

SUMMARY
Crystallography is a major technique for determining large RNA structures. Obtaining diffraction-quality crystals has been the bottleneck. Although several RNA crystallization methods have been developed, the field strongly needs additional approaches. Here we invented an in crystallo selection strategy for identifying mutations that enhance a target RNA’s crystallizability. The strategy includes constructing an RNA pool containing random mutations, obtaining crystals, and amplifying the sequences enriched by crystallization. We demonstrated a proof-of-principle application to the P4–P6 domain from the Tetrahymena ribozyme. We further determined the structures of four selected mutants. All four establish new crystal lattice contacts while maintaining the native structure. Three mutants achieve this by relocating bulges and one by making a helix more flexible. In crystallo selection provides opportunities to improve crystals of RNAs or RNA-ligand complexes. Our results also suggest that mutants may be rationally designed for crystallization by “walking” a bulge along the RNA chain.
Keywords: X-ray crystallography, structural biology, in vitro selection, group I intron, crystal lattice contacts
eTOC Blurb
To improve the odd of determining RNA structures using crystallography, Shoffner et al. invent an in crystallo selection method. This method identifies mutants with enhanced crystal incorporation by applying crystallization as a selection pressure to a very large number of variants simultaneously.
INTRODUCTION
RNA molecules often fold into defined three-dimensional structures to carry out their functions. While crystallography has had considerable success in determining atomic structures of RNAs, it is challenging to obtain crystals for large RNAs. Even when an RNA does crystallize, the crystals often do not diffract or diffract poorly. This challenge may be attributed to several features of RNAs. They have polyanionic backbones. Therefore, RNAs must overcome charge-charge repulsion, often via counterions, in order to fold properly and pack orderly into crystals. Indeed, structural stability has been shown to be an important factor in obtaining diffraction-quality crystals (Juneau and Cech, 1999; Juneau et al., 2001). Furthermore, unlike proteins, RNAs often fold with most of their side chains (bases) facing inwards, e.g., in double-stranded helices, making it harder to establish unique lattice contacts.
Great efforts in the field of RNA structural biology have been devoted to develop methods for facilitating crystallization (Ke and Doudna, 2004; Zhang and Ferre-D’Amare, 2014). Homologues are routinely explored in crystallization trials. Crystallization screens tailored for RNAs and RNA-protein complexes have been designed (Doudna et al., 1993; Scott et al., 1995). In vitro selection has been employed to identify mutations that improve structural stability and aid crystallization (Guo and Cech, 2002; Guo et al., 2004; Juneau and Cech, 1999; Juneau et al., 2001). Knowledge of RNA tertiary interactions, such as the GAAA tetraloop/tetraloop receptor interactions, has been put to use in engineering potential lattice contacts (Ferre-D’Amare et al., 1998; Golden et al., 1997; Reiter et al., 2010). Protein-binding sites may be engineered into RNAs so that proteins such as U1A and antibodies are included in crystallization and may bridge lattice interactions (Ferre-D’Amare and Doudna, 2000; Koldobskaya et al., 2011; Shechner et al., 2009; Ye et al., 2008). Many RNAs, such as the T-box riboswitch, naturally contain the Kink-turn structural motif and thereby can be cocrystallized with Kink-turn binding proteins (Zhang and Ferre-D’Amare, 2013). These methods have allowed successful structure determination for a substantial number of examples. Nevertheless, crystallization remains the major bottleneck. There is a great need to expand the RNA crystallography toolbox.
Unlike proteins, RNAs can carry functions while fully encoding their “genetic” information. This property led to the RNA World hypothesis (Gesteland et al., 2005) and the invention of in vitro selection methods (Tuerk and Gold, 1990; Wilson and Szostak, 1999). In vitro selection is also called Systematic Evolution of Ligands by EXponential amplification (SELEX). The method starts with a diverse pool of sequences that is subjected to iterative rounds of selection based on biophysical or functional properties, RT-PCR amplification of the “winner” sequences, and transcription to produce the next-generation library. Random mutagenesis may be included between rounds of selection to achieve “in vitro evolution.” In vitro selection is a powerful tool for both biological discoveries and biotechnological applications, which range from identifying binding sites of RNA-binding proteins to developing nucleic acid aptamers with high affinities for specific target molecules (Nimjee et al., 2017).
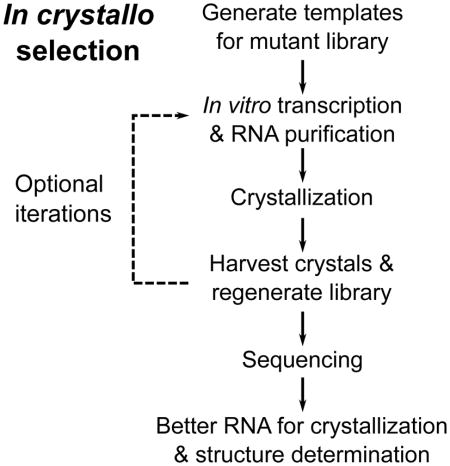
Here we invented an in crystallo selection strategy to identify RNA variants with improved abilities to incorporate into a crystal lattice (Figure 1A). The strategy is in vitro selection using crystallization as the selection criterion. The method includes introduction of random mutations to a target RNA molecule and crystallization of the library mixture. The latter step is somewhat counterintuitive as it is generally believed in crystallography that crystallization requires the subject macromolecules to be chemically and conformationally homogeneous (Doudna, 1997; Ferre-d’Amare and Doudna, 1997). As a proof-of-concept, we applied this schema to the wild-type (WT) 160-nt P4–P6 domain from the Tetrahymena self-splicing group I intron, which contains extensive secondary and tertiary structures but is non-trivial to crystallize (Cate et al., 1996). Crystal structures of selected mutants show how they engage in new lattice interactions.
Figure 1. In crystallo selection of P4–P6 variants.

(A) Schematic of the in crystallo selection strategy. Dashed line represents iterative selections that were performed but not used in structure determination. (B) Summary of selected mutants. (C) Mutations projected on the P4–P6 secondary structure. Green boxes show mutants that are structurally characterized. Red boxes show all other mutation sites. Bases highlighted with blue background fall within 5Å of a neighboring molecule in the crystal lattice. Note that unpaired residues at the bend between P5 and P5a are drawn in a way reflecting their direct stacking on top of the P5 and P5a stems. Such drawing makes it easier to understand the structural changes induced by the M2 mutations.
RESULTS
Design and implementation of the in crystallo selection strategy
We worked out an in crystallo selection procedure enabling direct selection for crystallizability (Figure 1A). We generated an initial mutant library of P4–P6 through 10 cycles of error-prone PCR, yielding a DNA sequence pool with most constructs expected to contain 0–2 mutations. We randomly picked 12 clones for sequencing and observed 7 mutant and 5 wild-type P4–P6 sequences. Following in vitro transcription and denaturing gel purification, the RNA library was annealed and subjected to crystallization by vapor diffusion. As the WT P4–P6 was known to crystallize in essentially the same crystal form under conditions containing either (+/−)2-Methyl-2,4-pentanediol (MPD) or polyethylene glycol 1000 (PEG1000), we compared selection conditions with these two precipitants. Large (around 200 μm) crystals appeared in both MPD and PEG1000 conditions within 4 days. Harvesting crystals from the drops yielded the initial pool of selection “winners.” We subjected the winner RNA libraries to a further round of selection, but only obtained large numbers of smaller crystals that were difficult to manipulate. Therefore, we opted to investigate the winner pools after a single round of selection.
We randomly picked a total of 24 clones from the MPD and PEG1000 pools for sequencing. The results revealed 11 mutants and 13 WT sequences (Figure 1B). The mutations distribute throughout the P4–P6 sequence and secondary structures (Figure 1C), except in the invariable RT-PCR primer annealing regions at the 5′- and 3′-ends. Even with the P4–P6 three-dimensional structures taken into account, it was not obvious how these mutations affect the ability of the RNA to join a crystal lattice.
Structure determination of P4–P6 mutants
To determine how these mutations might alter the RNA structure and crystal-lattice contacts, we crystallized individual mutants and collected diffraction data for four of them (Figure 1C and Table 1). For each construct we re-screened crystallization in both MPD and PEG1000 conditions, with or without spermine or cobalt hexamine as additives. M1–M4 and the WT P4–P6 control crystallized within 1–5 days. There were no discernible differences in their crystallization kinetics. We noticed some morphology differences among these crystals (Figure S1). Whereas WT and M1 crystals tended to grow in diamond shapes, the M2, M3, and M4 crystals were usually elongated, like prisms. As the WT structure was solved using crystals treated with isopropanol, we examined the effects of soaking crystals in mother liquor supplemented with 5%, 10%, or 15% (v/v) isopropanol.
Table 1.
Crystallographic data and refinement statistics.
| Crystals | M1 | M1 | M2 | M3 | M4 |
|---|---|---|---|---|---|
| Growth conditions | PEG1000 | MPD | MPD | MPD | PEG1000 |
| Soaking | Soaked with isopropanol | No | No | No | Soaked with isopropanol |
| Data collection | |||||
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 |
| Cell dimensions | |||||
| a, b, c (Å) | 74.7, 130.0, 146.4 | 75.5, 133.0, 147.5 | 75.6, 132.9, 146.8 | 76.7, 130.9, 146.4 | 74.3, 129.5, 146.1 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å)a | 66.5 – 3.14 (3.22 – 3.14) | 67.2 – 3.14 (3.22 – 3.14) | 64.2 – 3.70 (3.8 – 3.70) | 66.1 – 3.95 (4.06 – 3.95) | 63.6 – 2.80 (2.87 – 2.80) |
| Rmeas (%)b | 9.5 (90.6) | 11.9 (60.0) | 7.4 (104.4) | 9.8 (133.3) | 9.9 (175.7) |
| Rp.i.m.(%)b | 2.7 (24.2) | 3.6 (19.9) | 3.3 (44.2) | 2.9 (36.0) | 2.9 (45.4) |
| I/σ | 16.3 (2.98) | 12.9 (3.62) | 13.0 (1.92) | 15.6 (2.09) | 15.0 (1.46) |
| CC1/2 | 99.6 (89.9) | 99.1 (93.9) | 99.7 (78.7) | 99.9 (86.2) | 99.5 (76.0) |
| Completeness (%) | 98.9 (87.9) | 95.9 (82.6) | 99.5 (96.9) | 99.5 (94.4) | 99.3 (90.9) |
| Redundancy | 12.9 (11.8) | 10.3 (8.1) | 5.4 (5.0) | 12.9 (12.4) | 12.9 (11.3) |
| Refinement | |||||
| Resolution (Å) | 63.8 – 3.14 | 67.2 – 3.14 | 64.2 – 3.70 | 66.1 – 3.95 | 63.6 – 2.80 |
| No. of unique reflections | 25,212 | 25,498 | 16,335 | 13,323 | 35,173 |
| % of reflections in test set | 10% | 10% | 10% | 10% | 5% |
| Rwork/Rfree | 0.211/0.229 | 0.250/0.270 | 0.199/.229 | 0.246/0.266 | 0.211/0.226 |
| No. of atoms | |||||
| RNA | 6,770 | 6,776 | 6,778 | 6,812 | 6,746 |
| Mg2+ | 20 | 22 | 23 | 9 | 19 |
| K+ | 0 | 0 | 0 | 0 | 1 |
Values in parentheses are statistics for the highest resolution bin.
The space group and unit cell dimensions of the mutant P4–P6 crystals (Table 1) closely matched those of the WT (PDB ID 1GID; P212121; a = 74.8 Å, b = 128.7 Å, c = 145.9 Å). The crystals diffracted to 2.80–3.95 Å resolution, either the same as the WT (M4) or moderately worse (M1–M3). We solved the structures by refining the WT coordinates against the mutant diffraction data (see the STAR Methods section for details). The overall structures of the M1–M4 RNAs aligned well with that of the WT, all with backbone C1′ r.m.s.d. under 1 Å (both molecules A and B in the asymmetric unit are included in the calculation, Figure S2). There are no large changes in intermolecular positioning and orientation, as indicated by difference distance matrix plots (Figures S2 and 5B).
Figure 5. M4 weakens the P6b helix, allowing the L6b loop to establish new lattice contacts.

(A) The A230U mutation site in the 2.8-Å-resolution M4 structure (with atoms colored red, green and blue), compared to the WT structure shown in silver. The structures were superimposed by aligning all molecule B atoms outside of residues 230–244. Blue mesh shows the immediately surrounding 2Fo-Fc map for M4 contoured at 1.2 σ level. (B) Difference distance matrix plot between M4 and WT structures. Both molecules A and B in the asymmetric unit are shown, separated by dashed lines. Mutated sites are marked with arrows. Calculated r.m.s. deviation for all C1′ atoms in the asymmetric unit is shown below the plot. (C) The P6b stem loops in M4 and WT structures superimposed and colored as in (A). 2Fo-Fc map contoured at 1.2 σ level is shown as gray mesh. (D) New crystal contacts established by M4. Blue mesh shows 2Fo-Fc map contoured at 0.6 σ. A schematic of the new lattice contacts involving U236 in L6b and U199 in J5a/5 from four different molecules.
Swapping base-pairs in M1 and M2 produces new lattice interactions
The M1 (U131A) mutation causes a local structural rearrangement that establishes new lattice contacts. We first solved the M1 structure using crystals grown from a solution containing 22% (v/v) PEG1000 and soaked in mother liquor supplemented with 10% (v/v) isopropanol. The electron density map indicated a substantial local conformational rearrangement relative to the WT (Figure 2A and 2B). In the WT crystal, U131 pairs with A192 in the middle of P5a helix and the adjacent U130 forms a single-nucleotide bulge (Figure 2C). In the lattice, molecules A and B (as defined by the deposited coordinates) are related by a pseudo-twofold symmetry. U130 and U131 are about 7 and 10 Å from the closest atoms in the other molecule, respectively (Figure 2C), too far to mediate direct lattice contacts. In the M1 structure, mutation of U131 to adenine disrupts the original base pair with A192, allowing U130 to rotate into the helix to restore the pairing, while pushing A131 out toward the neighboring molecule (Figure 2D). Although the electron density map does not unambiguously define the position of the whole A131 base, we estimate that the N1 imine of A131 base in molecule A is within hydrogen-bonding distance to the 2′ hydroxyl of U244 in molecule B (Figure 2D), and that the N6 amino and N7 nitrogen of A131 in molecule B contact the 2′ hydroxyl of U245 in molecule A. Therefore, we conclude that U131A establishes new lattice contacts by shifting the bulge by one residue to a closer location and better orientation toward the neighboring molecule and by replacing the pyrimidine with a larger purine base.
Figure 2. Generation of a new lattice contact in mutant M1.

(A) Model of WT P4–P6 (grey) positioned as a rigid body in the 2Fo-Fc map (blue) of the M1 mutant contoured at 1.5σ, showing residues 130 and 131 do not fit the electron density. (B) The M1 structure contains a local structural rearrangement that explains the 2Fo-Fc map. (C) Stereoscopic view of WT P4–P6 crystal, showing P5 region of molecule A in green and P6 region of molecule B in red. (D) The 3.14 Å resolution M1 structure shows that swapping of base-pairs in molecule A (green) positions its A131 in contact with neighboring molecule B (red). Blue mesh shows σA-weighted 2Fo-Fc map contoured at 0.8σ level. In all figures showing detailed lattice contacts, we color a mutated region undergoing local structural changes green and the molecule receiving the new contact red.
To investigate whether the conformational and lattice rearrangements between WT and M1 structures might result from differences in crystal preparation, we solved the structure of M1 using crystals grown with MPD as the precipitant and frozen without soaking. We observed electron density consistent with base-pairing between U130 and A192 as well as relocation of A131 to contact the neighboring molecule (Figure S3), similar to the M1 structure under PEG1000 condition and soaked with isopropanol. This result supports the notion that the new lattice interactions are stable under different conditions and thereby are caused by the mutation identified using in crystallo selection.
We observed a similar theme in the structure of M2, which contains A125U and G126U. The electron density in this region of the M2 structure showed repositioning of residues 124 and 125 (compare Figures 3A and 3B). In the WT structure, G126 forms a non-canonical base pair with A196, stacking on top of the P5a stem (Figure 3C). C124 stacks on top of G126 and is further stabilized by hydrogen bonds with C197 and G201. This conformation forces the intervening A125 to the exterior of the molecule. In molecule B, the A125 C8 carbon is 3.0 Å from the U205 O3′ and 3.5 Å from the A206 OP1 oxygen on a symmetry-related molecule A (Figure 3C). These contacts are closer than C-O van der Waals distance of 3.7 Å and therefore are potential steric clashes or (mostly weak) C-H…O hydrogen bonds. In molecule B of the M2 structure, G126U replaces the G-A pair with a Watson-Crick U-A pair (Figure 3D). Importantly, substitution of A125 with uracil allows this smaller base to assume the position previously occupied by C124. The net result is that C124 is flipped out of the structure and points towards the 5′ and 3′ tails of a symmetry-related molecule A, creating an opportunity for the C124 N4 amino group to hydrogen-bond with the U106 O4 or C260 2′-OH of the symmetry-related chain. Similar structural rearrangements appear to occur in molecule A, with analysis limited by the lower quality of electron density in that region. Residues 124–126 in molecule A are not involved in lattice contacts in either WT or M2 crystals. In sum, the M2 structure demonstrates that repositioning of the bulge from position A125 to C124 creates new intermolecular hydrogen bonds and ameliorates a potential clash, while maintaining the core structure of the RNA.
Figure 3. Rearranged lattice interaction in the M2 mutant.

(A) Structure of WT P4–P6 (grey) fits poorly to the 2Fo-Fc map from the M2 mutant at 1.2σ. (B) Fitting of the M2 mutations to the electron density in (A) reveals the conformational rearrangement in the mutant RNA structure. (C) Stereo view of the J5/5a hinge of WT P4–P6 (molecule B) in green and symmetry-related molecule A in red. (D) The same region in the 3.70 Å resolution M2 structure. Electron density map (2Fo-Fc, 0.8σ) in blue showing the new position of C124.
An insertion mutation creates a base-stacking lattice contact
M3, which contains G134A and U185AA, involves a region critical for tertiary interactions of P4–P6. U185AA replaces U185 in the A-rich bulge (in sequence AAUAA) with two adenines (annotated in M3 as A185 and A185*), creating a stretch of six adenines. U185AA may also be viewed as U185A substitution plus A185* insertion. In the WT P4–P6 structure, this region of the RNA adopts a unique “corkscrew turn” conformation around two magnesium ions and is buttressed by a Hoogsteen base pair between A187 and U135 (Figures 4A and S4A). This base pair is then capped by a Watson-Crick pairing of C189 and G134. In this conformation, the A-rich bulge does not make substantial lattice interactions with a neighboring molecule.
Figure 4. Lattice contact created by a base-stacking interaction in mutant M3.

(A) Stereo view of the A-rich bulge (green) and J5/5a region of a symmetry-related molecule in the WT structure (red). (B) The M3 structure at 3.95 Å resolution shows that in molecule B inserted A185* and A186 take the approximate positions of A186 and A187 in the WT structure. Such a shift of register leads A187 to bulge out and produce new base stacking with the bulged A125 of a symmetry-related molecule A. Blue mesh shows 2Fo-Fc map contoured at 1.0 σ level.
In the M3 structure, the A185* insertion produces a local rearrangement that establishes a new lattice contact. The other two mutations, U185A and G134A, do not cause a large shift of these residues. In molecule B, A185* occupies the position of A186 in WT, pushing A186 to the position of A187 in WT. A186 forms a Watson-Crick pair with U135 in M3, replacing the A187-U135 Hoogsteen pair in WT. The M3 electron density maps of molecule B reveal that A187 bulges away from the structure to stack with the base of A125 that is flipped out of a symmetry-related molecule A (Figures 4B and S4B). In molecule A, A185* and A186 both bulge out in the same direction as A185, with the A185* base stacking on A185 (Figure S4C). The positions of A187 and further residues are largely unchanged compared to those of the WT structure. The conformational changes of the RNA chain are accompanied by repositioning of the two magnesium ions. This conformation is different from those of the M3 molecule B and the WT. Despite of the substantial local structural changes caused by the U185AA mutation, the overall structures of both molecules A and B remain similar, demonstrating remarkable structural plasticity of large RNAs. However, the M3 changes in molecule A do not establish new lattice contacts. Overall, the M3 structure demonstrates that a single-nucleotide insertion can generate a lattice interaction by complementing a dangling residue on an opposing molecule through a base-stacking interaction.
M1, M2 and M3 show a common theme that a bulge can migrate along the RNA chain by one or more residues to bridge a new crystal lattice contact. In M1 and M2, the bulge migration occurs along a helical strand, whereas in M3, the bulge walks along a strand involved in tertiary folding.
A mutation weakens the P6b stem, allowing the L6b loop to make new lattice contacts
We additionally solved the structure of M4, containing an A230U mutation in the P6b stem. While this mutation breaks the Watson-Crick pair with U244, we did not observe disruption of the stem. U230 assumes the position of A230 as in the WT structure (Figure 5A). However, with the smaller base, U230 stacks less extensively with neighboring bases and forms only one hydrogen bond with U244. We used difference distance matrix plot to compare the M4 and WT structures and identified a conformational change in the lower P6b stem loop (Figure 5B). The weakened base stacking and pairing at U230 allow the lower P6b stem of molecule B to shift by about 1 Å (Figure 5C). The L6b loops in both molecules A and B adopt conformations different from those of the WT and M1–M3 structures, so that the U236 base from molecule A stacks with U236 from a symmetry-related molecule B and also interacts with U199 from another molecule B′ (Figure 5D). In the WT structure, the occupancy for the L6b residues was set to 0, indicative of poor electron density and low confidence in their actual conformation. In the M1, M2 and M3 structures, there is continuous electron density for the backbone of L6b loops, but little support for direct crystal contacts. In M4, on the immediate upper side of A230U is a region of P4–P6 that makes extensive lattice contacts (Figure 1C). In a pseudo-twofold symmetric interaction between molecule A and a symmetry-related molecule B, 2′-OH of U228 forms a hydrogen bond with 2′-OH of U247, 2′-OH of G227 hydrogen-bonds to U249 phosphate, and U249 base-stacks with another U249. These lattice contacts, common to the WT and all mutant P4–P6, hold the molecules in the crystal lattice while letting the lower P6b stem explore new contacts. Thus, we conclude that M4 establishes new crystal contacts by weakening a helical stem and giving the adjacent stem loop flexibility to shift.
The new contacts in the context of crystal lattice
Molecules A and B in the asymmetric unit interact with each other via an extensive interface, effectively forming a dimer in the crystal lattice (Figure 6). The M1 mutation in each molecule establishes a new lattice contact, further strengthening the “dimerization interface”. Each dimer contacts neighboring dimers related by a unit translation along the a axis. At this “a-lateral interface”, molecule B contacts a symmetry-related molecule A through mostly backbone-backbone interactions. Both M2 and M3 mutations in molecule B introduce new contacts to this interface. In contrast, neither M2 nor M3 in molecule A add new lattice interactions. The “string” of P4–P6 dimers stacked along the a axis binds other strings to form a three-dimensional lattice via additional interfaces. The M4 mutation allows the L6b loop to establish new lattice contacts that involve four P4–P6 dimers from four strings, which are expected to strengthen the string-string interactions. The new lattice contacts introduced by M2, M3 and M4 are likely to favor addition of molecules from solution to the crystal along certain directions, thereby causing the observed morphological changes (Figure S1).
Figure 6. The new lattice contacts in crystals.

The left panel is a space-filling model of the WT P4–P6 molecules viewed along the b axis. The two unique molecules, A and B, are colored green and cyan, respectively. These two molecules effectively form a dimer via an extensive interface. Symmetry-related A and B molecules are indicated in dark and light gray, respectively. The right panel is a view of the RNA molecules in the left panel with a 90° rotation along the c axis. The P4–P6 dimer on the left side of the left panel is removed to illustrate the location of the contacts introduced by M2 (purple) and M3 (magenta, represented by residues 185–187) at the a-lateral interface on molecule B. Also visible in the right panel is the M1 (blue) mutation that establishes new contacts at the dimerization interface. The M4 (red) mutation allows the L6b loops (orange) in both molecules A and B to establish a new contact that involves four P4–P6 dimers (see also Figure 5D).
DISCUSSION
Our study provides the proof-of-concept that in crystallo selection is a viable strategy for enhancing the crystallizability of biological macromolecules. Our exercise revealed several features. First, all four winner mutants we structurally characterized establish new lattice contacts, suggesting that their mutations make it more favorable for them to be incorporated into the crystal lattice by lowering the free-energy change during this process. Second, all four structures are essentially identical to WT, indicating that in crystallo selection requires the mutations in the winners not to disrupt the folding or cause large conformational changes. By inspecting the secondary and three-dimensional structures, we expect most other selected P4–P6 mutants also to fold the same as WT. Thus, when a mixed population of molecular variants is subjected to crystallization, only those variants with sufficiently similar conformation are admitted to the growing crystal lattice. This is a welcoming benefit for studying structure-and-function relationship of target RNA molecules. Furthermore, this feature allows in crystallo selection to be used for identifying and characterizing variants that do not change the overall structure but improve other desired properties.
We observe local improvements of the electron density maps in the vicinity of the selected mutations relative to the WT structure. For example, in WT P4–P6 the solvent-exposed, bulged residue U130 is disordered and hence reported with occupancy set to zero (Cate et al., 1996). In the M1 structure we clearly identify both U130 and the bulged A131, which are stabilized by base-pairing with A192 and by the new lattice interaction, respectively. This suggests that in crystallo selection may be an effective strategy for stabilizing loops or other disordered regions within the context of the crystal lattice.
While we characterize M1–M4 individually, we notice that both M2 and M3 induce changes involving A125. M2 mutates A125 to a uracil that takes over the position of C124. In molecule B, such a local structural rearrangement allows P4–P6 to avoid a potentially unfavorable interaction with a neighboring molecule and instead positions C124 for new lattice contacts (Figure 3). A125 in molecule A is stacked on the new lattice contact induced by the M3 mutations (Figure 4). Furthermore, in the previous structure of P4–P6 ΔC209 mutant at 2.25 Å resolution (PDB ID 1HR2) (Juneau et al., 2001), the deletion results in A125 forming favorable lattice contacts not seen in the WT crystals—the base of A125 in molecule A forms a hydrogen bond with the U185 phosphate in a symmetry-related molecule B and the 2′-OH of A125 in molecule B hydrogen-bonds to the U205 phosphate of a symmetry-related molecule A. These new contacts are reflected by comparative SHAPE analysis of P4–P6 ΔC209 in solution and in crystals (Vicens et al., 2007). Thus, A125 may be considered a hotspot in the P4–P6 RNA for engineering new lattice contacts. In the larger, catalytically active Tetrahymena ribozyme, A125 mediates an inter-domain interaction by stacking with the A324 base in the L9 loop (Guo et al., 2004).
None of the mutants characterized here diffracted to higher resolution than WT P4–P6 when crystallized individually. We speculate that the lack of improvement in resolution may result from the complexity of P4–P6 crystal form, in which two molecules occupy the asymmetric unit. When selecting for mutant molecules that can successfully join the growing crystal lattice from a mixed population, each mutant needs only occupy one position in the asymmetric unit in order to become a winner. However when crystallized individually, a mutant has to fill both positions, perhaps negatively affecting crystal quality. For example, M2 and M3 in molecule A do not establish new lattice contacts but instead cause structural changes that may have compromised packing (Figure S4). Therefore we anticipate that the in crystallo selection approach will garner superior results with crystal forms containing a single molecule per asymmetric unit.
In crystallo selection is most applicable to situations in which crystals of a RNA, RNA-protein, or RNA-ligand complex have been obtained but do not diffract well. Indeed, in our experience working with the Tetrahymena group I intron and other large RNAs, most crystal forms do not diffract well (Golden et al., 1997). Therefore, we expect in crystallo selection to be widely useful. Most other crystallization methods, as summarized in the Introduction, focus on obtaining crystals ab initio. Thus, in crystallo selection and other methods are very complementary to each other and can be used in combination. Further, in protein crystallography, “cross-seeding” using microcrystals of a distantly homologous protein or even non-macromolecular solid substances can produce crystals for a protein (Abuhammad et al., 2017). We imagine that in crystallo selection may be used to obtain novel crystal forms by cross-seeding.
Our P4–P6 mutant structures highlight principles of RNA crystal packing that may be used to rationally design crystallization constructs. In the M1–M3 structures, bulged residues are repositioned or introduced to form hydrogen bonds or base-stacking across different molecules, while the local base pairing and the overall structure are preserved. As such, bulged RNA bases can be “walked” along the chain to a desired position without destabilizing the core packing of the molecule. Based on the M1 structure, we propose that existing bulged residues in a secondary structure may be repositioned in either direction along a helix, so that some of these variants might fortuitously create new lattice contacts. Similarly, bulged residues may be inserted. Such bulge residues add or alter features to the plain surface of an RNA helix. Indeed, we identified a couple of RNA structures in the PDB (ID: 2GIS and 1KXK) in which such a strategy appears to be readily applicable (Montange and Batey, 2006; Zhang and Doudna, 2002) (Figure S5). Our M2 and M3 structures suggest that the bulge engineering strategy may be applied to unpaired surface regions of the target RNA. Admittedly, in the absence of three-dimensional structures, these constructs are hard to design. However, chemical or nuclease footprinting can identify solvent-accessible regions. Additionally, sequence variation from different species may provide guidance. For instant, the U185 mutated in M3 is the only residue in the A-rich bulge that is not completely conserved. The “bulge walking” approach may be used both for crystallization ab initio and for improving existing RNA crystals.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Feng Guo (fguo@mbi.ucla.edu).
METHOD DETAILS
Construction of P4–P6 mutant library
The library was constructed by error-prone PCR using forward primer 5′-CAGTGAATTCAATTAATACGACTCACTATAGGAAT-3′ (underlined, EcoRI site; italic, T7 promoter; bold, transcribed P4–P6 sequence) and reverse primer 5′-GCAGGTCGACTCTAGACTCTTC-3′ (underlined, XbaI site for cloning; italic, EarI site for linearization). Error-prone PCR reactions contained 10 mM Tris-HCl pH 8.8, 50 mM KCl, 4.76 mM MgCl2, 0.56 mM dATP, 0.9 mM dCTP, 0.20 mM dGTP, 1.40 mM dTTP, 0.5 MnCl2, 0.5 μM each primer, 3 ng/μl WT P4–P6 template, and 0.08 U/μl Taq DNA polymerase (Roche). Reactions were initially heated at 94°C for 5 min, followed by 10 cycles of 94°C for 1 min, 52°C for 1 min, 72°C for 3 min, and a final 10-min incubation at 72°C. The PCR product was purified on a 2% agarose gel, digested with EcoRI and XbaI, and ligated into pUC19. The ligation reaction was transformed into XL-1 Blue MRF’ electroporation-competent E. coli cells (Agilent). We estimated that the library contains 10 million independently transformed clones.
RNA transcription and purification
RNA samples for in crystallo selection and for individual crystallization were generated using in vitro transcription. We first isolated mega/maxiprep plasmid DNA in the pUC19 vector containing the RNA sequence preceded by a T7 promoter and followed by an EarI restriction site. Linear templates for runoff transcription were prepared by overnight digestion with EarI followed by ethanol precipitation and resuspension in water. 130 μg of template DNA was then added to a 5 ml transcription reaction containing 40 mM Tris pH7.5, 25 mM MgCl2, 4 mM DTT, 20 mM spermidine, 3 mM each NTP, 350 μl purified T7 RNA polymerase, and 2 μg inorganic pyrophosphatase (Sigma-Aldrich). After 4 hr of incubation at 37°C, reactions were ethanol-precipitated and the RNA purified over a 6% polyacrylamide denaturing gel using a BIO-RAD tube gel apparatus. To elute RNA from the tube gel we used 0.5X TBE buffer supplemented with 3 M urea. Fractions were analyzed by UV absorbance and denaturing PAGE, and pure fractions were pooled, buffer exchanged into 10 mM Na HEPES pH 7.5, and concentrated using an Amicon centrifugal filter device.
In crystallo selection
Purified P4–P6 mutant library was annealed at 6 mg/ml by first preparing a solution of RNA in 10 mM NaCl, 5 mM MgCl2, and 5 mM Na HEPES pH 7.5. The RNA was heated to 55°C for 10 min followed by slow cooling of the heat block to around 40°C. The solution was then supplemented with 20 mM MgCl2 and allowed to cool further to room temperature. The annealed RNA library was screened for crystallization at room temperature by hanging drop vapor diffusion by mixing 2 μl RNA with 1 μl of well solution containing 24–30% (v/v) MPD or 22% PEG1000 with 50 mM sodium cacodylate pH 6.0 and 0.5 mM spermine. Crystals were observed in MPD and PEG1000 conditions within 4 days and grew to at least 200 μm in size. A large crystal from each condition was extensively washed in mother liquor and dissolved in 5 μl water. Reverse transcription was performed using 4 μl RNA, primer with sequence 5′-TGAACTGCATCCATATCAACAG-3′, and SuperScript II reverse transcriptase (ThermoFisher Scientific) at 42°C for 50 min. The cDNA product was amplified with PfuTurbo DNA polymerase (Agilent) and primers 5′-CAGTGAATTCAATTAATACGACTCACTATAGGAATTGCGGGAAAGGGGTC-3′ (underlined, EcoRI site; italic, T7 promoter; bold, part of transcribed P4–P6 sequence), and 5′-CGACTCTAGACTCTTCGTGAACTGCATCCATATCAACAG-3′ (underlined, XbaI site for cloning; italic, EarI site; bold, P4–P6 sequence). The PCR product was restriction digested, ligated into pUC19, and transformed as described in library construction.
Crystallization and structure determination
Mutant RNA was transcribed, purified, and annealed as described in in crystallo selection. M1 crystals prepared from PEG1000 solutions were produced at room temperature via hanging drop vapor diffusion by mixing 1 μl annealed RNA solution at 6 mg/ml with 2 μl of a well solution containing 22% (v/v) PEG1000, 50 mM sodium cacodylate pH 6.1, and 0.5 mM spermine. Single crystals were harvested and briefly soaked in a fresh well solution containing 10% isopropanol before flash frozen in liquid nitrogen. For M1 crystals obtained by MPD, 1 μl of a 6 mg/ml annealed RNA solution was mixed with 2 μl of a well solution containing 26% (v/v) MPD, 50 mM sodium cacodylate pH 6.0, and 1 mM cobalt hexamine. Crystals were flash-frozen without isopropanol treatment.
Crystals for M2 were grown by mixing 1 μl of 6 mg/ml RNA with 2 μl of well solution composed of 28% (v/v) MPD, 50 mM sodium cacodylate pH 6.0, and 0.5 mM spermine. M3 was crystallized under the same condition as M2 except that the MPD concentration in well solution was 31% (v/v). M2 and M3 crystals were frozen without isopropanol treatment.
M4 was crystallized from a drop containing 1 μl annealed RNA at 6 mg/ml and 2 μl of well solution composed of 22% (v/v) PEG1000, 50 mM sodium cacodylate pH 6.1, and 0.5 mM spermine. Prior to freezing, crystals were sequentially soaked in mother liquor solutions supplemented with 5%, 10%, and finally 15% (v/v) isopropanol.
Diffraction data were collected at 100K at the Advanced Photon Source beamline 24-ID-C using an energy of ~12.6 keV. Data were indexed, integrated, and scaled using XDS (Kabsch, 2010). To solve a structure, we started with the WT P4–P6 coordinates (PDB code 1GID) and deleted the mutated residues and an additional residue on the 5′ and 3′ sides of the mutation(s). We then generated an initial solution by performing rigid-body refinement of the deleted model to the data in PHENIX (Adams et al., 2010) using the default settings, which generally resulted in working and free R factors in the 30–35% range. Next we inspected the difference election density map; if additional residues near the site of mutation appeared to adopt an alternative conformation, we further reduced the phase bias of the search model by deleting those residues and repeating the rigid-body refinement. Magnesium ions that did not fit the electron density were removed. Once an acceptable starting model was obtained, we reset all atomic occupancies to 1 and manually modeled in the missing and mutated residues using Coot (Emsley et al., 2010). The model was further refined with BUSTER (Global Phasing Limited) (Smart et al., 2012), using the TLSbasic macro with target restraints from the initial deleted 1GID model and without B-factor refinement. Group B factors (one parameter per residue) were subsequently refined in PHENIX and the model was manually adjusted in Coot. These final refinement steps were iterated until an acceptable solution was obtained. All structure drawings were produced using PyMOL (Schrödinger, LLC).
Difference distance matrix plots
To calculate the difference distance matrices we used the distance_difference function of the Computational Crystallography Toolbox included with the Phenix distribution. We modified PDB files for the program to work with RNA structures. These PDB files contained only the C1′ atom records, in which the atom type was renamed to “CA” (α-carbon of a protein chain) and residue type changed to “GLY.” We also altered the Python script to output the difference distance matrix array, rather than generate a plot. We then plotted the matrix in GraphPad Prism (version 7).
QUANTIFICATION AND STATISTICAL ANALYSIS
Standard crystallography analysis methods were used, as described in the Method Details.
DATA AND SOFTWARE AVAILABILITY
Coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 6BJX, 6D8L, 6D8M, 6D8N, and 6D8O.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Taq DNA polymerase | Roche | Cat#11146165001 |
| EarI restriction endonuclease | New England Biolabs | Cat#R0528L |
| T7 RNA polymerase | This study | Prepared in lab |
| Inorganic pyrophosphatase | Sigma-Aldrich | Cat#I1643 |
| (+/−)-2-Methyl-2,4-pentanediol | Hampton Research | Cat#HR2-627 |
| Polyethylene glycol 1000 (50% w/v) | Hampton Research | Cat#HR2-523 |
| SuperScript II reverse transcriptase | ThermoFisher Scientific | Cat#18064014 |
| PfuTurbo DNA polymerase | Agilent | Cat#600250 |
| Deposited Data | ||
| Crystal structure of the WT P4–P6 domain | Cate et al., 1996 | PDB: 1GID |
| Crystal structure of P4–P6 mutant ΔC209 | Juneau et al., 2001 | PDB: 1HR2 |
| Crystal structure of P4–P6 mutant U131A (M1) grown under PEG1000 conditions and soaked with isopropanol | This study | PDB: 6BJX |
| Crystal structure of P4–P6 mutant U131A (M1) grown under MPD conditions and frozen without soaking | This study | PDB: 6D8L |
| Crystal structure of P4–P6 mutant A125U/G126U (M2) | This study | PDB: 6D8M |
| Crystal structure of P4–P6 mutant G134A/U185AA (M3) | This study | PDB: 6D8N |
| Crystal structure of P4–P6 mutant A230U (M4) | This study | PDB: 6D8O |
| Structure of the S-adenosylmethionine riboswitch | Montange and Batey, 2006 | PDB: 2GIS |
| Structure of the group II intron domains 5 and 6 | Zhang and Doudna, 2002 | PDB: 1KXK |
| Experimental Models: Organisms/Strains | ||
| E. coli: XL-1 Blue MRF’ electroporation-competent cells: Δ(mcrA)183 Δ(mcrCB-hsdSMR-mrr)173 endA1 supE44 thi-1 recA1 gyrA96 relA1 lac [F’ proAB lacIqZΔM15 Tn10 (Tetr)] | Agilent | Cat#200158 |
| Oligonucleotides | ||
| Error-prone PCR forward primer (5′-CAGTGAATTCAATTAATACGACTCACTATAGGAAT-3′) | This study | Synthesized in lab |
| Error-prone PCR reverse primer (5′-GCAGGTCGACTCTAGACTCTTC-3′) | This study | Synthesized in lab |
| Reverse transcription primer (5′-TGAACTGCATCCATATCAACAG-3′) | This study | Synthesized in lab |
| Library amplification forward primer (5′-CAGTGAATTCAATTAATACGACTCACTATAGGAATTGCGGGAAAGGGGTC-3′) | This study | Synthesized in lab |
| Library amplification reverse primer (5′-CGACTCTAGACTCTTCGTGAACTGCATCCATATCAACAG-3′) | This study | Synthesized in lab |
| Software and Algorithms | ||
| XDS | Kabsch, 2010 | http://xds.mpimf-heidelberg.mpg.de/ |
| PHENIX | Adams et al., 2010 | https://www.phenix-online.org/ |
| Coot | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| BUSTER | Smart et al., 2012 | https://www.globalphasing.com/buster/ |
| PyMOL | Schrödinger, LLC | https://pymol.org |
| Prism (version 7) | GraphPad Software | https://www.graphpad.com |
Supplementary Material
Highlights.
In crystallo selection method is invented for enhancing an RNA’s crystallizability
The strategy is successfully applied to the P4–P6 domain as a proof-of-principle
All 4 selected mutants we structurally characterized establish new crystal contacts
Three mutants make new contacts by relocating bulges along the RNA chain
Acknowledgments
We would like to thank Duilio Cascio and Michael Collazo in the UCLA-DOE crystallization and X-ray diffraction facilities for technical support. We thank Igor Kourinov at the Advanced Photon Source sector 24 (NE-CAT) for assistance with data collection; NE-CAT is supported by funding from the NIH (P41 GM103403 and S10 RR029205), and DOE (DE-AC02-06CH11357). This work is supported by the National Science Foundation (grant No. MCB-1616265) to F.G., and G.M.S. acknowledges support from the Ruth L. Kirschstein National Research Service Award GM007185 and the Whitcome Fellowship. T.R.C. is an investigator of the Howard Hughes Medical Institute.
Footnotes
AUTHOR CONTRIBUTIONS
F.G. and T.R.C. conceived the project over 15 years ago. F.G. and E.P. performed the in crystallo selection. G.M.S. rekindled the project. G.M.S., R.W. and F.G. prepared and crystallized the P4–P6 mutants and determined their structures. G.M.S. and F.G. wrote the manuscript with input from T.R.C.
DECLARATION OF INTERESTS
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abuhammad A, McDonough MA, Brem J, Makena A, Johnson S, Schofield CJ, Garman EF. “To cross-seed or not to cross-seed”: a pilot study using metallo-β-lactamases. Cryst Growth Des. 2017;17:913–924. [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cate JH, Gooding AR, Podell E, Zhou K, Golden BL, Kundrot CE, Cech TR, Doudna JA. Crystal structure of a group I ribozyme domain: principles of RNA packing. Science. 1996;273:1678–1685. doi: 10.1126/science.273.5282.1678. [DOI] [PubMed] [Google Scholar]
- Doudna JA. Preparation of homogeneous ribozyme RNA for crystallization. Methods Mol Biol. 1997;74:365–370. doi: 10.1385/0-89603-389-9:365. [DOI] [PubMed] [Google Scholar]
- Doudna JA, Grosshans C, Gooding A, Kundrot CE. Crystallization of ribozymes and small RNA motifs by a sparse matrix approach. Proc Natl Acad Sci U S A. 1993;90:7829–7833. doi: 10.1073/pnas.90.16.7829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferre-d’Amare AR, Doudna JA. Establishing suitability of RNA preparations for crystallization. Determination of polydispersity. Methods Mol Biol. 1997;74:371–377. doi: 10.1385/0-89603-389-9:371. [DOI] [PubMed] [Google Scholar]
- Ferre-D’Amare AR, Doudna JA. Crystallization and structure determination of a hepatitis delta virus ribozyme: use of the RNA-binding protein U1A as a crystallization module. J Mol Biol. 2000;295:541–556. doi: 10.1006/jmbi.1999.3398. [DOI] [PubMed] [Google Scholar]
- Ferre-D’Amare AR, Zhou K, Doudna JA. A general module for RNA crystallization. J Mol Biol. 1998;279:621–631. doi: 10.1006/jmbi.1998.1789. [DOI] [PubMed] [Google Scholar]
- Gesteland RF, Cech TR, Atkin JT. The RNA World. 10. Cold Spring Harbor Laboratory Press; 2005. [Google Scholar]
- Golden BL, Podell ER, Gooding AR, Cech TR. Crystals by design: a strategy for crystallization of a ribozyme derived from the Tetrahymena group I intron. J Mol Biol. 1997;270:711–723. doi: 10.1006/jmbi.1997.1155. [DOI] [PubMed] [Google Scholar]
- Guo F, Cech TR. Evolution of Tetrahymena ribozyme mutants with increased structural stability. Nat Struct Biol. 2002;9:855–861. doi: 10.1038/nsb850. [DOI] [PubMed] [Google Scholar]
- Guo F, Gooding AR, Cech TR. Structure of the Tetrahymena ribozyme: base triple sandwich and metal ion at the active site. Mol Cell. 2004;16:351–362. doi: 10.1016/j.molcel.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Juneau K, Cech TR. In vitro selection of RNAs with increased tertiary structure stability. RNA. 1999;5:1119–1129. doi: 10.1017/s135583829999074x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juneau K, Podell E, Harrington DJ, Cech TR. Structural basis of the enhanced stability of a mutant ribozyme domain and a detailed view of RNA--solvent interactions. Structure. 2001;9:221–231. doi: 10.1016/s0969-2126(01)00579-2. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke A, Doudna JA. Crystallization of RNA and RNA-protein complexes. Methods. 2004;34:408–414. doi: 10.1016/j.ymeth.2004.03.027. [DOI] [PubMed] [Google Scholar]
- Koldobskaya Y, Duguid EM, Shechner DM, Suslov NB, Ye J, Sidhu SS, Bartel DP, Koide S, Kossiakoff AA, Piccirilli JA. A portable RNA sequence whose recognition by a synthetic antibody facilitates structural determination. Nat Struct Mol Biol. 2011;18:100–106. doi: 10.1038/nsmb.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montange RK, Batey RT. Structure of the S-adenosylmethionine riboswitch regulatory mRNA element. Nature. 2006;441:1172–1175. doi: 10.1038/nature04819. [DOI] [PubMed] [Google Scholar]
- Nimjee SM, White RR, Becker RC, Sullenger BA. Aptamers as Therapeutics. Annu Rev Pharmacol Toxicol. 2017;57:61–79. doi: 10.1146/annurev-pharmtox-010716-104558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter NJ, Osterman A, Torres-Larios A, Swinger KK, Pan T, Mondragon A. Structure of a bacterial ribonuclease P holoenzyme in complex with tRNA. Nature. 2010;468:784–789. doi: 10.1038/nature09516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott WG, Finch JT, Grenfell R, Fogg J, Smith T, Gait MJ, Klug A. Rapid crystallization of chemically synthesized hammerhead RNAs using a double screening procedure. J Mol Biol. 1995;250:327–332. doi: 10.1006/jmbi.1995.0380. [DOI] [PubMed] [Google Scholar]
- Shechner DM, Grant RA, Bagby SC, Koldobskaya Y, Piccirilli JA, Bartel DP. Crystal structure of the catalytic core of an RNA-polymerase ribozyme. Science. 2009;326:1271–1275. doi: 10.1126/science.1174676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart OS, Womack TO, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C, Bricogne G. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr D Biol Crystallogr. 2012;68:368–380. doi: 10.1107/S0907444911056058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- Vicens Q, Gooding AR, Laederach A, Cech TR. Local RNA structural changes induced by crystallization are revealed by SHAPE. RNA. 2007;13:536–548. doi: 10.1261/rna.400207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DS, Szostak JW. In vitro selection of functional nucleic acids. Annu Rev Biochem. 1999;68:611–647. doi: 10.1146/annurev.biochem.68.1.611. [DOI] [PubMed] [Google Scholar]
- Ye JD, Tereshko V, Frederiksen JK, Koide A, Fellouse FA, Sidhu SS, Koide S, Kossiakoff AA, Piccirilli JA. Synthetic antibodies for specific recognition and crystallization of structured RNA. Proc Natl Acad Sci U S A. 2008;105:82–87. doi: 10.1073/pnas.0709082105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ferre-D’Amare AR. Co-crystal structure of a T-box riboswitch stem I domain in complex with its cognate tRNA. Nature. 2013;500:363–366. doi: 10.1038/nature12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ferre-D’Amare AR. New molecular engineering approaches for crystallographic studies of large RNAs. Curr Opin Struct Biol. 2014;26:9–15. doi: 10.1016/j.sbi.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Doudna JA. Structural insights into group II intron catalysis and branch-site selection. Science. 2002;295:2084–2088. doi: 10.1126/science.1069268. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.