

Abstract
IL-1R1 deficiency in mice causes severe susceptibility to Mycobacterium tuberculosis (Mtb). Mice and macrophage cultures lacking IL-1R1 display increased bacterial growth, suggesting that phagocytes may require IL-1R1-dependent anti-microbial signals to limit intracellular Mtb replication directly. However, the myeloid-cell-intrinsic versus extrinsic requirements for IL-1R1 to control Mtb infection in mice have not been directly addressed. Utilizing single cell analysis of infected cells, competitive mixed bone marrow chimeras and IL-1R1 conditional mutant mice, we show here that IL-1R1 expression by pulmonary phagocytes is uncoupled from their ability to control intracellular Mtb growth. Importantly, IL-1R1-dependent control was provided to infected cells in trans by both non-hematopoietic and hematopoietic cells. Thus, IL-1R1-mediated host resistance to Mtb infection does not involve mechanisms of cell-autonomous anti-microbicidal effector functions in phagocytes but requires the cooperation between infected cells and other cells of hematopoietic or non-hematopoietic origin to promote bacterial containment and control of infection.
INTRODUCTION
Mycobacterium tuberculosis (Mtb), the causative agent of Tuberculosis (TB), is an intracellular bacterial pathogen and now accounts for the highest mortality due to a single infectious agent worldwide (1). Mtb primarily infects pulmonary innate immune cells and host resistance to Mtb depends on anti-microbial effector functions of infected macrophages that limit intracellular bacterial growth (2, 3).
IL-1α and IL-1β are pro-inflammatory cytokines that share the signaling receptor IL-1R1, which is ubiquitously expressed in most organs and cell types (4, 5). Inflammatory responses mediated by IL-1 are associated with host resistance to both intracellular and extracellular bacteria. Mice deficient in IL-1α, IL-1β, both IL-1 species or IL-1R1 are highly susceptible to Mtb infection and have a 100–1000 fold increase in bacterial lung burden compared to WT animals (6–9). Macrophage cultures lacking IL-1R1 expression or IL-1 cytokine production and where IL-1β has been neutralized in vitro exhibit increased bacterial replication (8, 10, 11). Likewise, bacterial control is enhanced when IL-1α or IL-1β are increased in vitro by exogenous addition or through endogenous mutations in components of the IL-1 signaling pathways (11, 12), suggesting that IL-1R1 signaling on infected cells is necessary for IL-1-mediated protection. In line with this, transplantation of Il1r1−/− bone marrow (BM) into irradiated WT recipient mice rendered mice susceptible to Mtb infection (13). However, it remains unclear whether IL-1R1-dependent host control initiates infected-cell-intrinsic, anti-microbial effector functions or infected-cell-extrinsic, indirect, secondary protective pathways mediated by IL-1R1 signaling in bystander cells.
Here we show, utilizing single cell analysis of infected cells, mixed BM chimeras and IL-1R1 conditional mutant mice, that IL-1R1 expression by infected innate immune cells is, in fact, dispensable for limiting intracellular Mtb growth. Surprisingly, IL-1R1-dependent control of bacterial replication was provided by both non-hematopoietic and hematopoietic cells in trans. Thus, IL-1-dependent control of Mtb infection occurs not via direct macrophage-autonomous anti-microbial effector functions but via indirect pathways that require cooperation of multiple cells, including those of non-hematopoietic origin, to limit Mtb infection in mice.
MATERIALS AND METHODS
Experimental animals
Il1r1−/− mice were purchased from Jackson Laboratories (JAX 3018) and backcrossed to WT C57BL/6 (Taconic) for 11 generations. WT B6.SJL (CD45.1/1), WT B6.SJL/C57BL/6 (CD45.1/2) mice were obtained through a supply contract between NIAID and Taconic. Mice with selective deletion of IL-1R1 expression in CD45+ cells (Cd45-CreposxIl1r1loxP/loxP) were generated by inter-crossing Il1r1loxP/loxP and Cd45-Cre strains (14, 15). Animals were maintained in an AALAC-accredited BSL2 or BSL3 facility and experiments performed in compliance with an animal study proposal approved by the NIAID Animal Care and Use Committee.
Mtb infections
For infections with H37Rv Mtb (100–200 CFU/mouse unless noted otherwise), mice were placed in a whole body inhalation system (Glas-Col, Terre Haute, IN) and exposed to aerosolized Mtb. Delivery doses were set by measuring lung CFU 2–24 hrs post exposure from 3–5 control mice. CFU were determined in tissue homogenates via digestion and dissociation using GentleMacs (Miltenyi Biotec, CA) or mechanical homogenization using Precellys Evolution (Precellys, Atkinson, NH). Lung homogenates and BALF were serially diluted in PBS/Tween-20 and cultured on Middlebrook 7H11 agar plates supplemented with oleic acid-albumin-dextrose-catalase (Difco, Detroit, MI) and CFU counted 21 days later. H37Rv-mCherry was generated and provided by Ryan Larson, Shahin Shafiani and Kevin Urdahl.
Macrophage differentiation and in vitro assay
Murine BM cells from either WT or Il1r1−/− mice were cultured for 6 days in media containing recombinant murine M-CSF (50 ng/ml, eBioscience/Thermofisher) to generate BMDM before infection with Mtb. To assess intracellular bacteria and score infected cells BMDM were harvested (20mM EDTA/PBS) 2hrs, 1 or 5 days post infection with H37Rv at a MOI:1 and used for imaging, FACS or attached to glass slides using a Cytospin. Slides were stained with acid fast to count bacilli using a 60× magnification oil immersion objective. Per group, 50–123 macrophages per view field in 5 view fields per experiment were scored blindly for intracellular Mtb.
Preparation of BM chimeric mice
WT or Il1r1−/− deficient mice were lethally irradiated (950 rad) and reconstituted with 107 donor BM cells of either WT or Il1r1−/− origin. For competitive mixed BM chimeric mice, C57BL/6 B6.SJL (CD45.1/1 or CD45.1/2) WT mice were lethally irradiated (950 rad) and reconstituted with a total of 107 donor BM cells from C57BL/6 CD45.1/2 or CD45.1/1 WT mice mixed at equal parts with BM cells from Il1r1−/− mice. Mice were placed on trimethoprim-sulfamethoxazole water for 4 weeks after irradiation and allotted time for immune reconstitution was 9–12 weeks.
Cell Isolation from lung tissue and flow cytometry
Lungs were digested and dissociated via GentleMACS and Lung Cell Isolation Buffer (Miltenyi Biotec, CA). Digested lung was passged through a 100 μm cell strainer and an aliquot removed for CFU. Cells were washed and purified with 37% Percoll. Cells for sorting, were washed, counted and subsequently surface stained in a BSL3 containment area under sterile conditions. The following cell populations were sorted to 90–97% purity, CD45.1+ (WT) or CD45.1– (Il1r1−/−) CD11b+ cells, CD11b+ Gr1high (neutrophils), CD11b+ Gr1low (myeloid) and plated for CFU counts. Antibodies against I-Ab (clone M5/114.15.2), Ly6G (1A8), CD11c (HL3 and N418), CD45.1 (A20), CD45.2 (104), TCRβ (H57–597), NK1.1 (PK136), CD11b (M1/70), CD45 (30-F11), Gr-1 (RB6, 8C5) and Fixable live/dead Cell Stain were obtained from eBioscience/Thermofisher, Biolegend or BD Pharmingen. Samples were acquired on a X50 Symphony flow cytometer or sorted on a FACS Aria (BD Biosciences, San Jose, CA) and analyzed using FlowJo software.
Statistical analyses
Statistical analysis of In vitro data used quantile-quantile analysis, 2×2 contingency table, goodness of fit, and Mann-Whitney tests in Mathematica while in vivo data analysis used Mann-Whitney,Wilcoxon matched pairs (Fi and Log-rank Mantel-Cox tests in Prism.
RESULTS
We and others have previously shown that bone marrow-derived macrophage (BMDM) cultures lacking IL-1R1 or where IL-1 has been neutralized display increased CFU and cell-free bacteria (10, 11, 16). To explore how IL-1R1 contributes to bacterial control and whether Il1r1−/− BMDM harbor more intracellular bacteria per infected cell, BMDM were infected with H37Rv and no difference in initial bacterial uptake was observed (Fig. S1). The intracellular distribution of bacilli was quantified by acid fast staining and microscopy five days later (Fig. 1A). A significantly lower proportion of macrophages remained uninfected (0 bacilli/per macrophage) in Il1r1−/− compared to WT cultures while the proportion of cells with the highest number of bacteria (≥25 bacilli/per macrophage) was similar between the two cultures (Fig. 1B). When we assessed the average number of bacilli per infected macrophage by excluding cells with 0 and ≥25 bacillary loads, BMDM that lacked IL-1R1 had significantly fewer bacteria per cell (Fig. 1C). Using a quantile-quantile analysis we found that the distribution of Mtb in WT and Il1r1−/− macrophages was significantly different (goodness of fit test, p < 0.001) and the major difference in the distributions was due to macrophages containing intermediate levels of bacilli (10–20 bacilli/cell) (Fig.1D). Indeed, we found a significantly lower proportion of Il1r1−/− macrophages that harbored 10–20 bacilli compared to WT cells (Fig. 1E). These findings suggest that the observed decrease in the average number of bacilli in infected Il1r1−/− BMDM was likely due to the lack macrophages infected with 10–20 bacilli. Importantly, the reduced number of bacilli per Il1r1−/− macrophage argues that while IL-1R1 signaling by infected cells in vitro is dispensable for limiting intracellular replication via cell-autonomous anti-microbial effector functions it is important to limit the fraction of infected cells.
Figure 1: Distribution of intracellular bacilli in Il1r1−/− BMDM macrophage cultures.

WT or Il1r1−/− BMDM were infected with H37Rv (MOI:1) Mtb in vitro. Number of acid fast bacilli per 503 WT and 1481 Il1r1−/− macrophages was assessed by microscopy on d5 pi. Data are pooled from 2 independent experiments. A Histogram plots from WT (white) and Il1r1−/− (black) BMDM B Proportion of uninfected macrophages (defined as 0 Mtb bacilli, left panel) and heavily infected (defined as 25 or more Mtb bacilli, right panel). P values were derived by 2×2 contingency table test. C Average Mtb burden within 1–24 range (Mann-Whitney test). D Quantile-quantile plot of Mtb distribution. Dotted lines indicate area of significant difference in the distributions. E Average Mtb burden for intermediate (10–20 bacilli) range (Mann-Whitney test).
To ask whether pulmonary macrophages and other infected innate immune cells in vivo require IL-1R1-mediated signals to limit cellular spread of infection and intracellular control of Mtb bacilli, we utilized a mCherry-labelled H37Rv strain that allows for single cell flow cytometric analysis of infected cells from the lungs of Mtb infected mice (Fig. 2A). Three weeks after low dose aerosol infection of Il1r1−/− mice, neutrophils, myeloid cells and AM from Il1r1−/− mice displayed an 8–12 fold increase in the proportion of infected cells when compared to the same cell type from WT animals (Fig. 2B, Supplemental Fig.2). These data are consistent with the increased proportion of infected Il1r1−/− BMDM and the high lung CFU, susceptibility to infection and uncontrolled bacterial replication previously reported in Il1r1−/− mice (7, 16). To investigate the requirement for direct IL-1R1 signaling into neutrophils, myeloid cells, and AM for limiting the frequency of Mtb infected innate immune cells we generated competitive mixed BM chimeric mice where half of the donor BM is WT and the other half is Il1r1−/− distinguishable by CD45 allelism (Fig. 2C, Supplemental Fig.3). In stark contrast to complete Il1r1−/− mice, the proportion of CD45.2/2 Il1r1−/− neutrophils infected with H37Rv-mCherry was comparable to CD45.1/1 WT neutrophils in mixed BM chimeric mice and similar to the propoprtion of non chimeric WT mice (Fig. 2D). The proportion of pulmonary myeloid and AM subsets infected with H37Rv-mCherry was also similar between WT and Il1r1−/− cells in chimeric mice (Fig. 2E) and total lung CFU of mixed BM chimeric mice were unaltered compared to WT chimeric controls (Fig. 2F).
Figure 2: IL-1R1 expression by infected innate immune cells is expendable for limiting Mtb dissemination and intracellular growth in vivo.
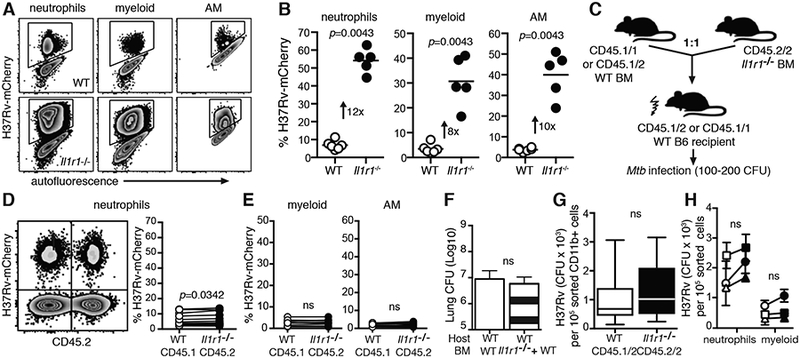
A Flow cytometric analysis for H37Rv-mCherry signal in pulmonary neutrophils, myeloid cells and AM from WT (top) or Il1r1−/− (bottom) mice 22 dpi B Frequency of H37Rv-mCherry infected cells from WT (white circles) or Il1r1−/− (black circles) cells. C Schematic of mixed BM chimeric approach. Analysis of donor CD45.1 (WT) and CD45.2 (Il1r1−/−) BM derived D neutrophils E myeloid cells and AM 22 days p.i. via and frequency of H37Rv-mCherry signal. Connecting lines depict individual animals. Data are representative of two (A,B,F) or three (D,E,G,H) independent experiments each, with 3–10 mice per group. F Lung CFU of mixed BM chimeras 21 dpi. FACS sorted WT or Il1r1−/− G CD11b+ cells or H neutrophils and myeloid cells from pooled mixed BM chimeric mice were plated for intracellular CFU enumeration on d21 pi. Connecting lines depict separate experiments with a pool of 5–10 mice each. Standard deviation shows technical variation of plated triplicates. Statistical analyses were assessed with Mann-Whitney (B, F) or Wilcoxon matched pairs test (D,E,G,H).
Next, we asked whether IL-1R1 expression on phagocytes was required to intiate anti-microbial effector functions and limit intracellular bacterial control in vivo. To this end, we again utilized mixed BM chimeric mice and sorted by flow cytometry total WT CD45.1/2 and Il1r1−/− CD45.2/2 CD11b+ cells three weeks after infection (Fig. 2G). Importantly, pulmonary Il1r1−/− CD11b+ cells contained equivalent numbers of CFU per sorted cells when compared to WT CD11b+ cells from the same animals (Fig. 2F). We next sorted neutrophils and myeloid cells to 90–97% purity (data not shown) to ask whether different phagocyte subsets exhibit different requirements for cell-autonomous IL-1R1 expression to limit intracellular bacterial growth. Similar to sorted total CD11b+ cells, neither neutrophil nor myeloid cells required IL-1R1 expression to control intracellular bacterial replication per cell as CFU count was comparable between sorted WT and Il1r1−/− cells (Fig. 2H). Taken together, the data from the mixed BM chimeric experiments demonstrate that cis IL-1R1 expression by infected pulmonary innate immune subsets is dispensable for IL-1R1-mediated host control of Mtb infection. Moreover, these data reveal that infected cells lacking IL-1R1 are protected against Mtb infection in trans by expression of IL-1R1 on bystander cells.
Since the data above indicated an infected-cell-extrinsic role for IL-1R1 signaling in host resistance to Mtb and because IL-1R1 is ubiquitously expressed, we next asked whether the observed trans-protection in mixed BM chimeras was due to IL-1R1 expression by WT BM-derived immune or recipient non-hematopoietic cells. To this end we generated chimeric mice that completely lacked IL-1R1 signals either in the donor BM or recipient compartments. Consistent with the increased susceptibility seen in Il1r1−/− mice (7, 14), mice lacking IL-1R1 in both donor and host compartments exhibited significantly decreased survival (Fig. 3A) and increased bacterial loads in the lungs (Fig. 3B). Importantly, chimeric mice where one compartment was of WT origin displayed similar survival and pulmonary CFU when compared to WT control recipients that received WT BM. These unexpected results clearly argue that IL-1R1 signaling in either hematopoietic or non-hematopoietic cells is sufficient to mediate IL-1-dependent host control and to provide IL-1R1-dependent trans-protection to infected innate immune cells. Our observations concerning survival and bacterial control are in contrast to similar chimeric experiments where IL-1R1 expression by BM-derived immune cells was found to be required for survival (13). This discrepancy could be due to differences in Il1r1−/− mouse strain background, Mtb strain and dosing, animal facilities and irradiation regiments employed. To control for caveats associated with lethal irradiation and mouse strain we next generated conditional Il1r1 deficient mice (Il1r1loxP/loxPxCd45-Crepos) that lack IL-1R1 expression on all hematopoietic CD45-expressing immune cells while CD45 negative non-hematopoietic cells retain IL-1R1 signaling (14, 15). Il1r1loxP/loxPxCd45-Crepos mice exhibited similar lung and BALF CFU when compared to mice without deletion (Il1r1loxP/loxPxCd45-Creneg) or to parental Il1r1loxP/loxP and Cd45-Cre strains (Fig. 4). Taken together, these data establish that IL-1-dependent host resistance is mediated by secondary signals acting in trans on infected innate immune cells and that these indirect IL-1R1-dependent signals can be provided by cells of non-hematopoietic origin to limit bacterial infection.
Figure 3: IL-1R1 expression by hematopoietic or non-hematopoietic compartments is sufficient for IL-1-mediated host resistance.
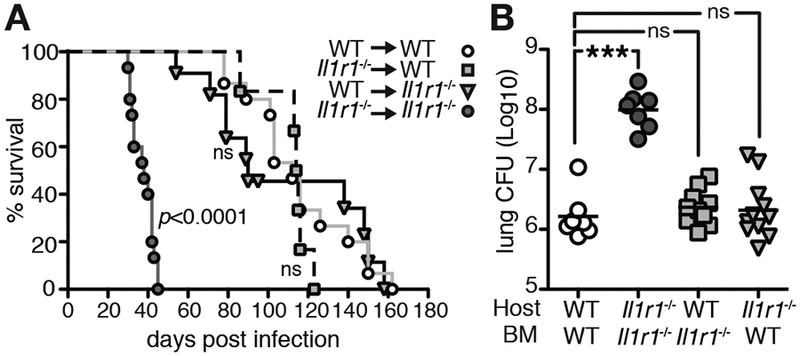
WT or Il1r1−/− mice were lethally irradiated and reconstituted with WT or Il1r1−/− BM and infected with H37Rv and A survival was monitored. Data is combined from three independent experiments with 3–5 mice per group. Statistical analyses were done by Log-rank Mantel-Cox test compared to WT/WT group. B Lung CFU 27 dpi. Data are from two independent experiments with 3–6 mice per group. Statistical analysis was assessed with the Mann-Whitney test and *** indicates p<0.0001.
Figure 4: Unimpaired host-resistance after conditional IL-1R1 deletion in CD45 expressing hematopoietic cells.
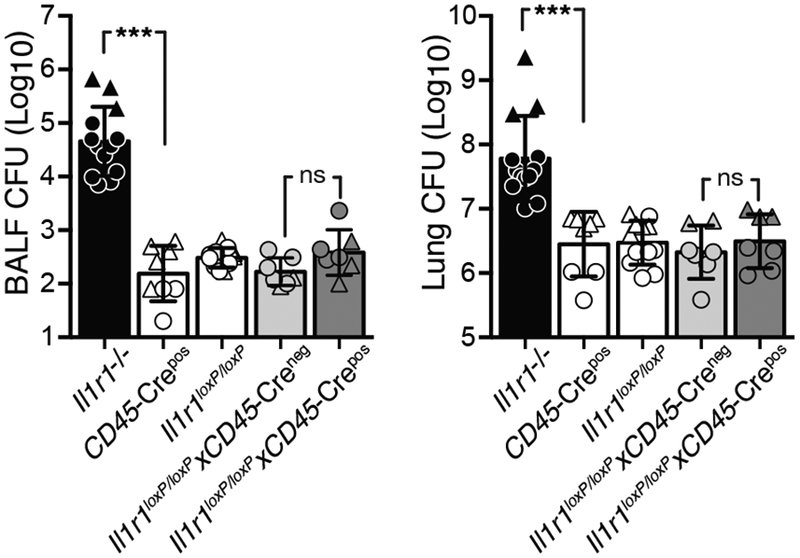
CFU in BALF and lung of Il1r1−/−, parental Cd45-Crepos and Il1r1loxP/loxP strains, Il1r1loxP/loxPxCd45-Creneg littermates and Cre deleted Il1r1loxP/loxPxCd45-Crepos mice 28 dpi with H37Rv. Data are pooled from two independent experiments and depicted as triangles (100–200 CFU) and circles (30 CFU infection), respectively, with 2–10 mice per group each. Statistical analysis was done with the Mann-Whitney test and *** indicates p<0.0001.
DISCUSSION
Our findings from both in vitro and in vivo studies argue that IL-1R1 expression mediates host resistance to Mtb by limiting the proportion of infected cells and dissemination rather than intracellular bacterial replication via anti-microbial effector functions by infected cells. Moreover, IL-1R1-dependent host resistance to Mtb is mediated by trans-protection of infected pulmonary phagocytes via bystander cells of both hematopoietic or non-hematopoietic origin.
Anti-microbial effector functions, such as phago-lysosomal fusion and acidification, nutrient starvation, free radical generation, autophagy, apoptosis, antigen presentation and major histocompatibility complex class II (MHCII) expression play critical cell-intrinsic roles in inhibiting intracellular bacterial growth (2, 3, 17–19). Competitive mixed BM chimeras coupled with cell isolation for intracellular bacterial load determination have been used to conclusively ascertain cell-intrinsic anti-microbial pathways in the lungs of infected mice (20). This experimental approach uniquely allows for dissecting cell-intrinsic versus cell-extrinsic requirements in vivo by directly comparing WT and gene deficient innate immune cells in an environment that is normalized and controlled for cell-extrinsic factors like inflammatory milieu, infectious dose, adaptive immunity and overall bacterial lung burden. It was first utilized to show that MHCII expression on infected cells was required to limit intracellular bacterial replication in vivo, demonstrating conclusively that T-cell-contact-dependent anti-microbial effector functions are necessary for Mtb intracellular control (20). Using a similar mixed BM chimera approach, the cell-intrinsic role of nitric oxide synthase 2 (Nos2) was exmined and it was shown that nitric oxide primarily limits detrimental inflammatory processes, instead of mediating cell-autonomous anti-microbial effects (21). The present study now adds novel insight into the requirements for intracellular bacterial control during Mtb infection in vivo. We demonstrate that IL-1R1-dependent host resistance to Mtb infection does not require cell-intrinsic anti-microbicidal activity since intracellular Mtb burden on a per cell basis was unchanged in competitive mixed BM chimeras. It remains possible that IL-1 regulates cell-autonomous anti-microbicidal effects in a paracrine manner. This finding is important as it will instruct future research efforts into the molecular mechanisms of IL-1R1-mediated host resistance to include cellular interactions and dissemination in addition to anti-microbial effects.
Cellular spread and dissemination of Mtb is thought to depend on high numbers of intracellular bacilli that trigger cytolytic bursting of infected macrophages at ≥25 Mtb per macrophage (22–25). The increased proportion of infected cells in the complete absence of IL-1R1 expression suggests a role for IL-1 signaling in limiting dissemination independently of the established Mtb burst size, since the average number of bacilli per Il1r1−/− BMDM was not increased. In contrast, the loss of macrophages from in vitro cultures harboring 10–20 bacteria together with the increase of infected cells in Il1r1−/− BMDMs and Il1r1−/− mice is consistent with cytolytic events and previously observed necrosis in Il1r1−/− mice (8, 16). Of note, control of cytolytic macrophage death in Mtb infection has been associated with prostaglandin E2 (PGE2) synthesis in vitro (26, 27), and we have previously shown that IL-1 promotes bacterial containment in vitro and in vivo via the induction of PGE2 (16). Thus, it is possible that IL-1 modulates host cell death leading to cytolysis at lower intracellular bacterial burden than the established burst size. In addition to the cytolytic burst-size model, efferocytosis provides another cellular mechanism for dissemination of bacilli to neighboring cells. Efferocytosis is the process by which dying cells are engulfed and removed by phagocytes and has previously been linked to both IL-1β and PGE2 during Mtb infection in vitro (11, 28). It remains unclear whether and how IL-1 regulates macrophage burst-size, cytolyic cell death or efferocytosis in vivo and future targeted studies are needed to more extensively explore the role of IL-1 in these processes.
Lastly, we found that IL-1R1 expression by cells of either hematopoietic or non-hematopoietic origin was sufficient and necessary to mediate trans-protection of infected cells, pulmonary bacterial control and host-resistance. Our findings suggest that IL-1R1-mediated host resistance to Mtb may be the result of a concerted effort of distinct cell types because trans-protection of infected phagocytes was mediated by cells of both hematopoietic and non-hematopoietic origin. IL-1R1 is ubiquitously expressed, with high expression levels in the lung (5, 29). Possible cell types that could mediate non-hematopoietic IL-1R1 signaling include: bronchiolar and alveolar epithelium, lymphatic and vascular endothelium, fibroblasts and neuroendocrine cells. Indeed, in vitro co-cultures of human macrophages and small airway epithelial cells showed that paracrine IL-1β interactions controlled Mtb growth (30). We have shown before that IL-1α and IL-1β are co-expressed by BM-derived pulmonary myeloid cells during Mtb infection in mice (7) and show here that BM-derived IL-1R1 signals are dispensable for host resistance. Thus, our findings are consistent with the notion that IL-1 mediates protection in a paracrine, rather than autocrine, fashion and argue that trans-protection occurs via secondary downstream mediators regulated by IL-1R1. We propose that such secondary mediators may be soluble, able to exert protective effects across distances and be induced in an IL-1-dependent manner by both immune and non-immune cells. For example, IL-1β induced secretion of antimicrobial peptides from epithelial cells was able to limit Mtb growth in co-cultured macrophages (30). PGE2 represents another candidate, since we have previously reported that IL-1-induced PGE2 confers resistance to Mtb infection (16) and it can be generated by cyclooxygenases in response to cytokine and hormonal stimuli by all cell types, including those of non-hematopoietic origin (31). Because PGE2 signals through four different subtypes of G-protein coupled receptors, studies involving singly-deficient and receptor combination-deficiencies are needed to determine whether PGE2 could mediate cell-autonomous antimicrobial effector functions. Thus, a systematic delineation of the cellular interaction partners required for IL-1R1-dependent host resistance to Mtb in vivo will be necessary to gain mechanistic insight into how IL-1-driven inflammation provides protection of infected cells in trans and reveal novel mechanisms that may lead to novel approaches for host-directed therapies.
Supplementary Material
Acknowledgements
We thank B. Hague for BSL3 FACS sorting and S. Oland for technical help.
This work was supported by the Intramural Research Program of NIH and by NIH grant R01 GM118553 (to V.V.G).
Abbreviations used in this article:
- BM
bone marrow
- BMDM
bone marrow derived macrophages
- Mtb
Mycobacterium tuberculosis
- MOI
multiplicity of infection
- WT
wild-type
- CFU
colony forming units
- dpi
d post infection
- AM
alveolar macrophage
- ns
not significant
- BALF
broncho-alveolar lavage fluid
REFERENCES
- 1.WHO. 2018. Tuberculosis Fact sheet N°104. http://www.who.int/mediacentre/factsheets/fs104/en/index.html.
- 2.Flannagan RS, Cosio G, and Grinstein S. 2009. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat Rev Microbiol 7: 355–366. [DOI] [PubMed] [Google Scholar]
- 3.Kaufmann SHE, and Dorhoi A. 2016. Molecular Determinants in Phagocyte-Bacteria Interactions. Immunity 44: 476–491. [DOI] [PubMed] [Google Scholar]
- 4.Dinarello CA 2018. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev 281: 8–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boraschi D, Italiani P, Weil S, and Martin MU. 2018. The family of the interleukin-1 receptors. Immunol Rev 281: 197–232. [DOI] [PubMed] [Google Scholar]
- 6.Juffermans NP, Florquin S, Camoglio L, Verbon A, Kolk AH, Speelman P, van Deventer SJ, and van Der Poll T. 2000. Interleukin-1 signaling is essential for host defense during murine pulmonary tuberculosis. J Infect Dis 182: 902–908. [DOI] [PubMed] [Google Scholar]
- 7.Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, Oland S, Gordon S, and Sher A. 2011. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity 35: 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, Kugler D, Hieny S, Caspar P, Nunez G, Schlueter D, Flavell RA, Sutterwala FS, and Sher A. 2010. Caspase-1 independent IL-1beta production is critical for host resistance to mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol 184: 3326–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamada H, Mizumo S, Horai R, Iwakura Y, and Sugawara I. 2000. Protective role of interleukin-1 in mycobacterial infection in IL-1 alpha/beta double-knockout mice. Lab Invest 80: 759–767. [DOI] [PubMed] [Google Scholar]
- 10.Jayaraman P, Sada-Ovalle I, Beladi S, Anderson AC, Dardalhon V, Hotta C, Kuchroo VK, and Behar SM. 2010. Tim3 binding to galectin-9 stimulates antimicrobial immunity. J Exp Med 207: 2343–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jayaraman P, Sada-Ovalle I, Nishimura T, Anderson AC, Kuchroo VK, Remold HG, and Behar SM. 2013. IL-1beta promotes antimicrobial immunity in macrophages by regulating TNFR signaling and caspase-3 activation. J Immunol 190: 4196–4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eklund D, Welin A, Andersson H, Verma D, Soderkvist P, Stendahl O, Sarndahl E, and Lerm M. 2014. Human gene variants linked to enhanced NLRP3 activity limit intramacrophage growth of Mycobacterium tuberculosis. J Infect Dis 209: 749–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Paolo NC, Shafiani S, Day T, Papayannopoulou T, Russell DW, Iwakura Y, Sherman D, Urdahl K, and Shayakhmetov DM. 2015. Interdependence between Interleukin-1 and Tumor Necrosis Factor Regulates TNF-Dependent Control of Mycobacterium tuberculosis Infection. Immunity 43: 1125–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abdulaal WH, Walker CR, Costello R, Redondo-Castro E, Mufazalov IA, Papaemmanouil A, Rothwell NJ, Allan SM, Waisman A, Pinteaux E, and Muller W. 2016. Characterization of a conditional interleukin-1 receptor 1 mouse mutant using the Cre/LoxP system. Eur J Immunol 46: 912–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang J, Hills D, Taylor E, Pfeffer K, Ure J, and Medvinsky A. 2008. Transgenic tools for analysis of the haematopoietic system: knock-in CD45 reporter and deletor mice. J Immunol Methods 337: 81–87. [DOI] [PubMed] [Google Scholar]
- 16.Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, Derrick SC, Shi R, Kumar NP, Wei W, Yuan X, Zhang G, Cai Y, Babu S, Catalfamo M, Salazar AM, Via LE, Barry CE 3rd, and Sher A. 2014. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 511: 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deretic V 2016. Autophagy in leukocytes and other cells: mechanisms, subsystem organization, selectivity, and links to innate immunity. J Leukoc Biol 100: 969–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hmama Z, Pena-Diaz S, Joseph S, and Av-Gay Y. 2015. Immunoevasion and immunosuppression of the macrophage by Mycobacterium tuberculosis. Immunol Rev 264: 220–232. [DOI] [PubMed] [Google Scholar]
- 19.Neyrolles O, Wolschendorf F, Mitra A, and Niederweis M. 2015. Mycobacteria, metals, and the macrophage. Immunol Rev 264: 249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Srivastava S, and Ernst JD. 2013. Cutting edge: Direct recognition of infected cells by CD4 T cells is required for control of intracellular Mycobacterium tuberculosis in vivo. J Immunol 191: 1016–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mishra BB, Lovewell RR, Olive AJ, Zhang G, Wang W, Eugenin E, Smith CM, Phuah JY, Long JE, Dubuke ML, Palace SG, Goguen JD, Baker RE, Nambi S, Mishra R, Booty MG, Baer CE, Shaffer SA, Dartois V, McCormick BA, Chen X, and Sassetti CM. 2017. Nitric oxide prevents a pathogen-permissive granulocytic inflammation during tuberculosis. Nat Microbiol 2: 17072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Behar SM, Divangahi M, and Remold HG. 2010. Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy? Nature Reviews Microbiology 8: 668–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee J, Repasy T, Papavinasasundaram K, Sassetti C, and Kornfeld H. 2011. Mycobacterium tuberculosis induces an atypical cell death mode to escape from infected macrophages. PloS one 6: e18367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Repasy T, Lee J, Marino S, Martinez N, Kirschner DE, Hendricks G, Baker S, Wilson AA, Kotton DN, and Kornfeld H. 2013. Intracellular bacillary burden reflects a burst size for Mycobacterium tuberculosis in vivo. PLoS Pathog 9: e1003190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stutz MD, Clark MP, Doerflinger M, and Pellegrini M. 2018. Mycobacterium tuberculosis: Rewiring host cell signaling to promote infection. J Leukoc Biol 103: 259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen M, Gan H, and Remold HG. 2006. A mechanism of virulence: virulent Mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. Journal of Immunology 176: 3707–3716. [DOI] [PubMed] [Google Scholar]
- 27.Divangahi M, Chen M, Gan H, Desjardins D, Hickman TT, Lee DM, Fortune S, Behar SM, and Remold HG. 2009. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nature Immunology 10: 899–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin CJ, Booty MG, Rosebrock TR, Nunes-Alves C, Desjardins DM, Keren I, Fortune SM, Remold HG, and Behar SM. 2012. Efferocytosis is an innate antibacterial mechanism. Cell host & microbe 12: 289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.consortium, G. The Genotype-Tissue Expression (GTEx) project. The Broad Institute of MIT and Harvard. 2017 https://www.gtexportal.org/home/.
- 30.Verway M, Bouttier M, Wang TT, Carrier M, Calderon M, An BS, Devemy E, McIntosh F, Divangahi M, Behr MA, and White JH. 2013. Vitamin D induces interleukin-1beta expression: paracrine macrophage epithelial signaling controls M. tuberculosis infection. PLoS Pathog 9: e1003407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Serhan CN, Chiang N, and Van Dyke TE. 2008. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nature reviews. Immunology 8: 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.