pH6.0 (#AR9661) or solution 2, pH9.0 (#AR9640). The epitope retrieval was followed with 10 minutes of endogenous peroxidase blocking using Bond peroxide blocking solution (#DS9800). Each set of primary and secondary antibody staining was completed sequentially, followed by incubation with TSA reagent for 15 minutes. Stained slides were then counterstained with Hoechst 33258 (#H3569) and mounted with ProLong® Diamond Antifade Mountant (#P36961, Life Technologies, Carlsbad, CA). Positive and negative controls (no primary antibody) and single stain controls were made for the 3-plex IF, with one primary antibody omitted to make sure that cross reactivity between the antibodies did not occur. The following primary antibodies were used: CD20 (1:100, clone MJ1, Leica), CD8 (1:100, clone 4B11, Leica), CD4 (1:100, clone 4B12, Leica), Ebi3 (1:100, clone HPA046635, Sigma), CK (1:500, clone Zo622, Agilent/DAKO), and IL12p35 (1:100, clone HPA001886, Sigma). For secondary: EnVision+ System- HRP, Labelled Polymer, Anti-mouse #K400 (1:50, Agilent/DAKO), EnVision+ System- HRP, Labelled Polymer, Anti-mouse #K4001 (1:50, Agilent/DAKO), ImmPRESS HRP Anti-Rabbit Polymer #MP-7401–15 (1:50, Vector Labs). CD8+ T cells in the tumor cell nests (i.e. directly adjacent to or in contact with tumor cells) and Ebi3+ immune cells were counted per 10x field of view (FOV), counting 3–6 FOVs per tumor sample (n=11 tumor samples). Average value of Ebi3+ cells/FOV across all tumor samples was estimated as 20 Ebi3+ cell/FOV. FOVs with the number of Ebi3+ cells < 20 were classified as Ebi3+ low and FOVs with the number of Ebi3+ cells ≥ 20 were classified as Ebi3+ high.
Lymphocyte isolation
Single-cell suspensions were prepared from tumors, spleens, and pancreatic lymph nodes. Spleens were mechanically disrupted using a plunger end of a 10ml syringe and resuspended in 1% FBS/PBS. For isolation of tumor-infiltrating lymphocytes, tumor tissue was minced into 1–2mm pieces and digested with collagenase IV (1.25 mg/mL; #LS004188, Worthington), 0.1% trypsin inhibitor from soybean (# T9128, Sigma), hyaluronidase (1 mg/mL; # LS 002592, Worthington), and DNase I (100 μg/mL; # LS002007, Worthington) in complete DMEM for 30 minutes at 37°C. Cell suspensions were passed through a 70 μm strainer and resuspended in RPMI media (Gibco). Lymphocytes were isolated from processed tumor tissues by OptiPrep (Sigma) density gradient centrifugation. MACS isolation of total lymphocytes (MACS Miltenyi #130–052-301) was performed on the lymphocyte-enriched fraction according to Miltenyi’s protocol, and the purity of MACS-enriched B cells and T cells was >85%. Cell sorting using a BD FACS ARIA II sorter was performed to isolate Bregs, conventional B cells (Bcon), and CD4+ and CD8+ T cells, and >95% purity of sorted cells was achieved.
Flow cytometry
For ex vivo stimulation, 100,000 cells were incubated with PMA (50 ng/mL; Sigma, #P8139) and ionomycin (200 ng/mL; Sigma, #I0634) in the presence of 1XGolgistop Brefeldin A (Biolegend) in complete RPMI medium for 5 hours at 37°C before staining. Cells were washed and blocked with anti-CD16/CD32 (Fc Block, BD Biosciences, 0.1 μg per 100,000 cells) for 5 minutes on ice and then stained with labeled antibodies against surface markers or cytokines on ice for 30 minutes in FACS buffer (PBS with 3% FCS and 0.05% sodium azide). For intracellular staining, cells were fixed, permeabilized and stained with labeled intracellular antibodies using the BD cytofix/cytoperm kit (BD Biosciences, Cat. No. 554714). Intracellular staining for Foxp3 was performed using a Foxp3 staining kit (eBioscience, Cat. No. 00–5523). The following monoclonal antibodies directed against mouse antigens were used for flow cytometry: Biolegend - CD45 (clone 30-F11), CD11b (clone M1/70), F4/80 (clone BM8), Ly6G (clone 1A8), Ly6C (clone HK1.4), CD206 (clone C068C2), MHCII (clone M5/114.15.2), CD3 (clone 17A2), CD4 (clone GK1.5), IFNγ (clone XMG1.2), TNFα (clone MP6-XT22), Foxp3 (clone FJK-16S), CD8 (clone 53–6.7), IL12p40 (clone C15.6), IL27p28 (clone MM27–7B1), CD19 (clone 6D5), CD45RB (clone C363–16A), CD21 (clone 7E9); Ebioscience - IL35p35 (clone 4D10p35); R&D - IL35EBi3 (clone 355022); BD Biosciences - CD1d (clone 1B1). Viability of cells were determined by staining with either 7-aminoactinomycin (Biolegend) or Live/Dead Aqua cell stain kit (Life technologies). All samples were acquired on LSR II and LSRII Fortessa (BD Bioscience) at UNC Flow Cytometry Core Facility and analyzed by FlowJo version 10.2 (Treestar, Inc.).
Anti–PD-1 treatment and CD8+ T-cell depletion
Mice were orthotopically injected into the pancreas with KPC cells. On day 7 post-injection, mice were intraperitoneally administered either 200 μg anti–PD-1 (BioXCell, clone RMP1–14) or IgG control (BioXCell, clone 2A3) diluted in 100 μL of sterile PBS. Three doses of antibody were given in total, on days 7, 9, and 11 post injection of KPC cells. For CD8+ T-cell depletion studies, 200 μg of anti-CD8 (BioXCell, clone 53–6.7) or an IgG isotype control (BioXCell, clone 2A3) diluted in 100 μl of sterile PBS were administered intraperitoneally daily starting 3 days prior to tumor cell injection and twice a week after tumor cell injection. Efficiency of CD8+ T-cell depletion was assessed by flow cytometry.
Statistical analysis
At least 9–21 mice were used in each group, with a minimum of 3 mice in each group per experiment, and the experiments were repeated a minimum 3 times to validate reproducibility. Group means were compared with Student’s t tests. Significance in variations between two groups was determined by an unpaired student t test (two-tailed). Statistical analyses were performed using GraphPad Prism software (version 7.0d), and data is presented as mean±SEM. p< 0.05 was considered statistically significant.
Results
IL35 is expressed by multiple immune cell subsets and promotes PDA growth
To understand the functional significance of IL35 in pancreatic tumor growth, we first investigated the source of IL35 expression in the tumor microenvironment. To do this, we employed well-characterized mouse models of PDA: a spontaneous model of pancreatic tumorigenesis KrasG12D/+;p48Cre/+ (KC), as well as KrasG12D;Trp53R172H;p48Cre/+ –expressing primary mouse pancreatic cancer cells (KPC), which give rise to pancreatic tumors upon orthotopic injection into the pancreata of syngeneic mice (12, 13, 32, 33, 36). We assessed the frequency of IL35+ immune cells using flow cytometry-based detection of cells that were double positive for both p35 and Ebi3 subunits (Fig. 1A). At 3 weeks post-orthotopic injection of KPC cells into the pancreata of wild-type (WT) mice, the abundance of IL35+ cells in tumors were similar to that of 4-month old KC mice (Fig. 1A-C). Both models displayed increased intrapancreatic and splenic accumulation of IL35+ cells compared with normal WT pancreas and spleen, which is in agreement with previously reported increases in IL35+ in cancer (18–23). We detected two dominant immune cell populations expressing IL35: CD19+ B cells and CD4+ T cells (Fig. 1A-C). Consistent with our previous report, the expression of IL35 in B cells was restricted primarily to Bregs (identified here as CD19+CD1dhiCD21hiCD5+ cells) (Fig. 1D) (13). Among CD4+ T cells, the expression of IL35 was primarily restricted to CD4+Foxp3+ and CD4+Foxp3– T cells (Fig. 1E and F). Altogether, these observations suggest that in the context of pancreatic cancer, CD4+ T cells and Bregs are the primary sources of the cytokine IL35.
Figure 1. IL35 is upregulated in CD4+ T cells and B cells in PDA.
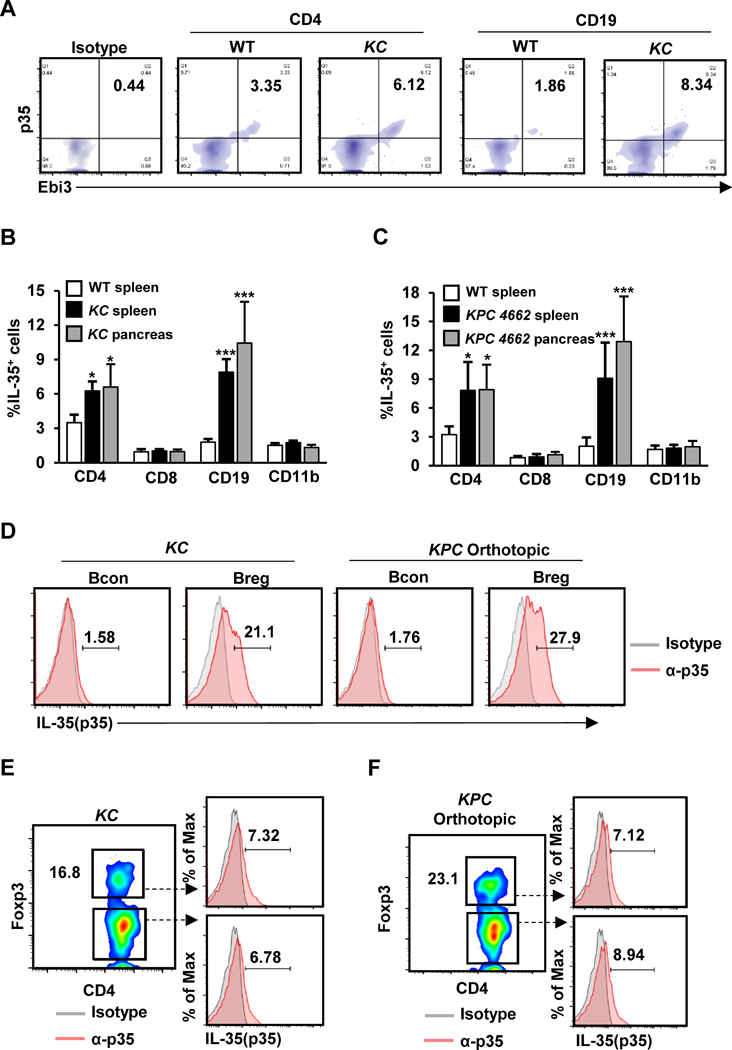
(A) Representative flow cytometry plots of CD4+ and CD19+ cells isolated from the spleens of WT or KC mice at 4 months of age. Cells were processed for intracellular staining with anti-p35 and anti-Ebi3. Percent of p35+Ebi3+CD4+ T cells and p35+Ebi3+CD19+ B cells is indicated. (B) Percentage of IL35+ splenic T cells, B cells, or myeloid cells in WT (n=12) or splenic and intratumoral IL35+ T cells, B cells, or myeloid cells KC mice (n=12). (C) Percentage of IL35+ splenic T cells, B cells, or myeloid cells in WT or splenic and intratumoral IL35+ splenic T cells, B cells, or myeloid cells in mice orthotopically injected with KPC 4662 cells and collected 3 weeks post-tumor cell injection. (D) Representative flow cytometry plots of intratumoral B conventional (Bcon; CD19+CD21+CD5–CD1d–) and B regulatory cells (Breg; CD19+CD21hiCD5+CD1dhi) from KC mice at 4 months of age or mice orthotopically injected with KPC cells and collected 3 weeks post-tumor cell injection. Cells were analyzed by staining with anti-p35 antibody. Percent of p35+ Bcon or Breg cells is indicated. (E) Representative flow cytometry plots of intratumoral CD4+Foxp3– T cells and CD4+Foxp3+ Treg cells from KC mice at 4 months of age. Cells were analyzed by staining with anti-p35 antibody. Percent of p35+ CD4+Foxp3– or Treg cells is indicated. (F) Representative flow cytometry plots of intratumoral CD4+Foxp3– T cells and Tregs from mice orthotopically injected with KPC cells and collected 3 weeks post-tumor cell injection. Cells were analyzed by staining with anti-p35 antibody. Percent of p35+ CD4+Foxp3– or Treg cells is indicated. Error bars indicate SEM. p-values were calculated using Student’s t-test (unpaired, two-tailed); NS – not significant, *p<0.05, ***p<0.001. Data represents 3 independent experiments.
Because several immune cell types could produce IL35, we elected to use mouse models that completely lack IL35 in all host cells in order to examine the role of host-derived IL35 in tumor growth. To that end, we used p35−/− and Ebi3−/− knockout animals, both of which lack IL35. GFP-labeled KPC 4662 cells were implanted into the pancreata of p35−/−, Ebi3−/−, or control syngeneic WT animals. The analysis of pancreata at 3 weeks post-injection revealed a significant reduction in tumor size in p35−/− and Ebi3−/− mice compared with WT mice (Fig. 2A and B). These results were recapitulated using additional syngeneic pancreatic cell line KPC 2173 (Fig. 2C). In addition to IL35, immunosuppressive cytokine IL10 is also produced by Bregs and Tregs in the PDA TME (13, 37). We previously showed that in contrast to IL35, the production of IL10 by B cells had no effect on pancreatic tumorigenesis, suggesting that the IL35 signaling axis is preferentially used in PDA (31). Here, KPC cells implanted into Il10−/− mice grew to a size comparable to that of KPC cells implanted into WT mice, suggesting that in the context of IL35 upregulation, absence of IL10 is not sufficient to alter pancreatic tumor growth (Fig. 2A-C).
Figure 2. IL35 regulation of tumor growth is accompanied by suppression of CD4+ effector T-cell activity and expansion of Tregs.
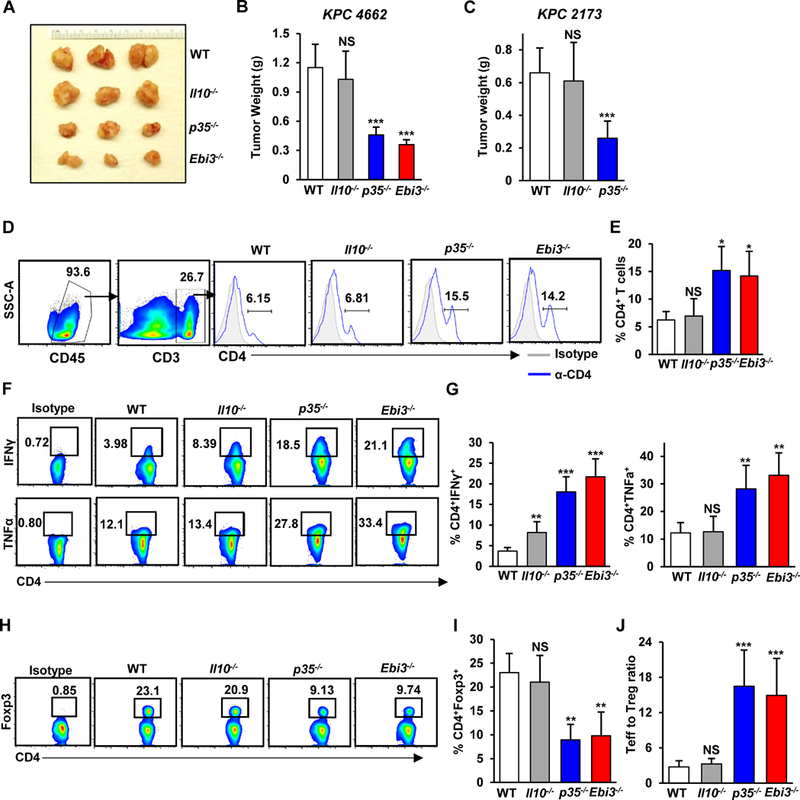
(A) Representative images of pancreatic tumors from WT, Il10−/−, p35−/−, and Ebi3−/− mice orthotopically injected with KPC 4662 cells and collected at 3 weeks post-injection of tumor cells. (B) Quantification of tumor weights from WT, Il10−/−, p35−/−, and Ebi3−/− (N=12; n=3/group) mice. (C) Quantification of tumor weights from WT (n=9), Il10−/− (n=9), and p35−/− (n=9) mice orthotopically injected with KPC 2173 cells and collected 3 weeks post-tumor cell injection. (D) Representative flow cytometry plots of gating strategy for identifying CD4+ T cells and frequency of intratumoral CD4+CD25– effector T cells isolated from WT, Il10−/−, p35−/−, and Ebi3−/− mice orthotopically injected with KPC 4662 cells and collected 3 weeks post-injection of tumor cells. Percent of CD45+CD4+ cells is indicated. (E) Quantification of the frequency of CD45+CD4+ T cells from WT, Il10−/−, p35−/−, and Ebi3−/− (n=3/group) mice. (F) Representative flow cytometry plots of CD4+IFNγ+ and CD4+TNFα+ T cells isolated from WT, Il10−/−, p35−/−, and Ebi3−/− mice orthotopically injected with KPC 4662 cells and collected 3 weeks post-injection of tumor cells. (G) Quantification of the frequency of CD4+IFNγ+ T cells (left graph, % of CD4+ T cells) and CD4+TNFα+ T cells (right graph, % of CD4+ T cells) from WT, Il10−/−, p35−/−, and Ebi3−/− (n=3/group) mice. (H) Representative flow cytometry plots of intratumoral CD4+Foxp3+ Treg cells isolated from WT, Il10−/−, p35−/−, and Ebi3−/− mice. (I) Quantification of the frequency of CD4+Foxp3+ (% of CD4+ T cells) from WT, Il10−/−, p35−/−, and Ebi3−/− (n=3/group) mice. (J) Mean CD4+ effector T cell to Treg ratio was calculated based on the percent positive lymphocyte population determined by flow cytometry. Error bars indicate SEM. p-values were calculated using Student’s t-test (unpaired, two-tailed); NS – not significant, *p<0.05, **p<0.01, ***p<0.001. Data represents 3–4 independent experiments.
IL35 modifies effector CD4+ T-cell and regulatory T-cell responses in PDA
To understand the underlying mechanism for reduced tumor growth in the absence of IL35, we examined whether IL35 contributes to the activation or recruitment of effector CD4+ T cells and Tregs. Deficiency in IL35 resulted in significantly increased tumor-infiltrating effector CD4+ T cells (Fig. 2D and E). We also observed significant increases in the frequency of intratumoral IFNɣ+ and TNFα+ CD4+ effector T cells in p35−/− and Ebi3−/− mice compared to control animals (Fig. 2F and G). We then analyzed the frequencies of intratumoral CD4+Foxp3+ Tregs. As shown in Fig. 2H and I, we observed a significant reduction in the frequency of tumor-infiltrating Tregs in animals deficient for IL35. The ratio of CD4+ effector T cells to Tregs was also significantly increased in the setting of IL35 deficiency (Fig. 2J). We did not observe significant differences in abundance or polarization of myeloid cells in the absence of IL35 (Supplementary Fig. S1A-F). Altogether, these data suggest that IL35 expression in PDA dampens the activity of effector CD4+ T cells and promotes the recruitment and/or proliferation of Tregs, overall contributing to the suppression of antitumor immune responses.
Because p35−/− and Ebi3−/− also lack cytokines IL12 and IL27, we assessed both of their expression in the PDA TME and functional relevance to pancreatic tumor growth. Flow cytometric analysis of immune cells in KC mice using antibodies against p40 and p28 subunits showed no appreciable expression of IL12 and IL27, respectively, in B and T lymphocytes (Supplementary Fig. S2A-D). Expression of p40 and p28 subunits in CD11b+ cells was reduced further in the cancer-associated microenvironment, which is consistent with the notion that pro-inflammatory functions of macrophages are suppressed in cancer (Supplementary Fig. S2A-D). We observed minor differences in tumor burden when KPC cells were injected into p40−/− animals, consistent with the notion that IL12 is known to exhibit mostly pro-inflammatory effector function (Supplementary Fig. S3A and B). IL27, however, has been previously linked to immunosuppression (17). To ascertain that the reduced tumor growth observed in IL35 null animals is not attributed to a loss of IL27 signaling, we implanted KPC cells into Il27ra−/− mice. As shown in Fig. 3A and B, we did not observe differences in tumor growth between control WT and Il27ra−/− animals, suggesting that IL27 signaling is not essential for potentiating immune responses in PDA. Consistent with our observation that the loss of IL12 or IL27 signaling was not consequential for tumor growth, we did not observe significant differences in effector T-cell infiltration or the activation or recruitment of Tregs to tumors in p40−/− or Il27ra−/− animals (Fig. 3C-G and Supplementary Fig. S3C-F). These observations suggest that the tumor growth effects observed in p35−/− and Ebi3−/− mice were due to IL35 deficiency.
Figure 3. IL27 signaling does not confer immunosuppression in PDA.
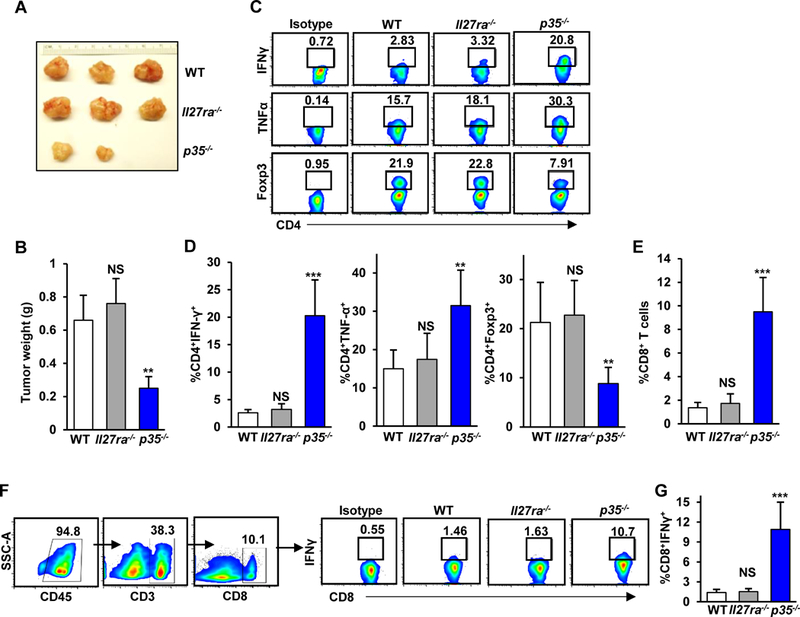
(A) Representative images of pancreatic tumors from WT, Il27ra−/−, and p35−/− mice orthotopically injected with KPC 4662 cells and collected at 3 weeks post-injection of tumor cells. (B) Quantification of tumor weights from WT, Il27ra−/−, and p35−/− (n=3/group) mice. (C) Representative flow cytometry plots of intratumoral CD4+IFNγ+, CD4+TNFα+, and CD4+Foxp3+ T cells isolated from WT, Il27ra−/−, and p35−/− mice. (D) Quantification of the frequency of intratumoral CD4+IFNγ+ T cells (left graph, % of CD4+ T cells), CD4+TNFα+ T cells (middle graph, % of CD4+ T cells), or CD4+Foxp3+ regulatory T cells (right graph, % of CD4+ T cells) from WT, Il27ra−/−, and p35−/− (n=3/group) mice. (E) Quantification of the frequency of intratumoral CD45+CD8+ T cells from WT, Il27ra−/−, and p35−/− (n=3/group) mice determined by flow cytometry. (F) Representative gating strategy for sorting CD8+ T cells and flow cytometry plots of intratumoral CD8+IFNγ+ T cells isolated from WT, Il27ra−/−, and p35−/− mice. Percent of CD8+ T cells is indicated. (G) Quantification of the frequency of intratumoral CD8+IFNγ+ T cells (% of CD8+ T cells) from WT, Il27ra−/−, and p35−/− (n=3/group) mice. Error bars indicate SEM. p-values were calculated using Student’s t-test (unpaired, two-tailed); NS – not significant, **p<0.01, ***p<0.001. Data represents 3 independent experiments.
IL35 deficiency increases CD8+ effector T-cell activity and efficacy of immunotherapy
Because we observed decreased Treg infiltration and concomitant improved function of CD4+ effector T-cell responses in the absence of IL35, we asked whether antitumor CD8+ T-cell responses might be affected as well. To this end, we quantified the extent of intratumoral infiltration and the activation of CD8+ T cells. A reduction in tumor growth in p35−/− and Ebi3−/− mice correlated with a significant increase in infiltrating CD8+ cytotoxic T cells (Fig. 4A and B). The ratio of CD8+ T cells to Tregs was significantly increased in the absence of IL35 (Fig. 4C). Tumor-infiltrating CD8+ T cells in IL35-deficient mice showed significant increases in IFNγ production compared with WT or Il10−/− mice, indicating an increase in the activation status of these cells (Fig. 4D and E). To interrogate the relationship of IL35+ T cells, B cells, and CD8+ T-cell infiltration in human disease, we examined primary tumor samples of patients with PDA. Using immunofluorescence, we detected both B cell– and T cell–specific expression of IL35 in tumors (Fig. 4F and Supplementary Fig. S4A and B). Consistent with the idea that IL35 may suppress CD8+ T-cell infiltration and activity, we found that presence of IL35+ immune cells negatively correlated with CD8+ T-cell infiltration into tumor cell nests (Fig. 4G and Supplementary Fig. S4C). These results suggest that IL35 in the PDA TME acts to restrain the infiltration and activation of tumor-directed CD8+ T cells.
Figure 4. IL35 suppresses cytotoxic CD8+ T-cell infiltration and activity in PDA.
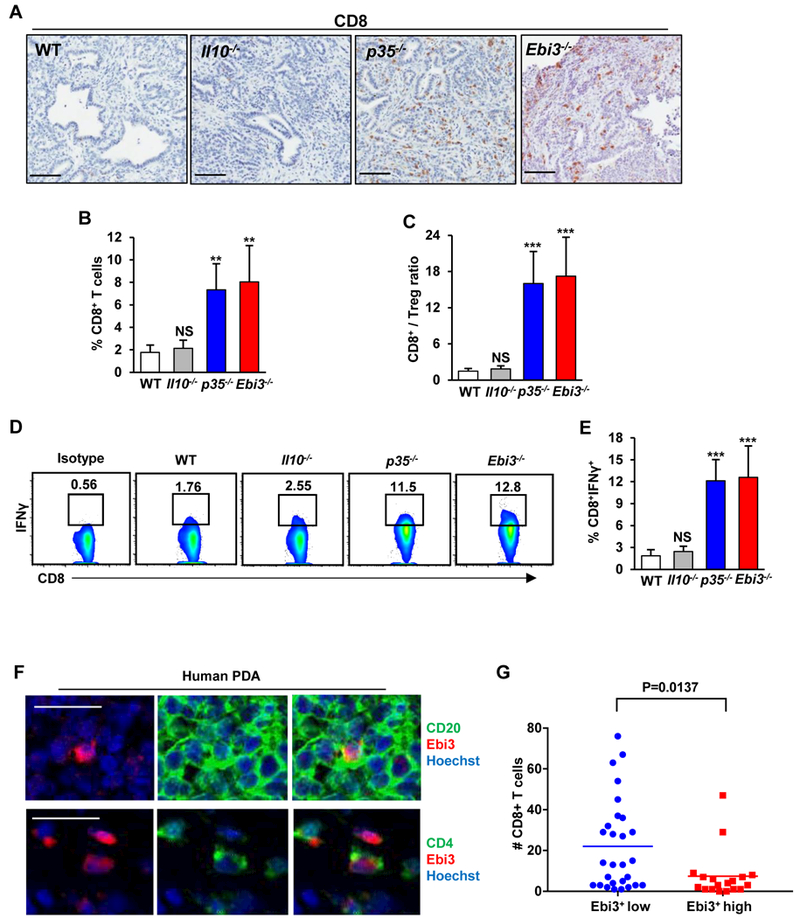
(A) Immunohistochemical detection of CD8+ T cells by DAB in tumor tissues from WT, Il10−/−, p35−/−, and Ebi3−/− mice orthotopically injected with KPC 4662 cells and collected at 3 weeks post-injection of tumor cells. Scale bars: 100 μm. (B) Quantification of the frequency of tumor infiltrating CD45+CD8+ T cells from WT, Il10−/−, p35−/−, and Ebi3−/− (n=3/group) mice determined by flow cytometry. (C) Mean CD8+ cytotoxic T cell to Treg ratio was calculated based on percent positive lymphocyte population determined by flow cytometry. (D) Representative flow cytometry plots of intratumoral CD8+IFNγ+ T cells isolated from WT, Il10−/−, p35−/−, and Ebi3−/− mice. Percent of CD8+ T cells is indicated. (E) Quantification of frequency of CD8+IFNγ+ T cells (% of CD8+ T cells) from WT, Il10−/−, p35−/−, and Ebi3−/− (n=12) mice. (F) Representative immunofluorescence staining. Top: Ebi3 (red) and CD20 (green); Bottom: Ebi3 (red) and CD4 (green) in samples of human PDA. Scale bars: 25 μm. (G) Number of CD8+ T cells in tumor cell nests as a function of low vs. high numbers of Ebi3+ immune cells. Each point is the number of tumor cell adjacent CD8+ T cells per 10x field of view (FOV). Data was derived from counting 3–6 FOV per tumor sample (n=11 tumor samples). Error bars indicate SEM. p-values were calculated using Student’s t-test (unpaired, two-tailed); NS – not significant, **p<0.01, ***p<0.001. Data represents 3–4 independent experiments.
Studies suggest that immunosuppression and poor CD8+ T-cell infiltration may be the primary cause of resistance to immunotherapy in pancreatic cancer (36, 38). Pre-existing intratumoral infiltration of CD8+ T cells has been found to be the best correlative marker for successful response to immunotherapy in melanoma (39, 40). We reasoned that because we observed a robust intratumoral presence of CD8+ T cells in the setting of IL35 deficiency, we could potentially achieve better control of tumor growth with checkpoint blockade. To this end, we treated WT and p35−/− animals bearing established PDA tumors with anti-PD-1 or isotype control antibodies (Fig. 5A). Consistent with previous reports, the treatment of WT mice with anti–PD-1 alone did not decrease tumor growth (Fig. 5B). However, compared to IL35 deficiency alone, the combination of anti–PD-1 with IL35 deficiency resulted in significant inhibition of tumor growth (Fig. 5B). We next wanted to understand which additional changes in T cell–mediated immune responses accompanied improved control of tumor growth in the setting of IL35 deficiency and anti–PD-1 combination. We observed no significant differences in either tumor-infiltrating effector CD4+ T-cell cytokine production or Foxp3+ Treg responses between p35−/− animals treated with control or anti–PD-1 (Fig. 5C-F). We next assessed the impact of anti–PD-1 treatment on intratumoral CD8+ T-cell populations in the context of IL35 deficiency. Compared to both WT and p35−/− tumors, an additional significant increase in the percent of infiltrating CD8+ T cells was observed (Fig. 5G). Intratumoral CD8+ T cells from anti–PD-1 treated p35−/− mice also displayed an activated phenotype, indicated by a significantly increased percentage of CD8+IFNγ+ cells, although this percent was comparable to p35−/− alone (Fig. 5H and I). To understand whether CD8+ T cells were responsible for the phenotypes observed, we analyzed additional cohorts of animals where CD8+ T cells were depleted from WT and p35−/− mice bearing PDAC tumors and treated with either anti–PD-1 or control (Fig. 5A and Supplementary Fig. S5A). Because only 2% of live cells in PDA tumors from WT mice were CD8+ T cells (Fig. 5G), it was not surprising that CD8+ T-cell depletion had little effect on WT PDA tumor size (Fig. 5B). However, the depletion of CD8+ T cells in p35−/− animals rescued tumor growth to a similar extent seen in WT animals, demonstrating that cytotoxic T cells are responsible for the observed decreases in tumor burden (Fig. 5B). These results were recapitulated using an independent KPC 2173 cell line (Supplementary Fig. S5B).
Figure 5. Deficiency in IL35 potentiates efficacy of anti-PD-1 checkpoint blockade.
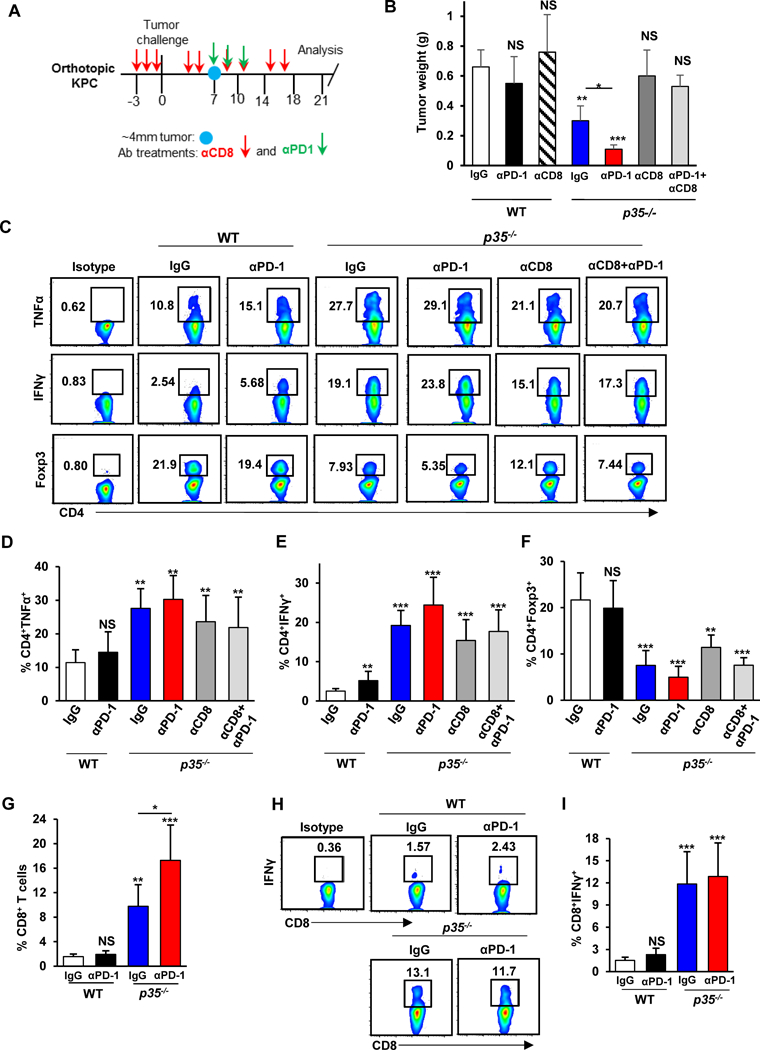
(A) Schematic of the antibody treatment regimen. Anti-CD8 or control IgG (red arrows) was administered for 3 days prior to tumor cell injection and then twice weekly on days 3, 5, 9, 11, 15, and 17. Administration of anti–PD-1 (green arrows) was initiated on day 7 after tumors reached approximately 4 mm in diameter (blue circle). Two more doses of anti–PD-1 were administered on days 9 and 11. Mice were sacrificed 3 weeks post-tumor cell injection. (B) Quantification of tumor weights from WT and p35−/− (n=3/group) mice orthotopically injected with KPC 4662 cells and treated with antibodies as described in (A). (C) Representative flow cytometry plots of intratumoral CD54+IFNγ+, CD4+TNFα+, and CD4+Foxp3+ T cells isolated from WT and p35−/− mice. Percent of CD4+ T cells is indicated. (D) Quantification of the frequency of intratumoral CD4+TNFα+ T cells (% of CD4+ T cells) from WT and p35−/− (n=3/group) mice. (E) Quantification of the frequency of intratumoral CD4+IFNγ+ T cells (% of CD4+ T cells) from WT and p35−/− (n=3/group) mice. (F) Quantification of the frequency of intratumoral CD4+Foxp3+ regulatory T cells (% of CD4+ T cells) from WT and p35−/− (n=9) mice. (H) Quantification of the frequency of tumor-infiltrating CD45+CD8+ T cells by flow cytometry from WT and p35−/− (n=3/group) mice. (I) Representative flow cytometry plots of CD8+IFNγ+ T cells isolated from WT and p35−/− mice. Percent of CD8+ T cells is indicated.(J) Quantification of the frequency of CD68+IFNγ+ T cells (% of CD8 T cells) from WT and p35−/− (n=3/group) mice. Error bars indicate SEM. p-values were calculated using Student’s t-test (unpaired, two-tailed); NS – not significant, *p<0.05, **p<0.01, ***p<0.001. Data represents 3 independent experiments.
Discussion
Effective response to immunotherapeutics, such as inhibition of checkpoint mechanisms on T cells, has been shown to correlate with pre-existing intratumoral effector T-cell infiltration (39, 40). Both human and murine PDAs fail to respond to checkpoint blockade therapy, as the majority of PDA tumors contain low numbers of infiltrating CD8+ T cells (36, 41–48). In the rare subset of PDA cases that contain T cells, infiltration does not correlate with increases in mutation load or neoepitope presence (38), suggesting that evasion of endogenous antitumor responses in PDA might be governed by the establishment of immunosuppressive networks. Our study provides evidence that IL35 serves as a crucial immunosuppressive node that attenuates the endogenous antitumor immune response to PDA. Expression of IL35 by immune cells in murine PDA was elevated, predominantly within the CD4+ T-cell and B-cell subsets. IL35 deficiency severely compromised tumor growth and correlated with increased intratumoral infiltration of effector CD4+ and CD8+ T cells, as well as decreased CD4+Foxp3+ regulatory T cells. Intratumoral CD8+ T cells and CD4+ effector T cells expressed elevated IFNɣ, suggesting that IL35 suppresses endogenous antitumor T-cell activity, and treatment with anti–PD-1 was effective in curbing PDA growth only in the context of IL35 deficiency. Together, our data suggest that the IL35 pathway is an attractive therapeutic target to augment effector T-cell infiltration and potentiate an endogenous antitumor response in order to improve the efficacy of immunotherapeutic agents in PDA.
The mechanisms that underlie the immune suppressive activity of IL35 in PDA remain poorly understood. Several immune cell types have been reported to produce IL35 in the context of autoimmunity and cancer. These include CD4+Foxp3+ Tregs, CD4+Foxp3– T cells, dendritic cells, and B cells (13, 24–28, 31, 49). We demonstrated that both PDA-associated B cells and CD4+ T cells were capable of producing IL35. Mechanistically, it is possible that immunosuppression in the PDA TME is established by concerted output of IL35 from both T and B cells or by a selective and dominant ability of either B cells or T cells to establish an IL35-driven immunosuppressive network. IL35 itself has been shown to support proliferation of Tregs, trigger de novo IL35 upregulation in Tregs and B cells, as well as promote conversion of effector CD4+ T cells into inhibitory IL35+Foxp3– T cells, indicating that a single source of IL35 might be sufficient to initiate establishment of immunosuppression. In support of this idea, a study in melanoma implicated IL35+ Tregs in promoting tumor growth (30). This, together with our findings that IL35+ B cells supported growth of pancreatic neoplasia (13), suggest that distinct IL35+ immune subtypes may confer a pro-tumorigenic advantage and highlight tumor-specific differences in how IL35 promotes cancer. Understanding the role of B cell– and T cell–specific contribution to the establishment of IL35-driven immunosuppression will be an important and necessary step towards creating precisely targeted therapies.
Our findings suggest that the mechanism by which host production of IL35 promotes pancreatic tumorigenesis was by limiting endogenous antitumor T-cell responses. It is not yet clear how IL35 controls infiltration and function of T-cell subsets in the setting of PDA, and it is possible that the suppressive effects exerted on CD8+ T cells is mediated directly or via modulation of CD4+ T effector cells or regulatory T cells. Any direct effects of IL35 on proliferative, migratory, or functional properties of target T cells likely depend on the expression of IL35 receptor chains, such as IL12Rb2/gp130 or WSX1/gp130 heterodimers or IL12Rb2/IL12Rb2 and gp130/gp130 homodimers (17). Of these, IL12Rb2 and WSX1 can be induced by inflammatory cues. Additional experiments are needed to characterize expression of IL35 receptor chains on T cells in the PDA TME and to understand how their expression regulates function and migration of target T cells in vivo. A study in a model of experimental autoimmune uveitis revealed that expression of p35 correlated with a decrease in chemokine receptor CXCR3, previously implicated in intratumoral trafficking of effector T cells (50, 51). These observations suggest that IL35 could directly regulate exclusion of T cells from the tumor parenchyma, and, at least partially, explain how IL35 suppresses endogenous antitumor CD8+ T-cell responses.
Mechanistic contribution of IL35 to immunotherapy resistance is not clear. A study by Turnis et al. suggests that IL35 may direct expression of the checkpoint molecules PD-1, LAG3, and TIM3 on CD8+ T cells, thus facilitating T-cell exhaustion (30). It is possible that downregulation of PD-1 expression on CD8+ T cells in PDA is transient and can be upregulated by additional mechanisms once the T cells infiltrate the tumor parenchyma. This possibility is supported by data suggesting progressive exhaustion of antitumor-targeted T cells and may explain why CD8+ T cells could still respond to anti–PD-1 therapy in the context of IL35 deficiency (52). Future studies aimed at understanding the crosstalk between IL35 expression and T cell exhaustion may provide further insight into the strategies that would harness the most effective combinations targeting immunosuppressive mechanisms in PDA.
Supplementary Material
Acknowledgments
We thank N. Kren for discussions, P. Patel and Animal Studies Core for help with mouse colony maintenance, Sebastien Coquery and Evan Trudeau for help with FACS. The UNC Flow Cytometry Core Facility, the UNC Translational Pathology Laboratory, the Animal Histopathology & Laboratory Medicine Core, and the UNC Lineberger Animal Studies Core are supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center.
Financial support: This work was supported by AACR-PanCAN Pathway to Leadership Grant 13–70-25-PYLA (YPG), UCRF (YPG), V Foundation for Cancer Research grant V2016–016 (YPG), and the WUSTL SPORE Career Enhancement Award grant 1P50CA196510–01A1 from the NCI (YPG).
Footnotes
Disclosure of potential conflict of interest: The authors declare no competing financial interests.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. Epub 2016/01/09. doi: 10.3322/caac.21332. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 2.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, Laheru D, Bahary N, Ramanathan RK, Tabernero J, Hidalgo M, Goldstein D, Van Cutsem E, Wei X, Iglesias J, Renschler MF. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369(18):1691–703. Epub 2013/10/18. doi: 10.1056/NEJMoa1304369. PubMed PMID: ; PMCID: PMC4631139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardiere C, Bennouna J, Bachet JB, Khemissa-Akouz F, Pere-Verge D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Ducreux M, Groupe Tumeurs Digestives of U, Intergroup P. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817–25. Epub 2011/05/13. doi: 10.1056/NEJMoa1011923. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 4.Vonderheide RH, Bayne LJ. Inflammatory networks and immune surveillance of pancreatic carcinoma. Curr Opin Immunol. 2013;25(2):200–5. Epub 2013/02/21. doi: 10.1016/j.coi.2013.01.006. PubMed PMID: ; PMCID: PMC3647365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18(16):4266–76. Epub 2012/08/17. doi: 10.1158/1078-0432.CCR-11-3114. PubMed PMID: ; PMCID: PMC3442232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pergamo M, Miller G. Myeloid-derived suppressor cells and their role in pancreatic cancer. Cancer Gene Ther. 2017;24(3):100–5. Epub 2016/12/03. doi: 10.1038/cgt.2016.65. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, McAllister F, Pasca di Magliano M. Immune cells in pancreatic cancer : Joining the dark side. Oncoimmunology. 2014;3:e29125. Epub 2014/08/02. doi: 10.4161/onci.29125. PubMed PMID: ; PMCID: PMC4108467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chao T, Furth EE, Vonderheide RH. CXCR2-Dependent Accumulation of Tumor-Associated Neutrophils Regulates T-cell Immunity in Pancreatic Ductal Adenocarcinoma . Cancer Immunol Res. 2016;4(11):968–82. Epub 2016/11/03. doi: 10.1158/2326-6066.CIR-16-0188. PubMed PMID: ; PMCID: PMC5110270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inman KS, Francis AA, Murray NR. Complex role for the immune system in initiation and progression of pancreatic cancer. World J Gastroenterol. 2014;20(32):11160–81. Epub 2014/08/30. doi: 10.3748/wjg.v20.i32.11160. PubMed PMID: ; PMCID: PMC4145756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wormann SM, Diakopoulos KN, Lesina M, Algul H. The immune network in pancreatic cancer development and progression. Oncogene. 2014;33(23):2956–67. Epub 2013/07/16. doi: 10.1038/onc.2013.257. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 11.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21(6):836–47. Epub 2012/06/16. doi: 10.1016/j.ccr.2012.04.024. PubMed PMID: ; PMCID: PMC3721510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, Vonderheide RH. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21(6):822–35. Epub 2012/06/16. doi: 10.1016/j.ccr.2012.04.025. PubMed PMID: ; PMCID: PMC3575028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pylayeva-Gupta Y, Das S, Handler JS, Hajdu CH, Coffre M, Koralov SB, Bar-Sagi D. IL35-Producing B Cells Promote the Development of Pancreatic Neoplasia . Cancer Discov. 2016;6(3):247–55. Epub 2015/12/31. doi: 10.1158/2159-8290.CD-15-0843. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, Gorjestani S, Liudahl SM, Truitt M, Olson P, Kim G, Hanahan D, Tempero MA, Sheppard B, Irving B, Chang BY, Varner JA, Coussens LM. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer . Cancer Discov. 2016;6(3):270–85. Epub 2015/12/31. doi: 10.1158/2159-8290.CD-15-0827. PubMed PMID: ; PMCID: PMC4783268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee KE, Spata M, Bayne LJ, Buza EL, Durham AC, Allman D, Vonderheide RH, Simon MC. Hif1a Deletion Reveals Pro-Neoplastic Function of B Cells in Pancreatic Neoplasia . Cancer Discov. 2016;6(3):256–69. Epub 2015/12/31. doi: 10.1158/2159-8290.CD-15-0822. PubMed PMID: ; PMCID: PMC4783189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jang JE, Hajdu CH, Liot C, Miller G, Dustin ML, Bar-Sagi D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer . Cell Rep. 2017;20(3):558–71. Epub 2017/07/21. doi: 10.1016/j.celrep.2017.06.062. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vignali DA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol. 2012;13(8):722–8. Epub 2012/07/21. doi: 10.1038/ni.2366. PubMed PMID: ; PMCID: PMC4158817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Niedobitek G, Pazolt D, Teichmann M, Devergne O. Frequent expression of the Epstein-Barr virus (EBV)-induced gene, EBI3, an IL-12 p40-related cytokine, in Hodgkin and Reed-Sternberg cells. J Pathol. 2002;198(3):310–6. Epub 2002/10/11. doi: 10.1002/path.1217. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Tao Q, Wang H, Wang Z, Wu F, Pan Y, Tao L, Xiong S, Wang Y, Zhai Z. Elevated IL-35 in bone marrow of the patients with acute myeloid leukemia. Hum Immunol. 2015;76(9):681–6. Epub 2015/10/04. doi: 10.1016/j.humimm.2015.09.020. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 20.Zeng JC, Zhang Z, Li TY, Liang YF, Wang HM, Bao JJ, Zhang JA, Wang WD, Xiang WY, Kong B, Wang ZY, Wu BH, Chen XD, He L, Zhang S, Wang CY, Xu JF. Assessing the role of IL-35 in colorectal cancer progression and prognosis. Int J Clin Exp Pathol. 2013;6(9):1806–16. Epub 2013/09/17. PubMed PMID: ; PMCID: PMC3759487. [PMC free article] [PubMed] [Google Scholar]
- 21.Jin P, Ren H, Sun W, Xin W, Zhang H, Hao J. Circulating IL-35 in pancreatic ductal adenocarcinoma patients. Hum Immunol. 2014;75(1):29–33. Epub 2013/10/15. doi: 10.1016/j.humimm.2013.09.018. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 22.Ma Y, Chen L, Xie G, Zhou Y, Yue C, Yuan X, Zheng Y, Wang W, Deng L, Shen L. Elevated level of interleukin-35 in colorectal cancer induces conversion of T cells into iTr35 by activating STAT1/STAT3. Oncotarget. 2016;7(45):73003–15. Epub 2016/09/30. doi: 10.18632/oncotarget.12193. PubMed PMID: ; PMCID: PMC5341959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang C, Li N, Li Z, Chang A, Chen Y, Zhao T, Li Y, Wang X, Zhang W, Wang Z, Luo L, Shi J, Yang S, Ren H, Hao J. Tumour-derived Interleukin 35 promotes pancreatic ductal adenocarcinoma cell extravasation and metastasis by inducing ICAM1 expression. Nat Commun. 2017;8:14035. Epub 2017/01/20. doi: 10.1038/ncomms14035. PubMed PMID: ; PMCID: PMC5253665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, Sehy D, Blumberg RS, Vignali DA. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450(7169):566–9. Epub 2007/11/23. doi: 10.1038/nature06306. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 25.Dixon KO, van der Kooij SW, Vignali DA, van Kooten C. Human tolerogenic dendritic cells produce IL-35 in the absence of other IL-12 family members. Eur J Immunol. 2015;45(6):1736–47. Epub 2015/03/31. doi: 10.1002/eji.201445217. PubMed PMID: ; PMCID: PMC4617619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Collison LW, Chaturvedi V, Henderson AL, Giacomin PR, Guy C, Bankoti J, Finkelstein D, Forbes K, Workman CJ, Brown SA, Rehg JE, Jones ML, Ni HT, Artis D, Turk MJ, Vignali DA. IL-35-mediated induction of a potent regulatory T cell population. Nat Immunol. 2010;11(12):1093–101. Epub 2010/10/19. doi: 10.1038/ni.1952. PubMed PMID: ; PMCID: PMC3008395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E, Ries S, Dang VD, Jaimes Y, Daridon C, Li R, Jouneau L, Boudinot P, Wilantri S, Sakwa I, Miyazaki Y, Leech MD, McPherson RC, Wirtz S, Neurath M, Hoehlig K, Meinl E, Grutzkau A, Grun JR, Horn K, Kuhl AA, Dorner T, Bar-Or A, Kaufmann SHE, Anderton SM, Fillatreau S. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature. 2014;507(7492):366–70. Epub 2014/02/28. doi: 10.1038/nature12979. PubMed PMID: ; PMCID: PMC4260166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang RX, Yu CR, Dambuza IM, Mahdi RM, Dolinska MB, Sergeev YV, Wingfield PT, Kim SH, Egwuagu CE. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat Med. 2014;20(6):633–41. Epub 2014/04/20. doi: 10.1038/nm.3554. PubMed PMID: ; PMCID: PMC4048323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Z, Liu JQ, Liu Z, Shen R, Zhang G, Xu J, Basu S, Feng Y, Bai XF. Tumor-derived IL-35 promotes tumor growth by enhancing myeloid cell accumulation and angiogenesis. J Immunol. 2013;190(5):2415–23. Epub 2013/01/25. doi: 10.4049/jimmunol.1202535. PubMed PMID: ; PMCID: PMC3578001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turnis ME, Sawant DV, Szymczak-Workman AL, Andrews LP, Delgoffe GM, Yano H, Beres AJ, Vogel P, Workman CJ, Vignali DA. Interleukin-35 Limits Anti-Tumor Immunity. Immunity. 2016;44(2):316–29. Epub 2016/02/14. doi: 10.1016/j.immuni.2016.01.013. PubMed PMID: ; PMCID: PMC4758699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pylayeva-Gupta Y Molecular Pathways: Interleukin-35 in Autoimmunity and Cancer. Clin Cancer Res. 2016;22(20):4973–8. Epub 2016/09/02. doi: 10.1158/1078-0432.CCR-16-0743. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 32.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, Wright CV, Hruban RH, Lowy AM, Tuveson DA. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4(6):437–50. Epub 2004/01/07. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 33.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–83. Epub 2005/05/17. doi: 10.1016/j.ccr.2005.04.023. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 34.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15(24):3243–8. Epub 2001/12/26. doi: 10.1101/gad.943001. PubMed PMID: ; PMCID: PMC312845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CV. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. 2002;32(1):128–34. Epub 2002/08/20. doi: 10.1038/ng959. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 36.Evans RA, Diamond MS, Rech AJ, Chao T, Richardson MW, Lin JH, Bajor DL, Byrne KT, Stanger BZ, Riley JL, Markosyan N, Winograd R, Vonderheide RH. Lack of immunoediting in murine pancreatic cancer reversed with neoantigen. JCI Insight. 2016;1(14). Epub 2016/09/20. doi: 10.1172/jci.insight.88328. PubMed PMID: ; PMCID: PMC5026128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X, Wang L, Mo Q, Dong Y, Wang G, Ji A. Changes of Th17/Treg cell and related cytokines in pancreatic cancer patients. Int J Clin Exp Pathol. 2015;8(5):5702–8. Epub 2015/07/21. PubMed PMID: ; PMCID: PMC4503155. [PMC free article] [PubMed] [Google Scholar]
- 38.Balli D, Rech AJ, Stanger BZ, Vonderheide RH. Immune Cytolytic Activity Stratifies Molecular Subsets of Human Pancreatic Cancer. Clin Cancer Res. 2017;23(12):3129–38. Epub 2016/12/23. doi: 10.1158/1078-0432.CCR-16-2128. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 39.Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer. 2014;2:3. Epub 2014/05/16. doi: 10.1186/2051-1426-2-3. PubMed PMID: ; PMCID: PMC4019906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–71. Epub 2014/11/28. doi: 10.1038/nature13954. PubMed PMID: ; PMCID: PMC4246418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, Sherry RM, Topalian SL, Yang JC, Lowy I, Rosenberg SA. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33(8):828–33. Epub 2010/09/16. doi: 10.1097/CJI.0b013e3181eec14c. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–65. Epub 2012/06/05. doi: 10.1056/NEJMoa1200694. PubMed PMID: ; PMCID: PMC3563263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer AR, Bajor DL, Clendenin C, Stanger BZ, Furth EE, Wherry EJ, Vonderheide RH. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol Res. 2015;3(4):399–411. Epub 2015/02/14. doi: 10.1158/2326-6066.CIR-14-0215. PubMed PMID: ; PMCID: PMC4390506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, Teichmann SA, Janowitz T, Jodrell DI, Tuveson DA, Fearon DT. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(50):20212–7. Epub 2013/11/28. doi: 10.1073/pnas.1320318110. PubMed PMID: ; PMCID: PMC3864274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67(19):9518–27. Epub 2007/10/03. doi: 10.1158/0008-5472.CAN-07-0175. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 46.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O’Dwyer PJ, Vonderheide RH. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–6. Epub 2011/03/26. doi: 10.1126/science.1198443. PubMed PMID: ; PMCID: PMC3406187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, Miller DK, Christ AN, Bruxner TJ, Quinn MC, Nourse C, Murtaugh LC, Harliwong I, Idrisoglu S, Manning S, Nourbakhsh E, Wani S, Fink L, Holmes O, Chin V, Anderson MJ, Kazakoff S, Leonard C, Newell F, Waddell N, Wood S, Xu Q, Wilson PJ, Cloonan N, Kassahn KS, Taylor D, Quek K, Robertson A, Pantano L, Mincarelli L, Sanchez LN, Evers L, Wu J, Pinese M, Cowley MJ, Jones MD, Colvin EK, Nagrial AM, Humphrey ES, Chantrill LA, Mawson A, Humphris J, Chou A, Pajic M, Scarlett CJ, Pinho AV, Giry-Laterriere M, Rooman I, Samra JS, Kench JG, Lovell JA, Merrett ND, Toon CW, Epari K, Nguyen NQ, Barbour A, Zeps N, Moran-Jones K, Jamieson NB, Graham JS, Duthie F, Oien K, Hair J, Grutzmann R, Maitra A, Iacobuzio-Donahue CA, Wolfgang CL, Morgan RA, Lawlor RT, Corbo V, Bassi C, Rusev B, Capelli P, Salvia R, Tortora G, Mukhopadhyay D, Petersen GM, Australian Pancreatic Cancer Genome I, Munzy DM, Fisher WE, Karim SA, Eshleman JR, Hruban RH, Pilarsky C, Morton JP, Sansom OJ, Scarpa A, Musgrove EA, Bailey UM, Hofmann O, Sutherland RL, Wheeler DA, Gill AJ, Gibbs RA, Pearson JV, Waddell N, Biankin AV, Grimmond SM. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531(7592):47–52. Epub 2016/02/26. doi: 10.1038/nature16965. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 48.Stromnes IM, Hulbert A, Pierce RH, Greenberg PD, Hingorani SR. T-cell Localization, Activation, and Clonal Expansion in Human Pancreatic Ductal Adenocarcinoma. Cancer Immunol Res. 2017;5(11):978–91. Epub 2017/10/27. doi: 10.1158/2326-6066.CIR-16-0322. PubMed PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chaturvedi V, Collison LW, Guy CS, Workman CJ, Vignali DA. Cutting edge: Human regulatory T cells require IL-35 to mediate suppression and infectious tolerance. J Immunol. 2011;186(12):6661–6. Epub 2011/05/18. doi: 10.4049/jimmunol.1100315. PubMed PMID: ; PMCID: PMC3110563. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50.Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, Muhitch JB, Ku AW, Frelinger JG, Odunsi K, Gajewski TF, Luster AD, Evans SS. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Commun. 2015;6:7458. Epub 2015/06/26. doi: 10.1038/ncomms8458. PubMed PMID: ; PMCID: PMC4605273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dambuza IM, He C, Choi JK, Yu CR, Wang R, Mattapallil MJ, Wingfield PT, Caspi RR, Egwuagu CE. IL-12p35 induces expansion of IL-10 and IL-35-expressing regulatory B cells and ameliorates autoimmune disease. Nat Commun. 2017;8(1):719. Epub 2017/09/30. doi: 10.1038/s41467-017-00838-4. PubMed PMID: ; PMCID: PMC5620058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stromnes IM, Schmitt TM, Hulbert A, Brockenbrough JS, Nguyen H, Cuevas C, Dotson AM, Tan X, Hotes JL, Greenberg PD, Hingorani SR. T Cells Engineered against a Native Antigen Can Surmount Immunologic and Physical Barriers to Treat Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2015;28(5):638–52. Epub 2015/11/04. doi: 10.1016/j.ccell.2015.09.022. PubMed PMID: ; PMCID: PMC4724422. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.