

Abstract
α-Synuclein (α-Syn) is a presynaptic protein that is accumulated in its amyloid form in the brains of Parkinson’s patients. Although its biological function remains unclear, α-syn has been suggested to bind to synaptic vesicles and facilitate neurotransmitter release. Recently, studies have found that α-syn induces membrane tubulation, highlighting a potential mechanism for α-syn to stabilize highly curved membrane structures which could have both functional and dysfunctional consequences. To understand how membrane remodeling by α-syn affects amyloid formation, we have studied the α-syn aggregation process in the presence of phosphatidylglycerol (PG) micellar tubules, which were the first reported example of membrane tubulation by α-syn. Aggregation kinetics, β-sheet content, and macroscopic protein-lipid structures were observed by Thioflavin T fluorescence, circular dichroism spectroscopy and transmission electron microscopy, respectively. Collectively, the presence of PG micellar tubules formed at a stochiometric (L/P =1) ratio was found to stimulate α-syn fibril formation. Moreover, transmission electron microscopy and solid-state nuclear magnetic resonance spectroscopy revealed the co-assembly of PG and α-syn into fibril structures. However, isolated micellar tubules do not form fibrils by themselves, suggesting an important role of free α-syn monomers during amyloid formation. In contrast, fibrils did not form in the presence of excess PG lipids (≥ L/P = 50), where most of the α-syn molecules are in a membrane-bound α-helical form. Our results provide new mechanistic insights into how membrane tubules modulate α-syn amyloid formation and support a pivotal role of protein–lipid interaction in the dysfunction of α-syn.
Keywords: membrane remodeling, protein-lipid interaction, Parkinson’s disease, transmission electron microscopy, Thioflavin T, circular dichroism
Graphical abstract

1. Introduction
α-Synuclein (α-syn) is a vertebrate specific protein, expressed primarily in neurons of the central nervous system [1]. It is found at the highest concentrations (~20–100 μM [2, 3]) in the presynaptic spaces at the ends of axons. Lower levels of the protein are observed in other cell types of the central nervous system, and the gene is also transcribed in other tissues [1]. Within neurons, α-syn is mostly cytosolic and enriched in presynaptic nerve terminals with some fraction associated with synaptic vesicles [4]. Despite extensive study, pinpointing specific biological roles for α-syn has proven difficult. Mice with the α-syn gene (SNCA) knocked out presented no obvious pathology, with only subtle perturbation of neuronal function [5]. There are two other α-syn-related proteins, β-syn and γ-syn [4]. For mice with all three synuclein genes knocked out, pathology was pronounced, altering synaptic structure and transmission and causing age-dependent neuronal dysfunction, implying a role for the synucleins in long-term maintenance of neurons [6]. Detailed investigation of neurotransmitter release from presynaptic vesicles has revealed a role for α-syn as a chaperone protein, aiding the rapid re-assembly of the SNARE complex of proteins [7, 8]. The SNARE complex is responsible for membrane fusion in the exocytosis process that releases the neurotransmitters into the synapse [9].
α-Syn is implicated in Parkinson’s disease (PD) as a pathogenic agent through disease-related mutations as well as gene duplication and triplication, which cause early-onset autosomal dominant PD [10–17]. Notably, cytosolic accumulation of insoluble amyloid fibrils of α-syn is the cellular hallmark for both sporadic and familial PD [18]. Gaining insights on how soluble α-syn transitions to aggregated forms will facilitate elucidation of molecular events in amyloid pathology. Because α-syn is both cytoplasmic and membrane-associated, full characterization of protein-membrane interactions is necessary. For example, the presence and the type of membranes can either stimulate or inhibit α-syn oligomerization and amyloid formation in vitro [19–23]. The relationship between membranes and α-syn amyloid formation is further strengthened by observations that early-onset mutants exhibit different membrane-binding properties and that prefibrillar α-syn can permeabilize membranes [24–31]. Furthermore, there is mounting cellular evidence that overexpression of α-syn is correlated with observations of Golgi fragmentation [32, 33], mitochondrial fission [34], and lysosomal malfunction [35, 36], supporting the involvement of protein-lipid interaction in pathogenesis. However, the exact molecular mechanisms by which α-syn promotes membrane disruption are not well understood [37–39]. The various cellular membranes have distinct chemical and physical characteristics, such as specific phospholipid composition and membrane curvature, which are especially important for modulating α-syn membrane interactions [40]. Indeed, membrane binding of α-syn is sensitive to vesicle size and the presence of anionic phospholipids [41, 42]. Thus, we are motivated to develop understanding of how specific phospholipids modulate protein structure, membrane binding, and aggregation propensity of α-syn.
Our recent work indicates that α-syn strongly influences the structure and properties of phospholipid bilayers including membrane thickness [43, 44] and formation of lipid tubular structures [45]. Other examples of α-syn–lipid structures such as bilayer discs [46–48] and cylindrical micellar tubules [49, 50] also have been reported in the literature. The biological relevance of membrane tubulation induced by α-syn is bolstered by the enhanced levels of several membrane curvature sensing/generating proteins (BAR domains) in mice with all three α-, β-, and γ-syn genes knocked out, implying up-regulation to compensate the loss of function from synuclein deficiency [51]. We are interested in the interplay between protein structure and membrane deformation and whether this process can influence amyloid formation. In a prior study, we showed that the presence of zwitterionic phosphatidylcholine tubules inhibited α-syn amyloid formation via a competing mechanism for the available pool of soluble protein [45]. While phosphatidylcholine is a major component of cellular membranes, it is well established that α-syn prefers to bind to negatively charged membranes composed of anionic lipids such as phosphatidic acid (PA), phosphatidylserine (PS), and phosphatidylglycerol (PG) [40]. Consequently, the ability for α-syn to tubulate membranes is strongly enhanced as shown for POPG, where thin, cylindrical micelle tubules are induced by α-syn [49, 50]. Here, we have extended our study to phosphatidylglycerol micellar tubules and their influence on α-syn aggregation, to uncover how protein-lipid structures can play a role in amyloid fibril formation.
2. Materials and methods
2.1. Chemicals
All lipids (POPG, (1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (sodium salt)), POPA (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate), POPS (1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine), and POPG-d31 (1-palmitoyl(D31)-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)) were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). Thioflavin T (ThT), 3-(N-morpholino) propanesulfonic acid (MOPS), and sodium chloride were purchased from Sigma-Aldrich (St. Louis, MO). Ultra-pure tris(hydroxymethyl)aminomethane (Tris) and guanidinium hydrochloride (GuHCl) were purchased from MP Biomedicals (Santa Ana, CA).
2.2. Lipid and vesicle preparation
Lipids (~30 mM) were stored in chloroform stock solutions. Lipid films were prepared by drying appropriate amounts of the stock solution in a stream of dry N2 followed by placement in a vacuum oven (<20 kPa) at 40 °C for at least 2 h to ensure removal of organic solvent. To make multilamellar vesicles (MLVs), buffer solution (20 mM MOPS, 100 mM NaCl, pH 7) was added to the dried lipid films and vortexed at least 3 times for 60 s. To form small unilamellar vesicles (SUVs), a probe-tip sonication method was used as previously described [45]. Vesicle sizes were determined by dynamic light scattering using a Dynapro NanoStar (Wyatt) and visualized by TEM. Each measurement contained ten 5-s acquisitions. A cumulant fit and a sphere model were used to obtain the average hydrodynamic radius (maximum intensity at radius ~ 40 nm). All buffers were filtered through a 0.22 μm filter (Millipore) before use.
2.3. α-Syn preparation and aggregation experiments
α-Syn was expressed and purified as previously described [52] with the following modification. Instead of applying the dialyzed fractions from the Hi-Prep DEAE column (GE Healthcare) directly onto MonoQ 16/10 (Amersham Biosciences), the protein was first treated with GuHCl to a final concentration of 4 M, buffer exchanged by a Hi-Prep Desalting column (GE Healthcare) into 20 mM Tris, pH 8 buffer, and then applied to Mono Q 16/10 column. Protein homogeneity was assessed by SDS-PAGE, visualized by silver-staining, as well as LC-MS. Samples for aggregation experiments were prepared by exchanging purified α-syn into pH 7 buffer (20 mM MOPS, 100 mM NaCl) using a pre-chilled PD-10 column (GE Healthcare). The samples were then filtered through YM-100 filters (Millipore) to remove any preformed aggregates immediately prior to aggregation. Final solutions contained 70 μM α-syn and specified lipids/vesicles. Aggregation experiments were performed in Eppendorf tubes or sealed quartz cuvettes at 37 °C with continuous shaking (600 rpm, VWR Mini-Micro 980140 shaker). For ThT time-tracking experiments, small aliquots were taken out and diluted 10-fold to incubate with 20 μM ThT for 15 min. ThT fluorescence was measured in a micro-quartz cuvette (Starna) at 25 °C using a Horiba Fluorolog-3 spectrofluorimeter (λex = 400 nm, λobs = 450–600 nm, 0.25 s integration time, 1 and 2 nm excitation and emission slit widths, respectively). CD time tracking measurements were performed on the same samples that contained 20 μM ThT using 1 mm quartz cuvettes and a Jasco J-715 spectropolarimeter (collection parameters were 198 to 260 nm, 1 nm data pitch, continuous scanning with 1 nm bandwidth, 100 nm/min, and 3 accumulations, 25 °C).
2.4. Transmission Electron Microscopy
TEM was performed using a JEOL JEM 1200EX transmission electron microscope (accelerating voltage 80 keV) equipped with an AMT XR-60 digital camera. A 3 μL sample droplet was applied onto grids (400-mesh formvar and carbon coated copper, Electron Microscopy Sciences) for 1 min, wicked-off by filter paper, followed by 3 μL H2O droplet to remove excess sample material, wicked-off by filter paper, then stained with a few drops of 1% (w∕v) aqueous uranyl acetate solution. Excess solution was absorbed with filter paper and air-dried. For OsO4 staining, 0.1% solution was used in a closed glass petri-dish for 30 min, then air-dried overnight in the hood.
2.5. Solid-state NMR
Purified α-syn was exchanged into water containing 0.01% NaN3 using a PD-10 desalting column (GE Healthcare) and lyophilized. The protein was then rehydrated into 20 mM MOPS, 100 mM NaCl buffer made in deuterium-depleted H2O. The final sample contained 800 μM each of α-syn and POPG-d31. After 3 days of incubation at 37 °C, the fibrils were spun-down using a TLA100.2 rotor at 100,000 rpm in a Beckman Coulter ultracentrifuge for 1 h, and the pellet was collected. Spectra were recorded at ambient temperature on a DMX500 spectrometer (Bruker Biospin Inc., Billerica, MA) equipped with a 4 mm 1H/X gradient MAS probe (Doty Scientific, Columbia SC) using a quadrupolar echo sequence, pulse length of 7 μs, echo delay time of 50 μs, delay time between transients 250 ms, spectral width 200 kHz. For the MLV control sample, 4,096 transients of 4 K complex time-domain data points were acquired, and 204,800 transients were acquired for α-syn and POPG (L/P = 1) sample.
2.6. Isolation of long micellar tubules
Samples containing α-syn and POPG were spun-down using a TLA100.2 rotor at 100,000 rpm in a Beckman Coulter ultracentrifuge for 1 h, and the pellet containing the micelle tubules was collected.
3. Results
3.1. Tubulation of anionic membranes by α-syn
To test whether tubulation of negatively charged membranes is a common property of α-syn, MLVs composed of anionic POPA, POPG, and POPS (Fig. 1A) were incubated with different concentrations of monomeric α-syn ([lipid] = 600 μM, lipid-to-protein (L/P) = 5, 20, and 50) for 15 min at RT. We were particularly interested in POPA because of its intrinsic preference for negative curvature due to its small headgroup. Further, POPA membrane tubulation induced by α-syn had not been evaluated before. The most extensively studied lipid in this context is POPG [49, 50, 53]. Lipid tubules were clearly visualized by TEM at all concentrations. Representative TEM images are shown in Figs. 1E, 1F, and 1G for L/P = 20. For comparison, TEM of the starting, large MLVs are also shown in Figs. 1B, 1C, and 1D. After adding α-syn, the micron-sized MLVs disappear and the dominant structures were tubules with approximate diameters of 7 nm, consistent with prior reports of cylindrical micelles induced by α-syn [49, 50]. The tubules appear like a network of flexible strings. No obvious morphological differences were seen amongst the three chemically diverse anionic lipids.
Figure 1.
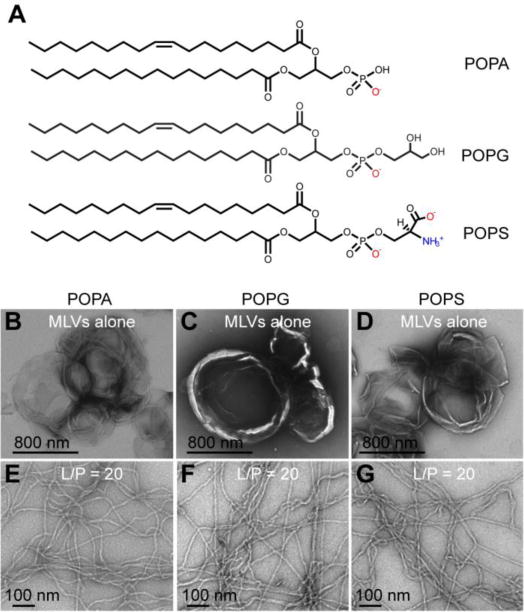
α-Syn tubulates anionic membranes. Chemical structures of POPA, POPG, and POPS (A). TEM images of POPA (B), POPG (C), and POPS (D) MLVs (600 μM) before and after (E-G) adding α-syn (30 μM, L/P = 20). Experiments were performed in pH 7 MOPS buffer with 100 mM NaCl at RT. Scale bars as noted.
3.2 Stability of POPG tubules
Because MLVs are not compatible for spectroscopic measurements such as CD or fluorescence, we used SUVs for the rest of this study. To examine the stability of POPG tubules induced by α-syn, a low vs. high lipid content condition (L/P = 1 and L/P = 50) was compared. α-Syn was added to POPG SUVs and incubated for 3 days at 37 °C. The protein concentration was fixed at 70 μM and the lipid concentration varied. Within 15 min of mixing (termed as T0), the samples were deposited and prepared for TEM analysis. Representative TEM images are shown on the top panels of Fig. 2. POPG SUVs alone are shown in Fig. 2A where only circular vesicles are observed. At L/P = 1, thin POPG micellar tubules with ~ 7 nm diameter appear (Fig. 2B). The tubules are curvy and entangled together. Some small circular particles are also seen which are reminiscent of those previously reported POPG nanoparticles [46, 48], but because SUVs were used here, the two species cannot be readily discerned. At higher POPG concentration, only wider, presumed bilayer tubes (~ 30 nm) were observed. These results reaffirm that membrane curvature generation by α-syn is positively correlated with protein concentration. To determine that monomers are involved in this process, a control was conducted with preformed α-syn fibrils which are not able to tubulate POPG vesicles (Figure S1).
Figure 2.
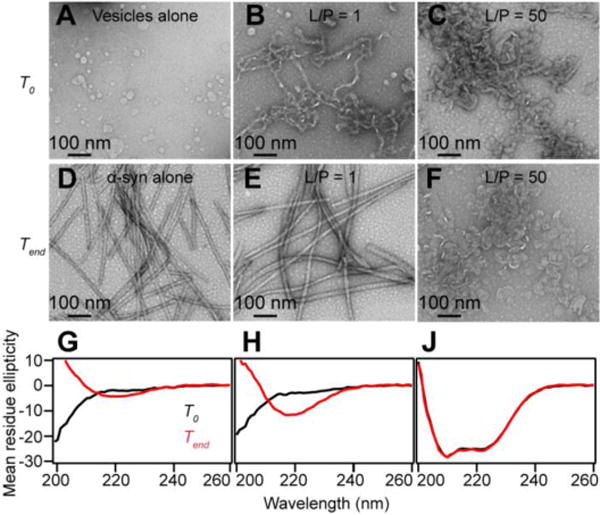
Stability of POPG tubules as assessed by TEM and CD. Representative TEM images of POPG SUVs alone (A), micelle tubules and bilayer tubes induced by α-syn at L/P = 1 (B) and L/P = 50 (C) before (marked as T0), and of α-syn in the absence (D) and presence of POPG after incubation at 37 °C with agitation (marked as Tend) for L/P = 1 (E) and L/P = 50 (F). Scale bars as noted. Corresponding CD spectra of α-syn alone (G), L/P = 1 (H), and L/P = 50 (J) at T0 (black) and Tend (red). Unit of mean residue ellipticity (deg cm2 dmol−1 × 10−3) is omitted for clarity. Experiments were performed with 70 μM α-syn in Eppendorf tubes with low (70 μM) and high (3.5 mM) concentrations of POPG with shaking at pH 7 (20 mM MOPS, 100 mM NaCl).
After 3 days of incubation (denoted as Tend, Fig. 2E and 2F), a noticeable difference can be seen from the low POPG content (L/P = 1), but not from the high POPG content sample. Thicker, and straight filaments (~15–20 nm) are now observed, nearly indistinguishable from the control sample, α-syn amyloid fibrils in the absence of lipids (Fig. 2D). Further, the POPG micellar tubules (~ 7 nm) observed at T0 are not found and thus, appear to have been consumed. To corroborate that the filaments visualized by TEM are indeed α-syn amyloid fibrils in the low POPG condition, CD spectroscopy was employed to probe for the presence of β-sheet secondary structure. The measured CD spectra confirmed that a transformation of protein conformation had occurred. At Tend, a characteristic negative maximum at 218 nm for β-sheet is exhibited (Fig. 2G). Compared to α-syn alone sample, the β-sheet content is enhanced in the presence of stoichiometric amount of POPG (Fig. 2H). In contrast, CD spectra of high POPG concentration overlaid for T0 and Tend (Fig. 2J) confirm no secondary structure change had occurred and that the bilayer tubes seen in the TEM images are stable structures under the conditions examined here. Instead of one negative maximum, two negative peaks at 208 and 222 nm were observed, signatures for an α-helical conformation stabilized by POPG [49]. These data suggest that POPG micellar tubules have led to α-syn amyloid formation, whereas the POPG bilayer tubes sequester α-syn in an α-helical state and inhibit amyloid formation.
3.3 Effect of POPG on α-syn amyloid formation kinetics
To investigate specifically on how POPG micellar tubules disappear and α-syn amyloid fibrils form, a more detailed aggregation kinetics experiment was performed. Aliquots were taken out at each time point (~ every 3–4 h) and evaluated by Thioflavin T (ThT) fluorescence, CD spectroscopy, and TEM. ThT and TEM were used to detect and visualize amyloid fibrils, respectively. CD measurements were used to assess secondary structural changes.
An example of ThT fluorescence enhancement during the aggregation of α-syn is shown in Fig. 3A. By plotting the integrated ThT intensity from 450 to 550 nm as a function of incubation time, the expected sigmoidal kinetics curve is generated (Fig. 3B). Here, data for two replicates exhibit a lag phase of ~ 43–50 h. In the presence of low lipid content (L/P = 1), faster aggregation is observed with a significantly shortened lag phase (~ 20 h). The growth phase slows but, the stationary phase is still reached sooner than α-syn alone. Consistent to our equilibrium incubation experiments (Fig. 2F), no ThT intensity change was observed at L/P = 50, indicating fibril formation is suppressed.
Figure 3.
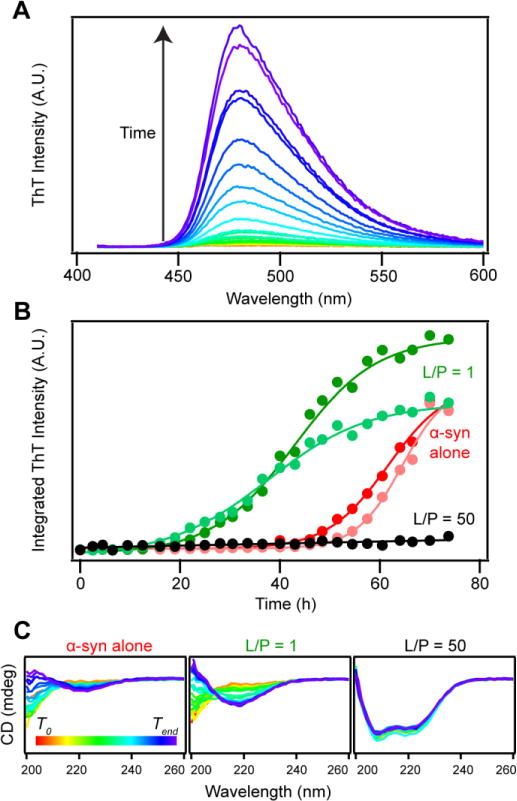
Effect of POPG on α-syn aggregation kinetics. (A) ThT fluorescence of α-syn (70 μM) in buffer (20 mM MOPS, 100 mM NaCl, pH 7) at different time points (red-to-purple indicates T0 to Tend). (B) Integrated ThT intensity of α-syn as a function of incubation time (37 °C) in the absence (pink and red) and presence of POPG (L/P = 1 (light and dark green) and L/P = 50 (black)). (C) Corresponding CD spectra of α-syn in buffer (left), L/P = 1 (middle), and L/P = 50 (right) at different time points. Color scheme from red to purple represents T0 to Tend of incubation. All panels have the same y-axes. Labels have been omitted for clarity.
The corresponding CD spectra are shown in Fig. 3C. Both L/P = 1 and α-syn alone exhibit the characteristic feature for β-sheet formation at 218 nm. Interestingly, a stronger peak is seen at L/P = 1, indicating more β-sheet content. In contrast, at L/P = 50 α-syn remains α-helical throughout the experiment.
3.4 Effect of POPG on α-syn amyloid fibril structure
To examine the macroscopic changes during aggregation, TEM images were collected at different time points (Fig. 4). α-Syn in the absence of POPG lipids served as the fibril reference. Small aggregates started to appear at 22 h whereas mature fibrils were apparent at 33.5 h. Interestingly, many amyloid fibrils were already detectable by TEM especially during the late lag phase, where ThT response is still negligible. This could be due to the amounts of fibrils present are below the ThT detection limit. Alternatively, it is plausible that the lack of a strong ThT response is due to differences in the molecular features of the fibrils during lag and stationary phases.
Figure 4.
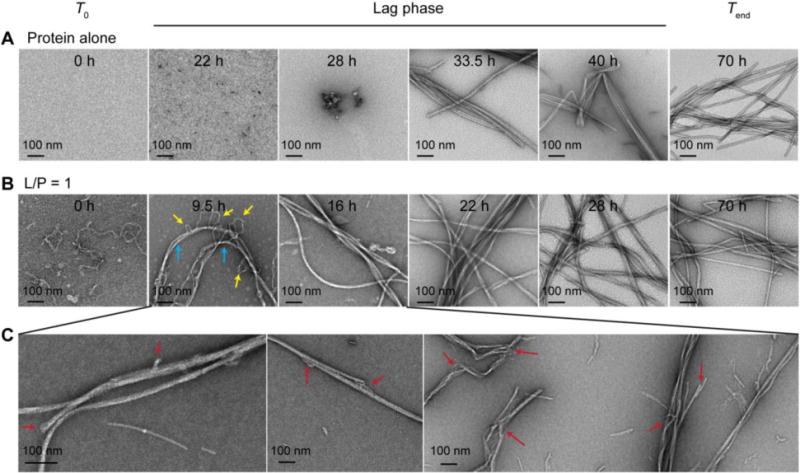
TEM visualization of α-syn fibril formation in the absence (A) and presence of stoichiometric POPG (B) at various time points (as indicated). Yellow and cyan arrows indicate micellar tubules and amyloid fibrils, respectively. (C) Gallery of structures found during the lag phase (9.5–16 h). Uncommon features not observed for α-syn alone are indicated by red arrows. Scale bars are as shown.
As previously observed at L/P = 1, the initial structures are mostly thin micellar tubules. After 9.5 h, longer fibrillar structures (as indicated by cyan arrows) are visualized (Fig. 4B). Micellar tubules (as indicated by yellow arrows) appeared to be wrapping around or inserting into the amyloid fibrils, suggestive of a co-assembly process. At 16 h, long and straight fibrils are observed with few curvy micellar tubules. At 22 h, the dominant structures are more laterally associated fibrils, which change little with prolonged incubation up to 70 h. While in agreement with the ThT kinetics data that α-syn amyloid formation is stimulated at L/P = 1, amyloid fibrils are seen by TEM before a measurable ThT intensity increase. A careful examination of multiple TEM images reveals distinct morphologies that were not observed for α-syn aggregated alone in buffer. As shown in Fig. 4C, unusual fibrillar states, such as split-ends and branching (indicated by arrows), were observed during the lag phase (< 20 h), suggesting that POPG lipids are directly participating in the fibril assembly process. Further, we hypothesize that the molecular composition and structure of the final fibrillar state for L/P = 1 is altered, which is supported by the intensity differences observed in the CD data. In contrast, there was no obvious morphological changes at high lipid content of L/P = 50 (Fig. S2).
Tracking by TEM revealed the possibility of incorporation of POPG micellar tubules into the mature fibrils and thus, could serve as a nucleation surface and promote fibril growth. To test this hypothesis, OsO4, a lipid-specific staining reagent was utilized. It positively stains for unsaturated lipids because it reacts with the double bond on the acyl chain. α-Syn fibrils formed at L/P = 1 indeed showed positive-stained filaments (Fig. S3), confirming the presence of lipids. In addition, using POPG-d31 with a perdeuterated palmitic acid chain and solid-state 2H-NMR, the presence of POPG is verified in the pelleted amyloid fibril samples (Fig. S4). The 2H spectra report asymmetry in motions of lipid molecules in lipid mesophases as well as a gradient in order of lipid methylene segments along the hydrocarbon chain. Spectra originating from lipids in a small tubular structure are expected to yield a band of resonances of about half the width compared to a lamellar phase. Indeed, Fig. S4 appears to be a superposition of a band of broadened resonances characteristic of a fluid lamellar phase in coexistence with a band of resonances of reduced width as expected for fluid lipids in tubules of small diameter. Analysis is complicated by broadening of resonances due to lipid-protein interaction and curvature of mesophases.
3.5. Stability of isolated POPG micellar tubules
One possible mechanism in which POPG micellar tubules can modulate α-syn amyloid formation is through tubule self-association, bringing surface-bound proteins together, spatially constraining them between tubules, and stabilizing intermolecular interactions towards amyloid formation. To test that hypothesis, we examined whether POPG micellar tubules would be stable upon removal from free α-syn and whether they could convert to amyloid fibrils on their own. POPG micellar tubules were pelleted by ultracentrifugation and then incubated at 37 °C with agitation for ~ 72 h. Samples before and after incubation were processed for TEM analysis (Fig. 5). While it is evident that the tubules have flattened (~30 nm) and appear more tape-like at the end of 3 days, no prototypical straight amyloid fibrils were found. This result suggests that the presence of unbound α-syn molecules play an important role in amyloid assembly.
Figure 5.
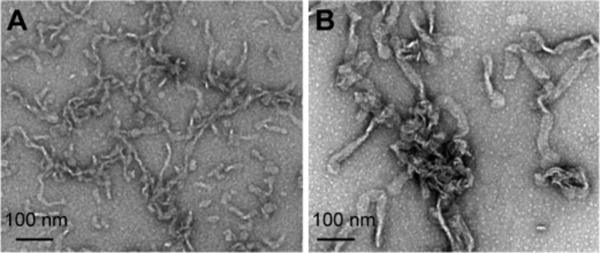
Stability of isolated POPG micellar tubules. Sample composed of POPG and α-syn at L/P = 1 was spun down using ultracentrifugation. (A) TEM image of the pellet fraction containing isolated POPG micelle tubules. (B) TEM image of sample shown in (A) after incubation at 37 °C with agitation for ~ 72h. Scale bars are as shown.
4. Discussion
4.1. Biological implications of membrane remodeling
The ability of α-syn to bend anionic membranes into structures with higher curvature is explained by the hydrophobic insertion mechanism, where the extended amphipathic helix of α-syn acts as a wedge, pushing phospholipid headgroups apart, thereby inducing thinning and spontaneous local curvature [54]. This deformation process could be related to its proposed function in the recycling of synaptic vesicles [7, 8], but because anionic lipids are generally in low abundance (synaptic vesicles contain ~12% POPS [55]), membrane curvature generation by α-syn is uncertain in vivo. However, if the local concentration of anionic lipids is increased under certain conditions, such as in aging for phosphatidic acid [56, 57], membrane remodeling and amyloid formation could potentially occur and lead to vicious consequences.
In a broader context, intimate relationships have also been observed for membranes and fibril formation of other amyloidogenic peptides linked to Alzheimer’s and type-II diabetes such as amyloid-β and Islet amyloid peptide (IAPP), respectively. For example, lipids influence Aβ fibril formation kinetics and morphology [58, 59] and membrane fragmentation was observed during fibril growth of IAPP [60–62]. Furthermore, membrane tubulation has also been documented for IAPP [63]. Taken together, interplay between membrane deformation and aggregated protein/lipid structures could underlie a common mechanism in the progression of amyloid-related diseases, offering a new avenue for potential treatment through the modulation of protein-membrane interactions, which has been recently suggested for α-syn [64].
4.2. Influence of POPG on α-syn amyloid formation
Our data show that α-syn forms amyloids faster in the presence of micellar tubules. Conversely, an excess of lipids inhibits α-syn amyloid formation, where α-helical structure is stabilized. A simple explanation of this observation is the relative surface availability for α-syn binding. When there is a sufficient reservoir of free α-syn, the POPG micellar tubules serve as a limited platform to accumulate α-syn molecules favoring aggregation. However, with higher amounts of POPG present, there is more membrane surface to accommodate α-syn, thus minimizing interprotein interactions and preventing amyloid fibril formation.
Interestingly, more β-sheet content was observed by CD for L/P = 1, suggesting that the presence of tubules can increase the amounts of fibrils formed. Alternatively, fibrils formed at L/P = 1 could adopt different conformations (i.e. polymorphic) resulting in more β-sheet content, such as longer β-strands or a larger amyloid core. TEM images showed no obvious differences between mature amyloid fibrils formed in the absence and presence of lipids, which suggest that it is fibril abundance. However, complex structures were revealed by TEM images taken during the lag phase, hinting at a different mechanism of amyloid fibril assembly and underlying molecular structures as evidenced by the uncommon features of split-ends and branching.
While it is difficult to pinpoint the exact mechanism of how the POPG micellar tubules stimulate α-syn amyloid formation, we consider the following scenario to rationalize our data. One possibility is that POPG micellar tubules recruit α-syn to the surface and effectively increase its local concentration, leading to nucleation and resulting in the observed stimulatory effect. During this process, the micellar structure could become vulnerable as molecules of α-syn self-associate and begin to dissociate from the lipid surface. Then, the exposed POPG molecules would be in competition with the amyloid fibrils for the free pool of α-syn. On the other hand, the fibrils themselves could interact with the micellar tubules due to electrostatics of N-terminal Lys residues outside the amyloid core [65–68], which could lead to fibril deposition on the lipid tubules. In addition, POPG molecules could reorganize and redistribute to more favorable sites on the amyloid fibrils. This would result in the gradual disappearance of the micellar tubules as seen in the TEM images during the lag phase. Images from the OsO4 staining experiment also suggest that the tubules have restructured during amyloid formation as α-syn fibrils appear uniformly decorated with POPG. The co-assembly of phospholipids and α-syn fibrils also has been previously reported by Hellstrand et al. [69]. Finally, as POPG lipids are incorporated into the fibrils, they could also function as secondary nucleation sites by recruiting more α-syn to the fibril surface through electrostatics. In this mechanism, soluble monomers are needed to drive amyloid formation which is supported by the observation that isolated tubules cannot form amyloid fibrils by themselves.
5. Conclusion
In the present study, the aggregation process of α-syn in the presence of POPG membranes was examined using ThT fluorescence, CD spectroscopy, and TEM. Collectively, the data showed that micellar POPG tubules formed at low stoichiometric ratio (L/P = 1) lead to the stimulation of α-syn fibril formation. Moreover, TEM and solid-state NMR provided evidence of co-assembly of lipids and α-syn into fibrillar structures. Isolated micellar tubules do not form α-syn fibrils by themselves, suggesting the important role of free α-syn monomers during amyloid formation. In contrast to L/P = 1, no fibrils were evident at or above L/P = 50, where α-helices of α-syn are stabilized. Our results suggest that membrane tubules stimulate α-syn amyloid formation and support a pivotal role of protein–lipid interaction in the dysfunction of α-syn in PD.
Supplementary Material
Highlights.
α-Syn remodels anionic PG, PA, and PS membranes into micellar tubules
Effect of POPG on α-syn amyloid formation was probed by CD, ThT fluorescence, and TEM
POPG micellar tubules shorten the lag phase and enhanced β-sheet formation
POPG micellar tubules are co-assembled into α-syn fibrils
POPG micellar tubes do not to convert into amyloids in the absence of free α-syn
Acknowledgments
This work is supported by Intramural Research Program at the National Institutes of Health, National Heart, Lung, and Blood Institute (NHLBI) and National Institute on Alcohol Abuse and Alcoholism. DLS and CD measurements were performed using instruments in the NHLBI Biophysics Core Facility. TEM and LC-MS were performed in the NHLBI Electron Microscopy and Biochemistry Cores, respectively.
Abbreviations
- α-Syn
α-Synuclein
- PD
Parkinson’s disease
- L/P
lipid-to-protein
- POPG
1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)
- POPA
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate
- POPS
1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine
- POPG-d31
1-palmitoyl(D31)-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)
- ThT
Thioflavin T
- GuHCl
guanidinium hydrochloride
- MOPS
3-(N-morpholino) propanesulfonic acid
- Tris
tris(hydroxymethyl)aminomethane
- TEM
transmission electron microscopy
- CD
circular dichroism
- MLV
multilamellar vesicle
- SUV
small unilamellar vesicle
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DAC, Kondo J, Ihara Y, Saitoh T. Molecular-cloning of cDNA-encoding an unrecognized component of amyloid in Alzheimer-disease. Proc Natl Acad Sci U S A. 1993;90:11282–11286. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilhelm BG, Mandad S, Truckenbrodt S, Krohnert K, Schafer C, Rammner B, Koo SJ, Classen GA, Krauss M, Haucke V, Urlaub H, Rizzoli SO. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science. 2014;344:1023–1028. doi: 10.1126/science.1252884. [DOI] [PubMed] [Google Scholar]
- 3.Iwai A, Masliah E, Yoshimoto M, Ge NF, Flanagan L, Desilva HAR, Kittel A, Saitoh T. The precursor protein of non-Aβ component of Alzheimers-disease amyloid is a presynaptic protein of the central-nervous-system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- 4.Clayton DF, George JM. The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998;21:249–254. doi: 10.1016/s0166-2236(97)01213-7. [DOI] [PubMed] [Google Scholar]
- 5.Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JMG, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking α-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- 6.Greten-Harrison B, Polydoro M, Morimoto-Tomita M, Diao L, Williams AM, Nie EH, Makani S, Tian N, Castillo PE, Buchman VL, Chandra SS. αβγ-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci USA. 2010;107:19573–19578. doi: 10.1073/pnas.1005005107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burre J, Sharma M, Sudhof TC. α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci USA. 2014;111:E4274–E4283. doi: 10.1073/pnas.1416598111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rizo J, Sudhof TC. SNAREs and Munc18 in synaptic vesicle fusion. Nature Rev Neurosci. 2002;3:641–653. doi: 10.1038/nrn898. [DOI] [PubMed] [Google Scholar]
- 10.Polymeropoulos MH, Lavedan C, Leroy E, Ide S, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos E, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson W, Lazzarini A, Duvoisin R, DiIorio G, Golbe L, Nussbaum R. Mutation in the α-synuclein gene identified in families with Parkinson s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 11.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson s disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 12.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Tortosa EG, del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 13.Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destee A. α-Synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 14.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. α-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841–841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 15.Lesage S, Anheim M, Letournel F, Bousset L, Honore A, Rozas N, Pieri L, Madiona K, Durr A, Melki R, Verny C, Brice A, S. French Parkinsons Dis Genet G51D α-synuclein mutation causes a novel Parkinsonian-pyramidal syndrome. Ann Neurol. 2013;73:459–471. doi: 10.1002/ana.23894. [DOI] [PubMed] [Google Scholar]
- 16.Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, Weir D, Thompson C, Szu-Tu C, Trinh J, Aasly JO, Rajput A, Rajput AH, Stoessl AJ, Farrer MJ. α-Synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord. 2013;28:811–813. doi: 10.1002/mds.25421. [DOI] [PubMed] [Google Scholar]
- 17.Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, Tienari PJ, Poyhonen M, Paetau A. A novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol Aging. 2014;35 doi: 10.1016/j.neurobiolaging.2014.03.024. [DOI] [PubMed] [Google Scholar]
- 18.Cookson MR. The biochemistry of Parkinson’s disease. Annu Rev Biochem. 2005;74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400. [DOI] [PubMed] [Google Scholar]
- 19.Zhu M, Li J, Fink AL. The association of α-synuclein with membranes affects bilayer structure, stability, and fibril formation. J Biol Chem. 2003;278:40186–40197. doi: 10.1074/jbc.M305326200. [DOI] [PubMed] [Google Scholar]
- 20.Zhu M, Fink AL. Lipid binding inhibits α-synuclein fibril formation. J Biol Chem. 2003;278:16873–16877. doi: 10.1074/jbc.M210136200. [DOI] [PubMed] [Google Scholar]
- 21.Galvagnion C, Buell AK, Meisl G, Michaels TCT, Vendruscolo M, Knowles TPJ, Dobson CM. Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat Chem Biol. 2015;11:229–U101. doi: 10.1038/nchembio.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galvagnion C, Brown JWP, Ouberai MM, Flagmeier P, Vendruscolo M, Buell AK, Sparr E, Dobson CM. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of α-synuclein. Proc Natl Acad Sci U S A. 2016;113:7065–7070. doi: 10.1073/pnas.1601899113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grey M, Dunning CJ, Gaspar R, Grey C, Brundin P, Sparr E, Linse S. Acceleration of α-synuclein aggregation by exosomes. J Biol Chem. 2015;290:2969–2982. doi: 10.1074/jbc.M114.585703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, Lansbury PT. α-Synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol. 2002;322:1089–1102. doi: 10.1016/s0022-2836(02)00735-0. [DOI] [PubMed] [Google Scholar]
- 25.Choi W, Zibaee S, Jakes R, Serpell LC, Davletov B, Crowther RA, Goedert M. Mutation E46K increases phospholipid binding and assembly into filaments of human α-synuclein. FEBS Lett. 2004;576:363–368. doi: 10.1016/j.febslet.2004.09.038. [DOI] [PubMed] [Google Scholar]
- 26.Fares MB, Ait-Bouziad N, Dikiy I, Mbefo MK, Jovicic A, Kiely A, Holton JL, Lee SJ, Gitler AD, Eliezer D, Lashuel HA. The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α-synuclein, and enhances its secretion and nuclear localization in cells. Hum Mol Genet. 2014;23:4491–4509. doi: 10.1093/hmg/ddu165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bussell R, Eliezer D. Residual structure and dynamics in Parkinson’s disease-associated mutants of α-synuclein. J Biol Chem. 2001;276:45996–46003. doi: 10.1074/jbc.M106777200. [DOI] [PubMed] [Google Scholar]
- 28.Jensen PH, Nielsen MS, Jakes R, Dotti G, Goedert M. Binding of α-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J Biol Chem. 1998;273:26292–26294. doi: 10.1074/jbc.273.41.26292. [DOI] [PubMed] [Google Scholar]
- 29.Jo E, Fuller N, Rand RP, St George-Hyslop P, Fraser PE. Defective membrane interactions of familial Parkinson’s disease mutant A30P α-synuclein. J Mol Biol. 2002;315:799–807. doi: 10.1006/jmbi.2001.5269. [DOI] [PubMed] [Google Scholar]
- 30.Khalaf O, Fauvet B, Oueslati A, Dikiy I, Mahul-Mellier AL, Ruggeri FS, Mbefo MK, Vercruysse F, Dietler G, Lee SJ, Eliezer D, Lashuel HA. The H50Q Mutation Enhances α-Synuclein Aggregation, Secretion, and Toxicity. J Biol Chem. 2014;289:21856–21876. doi: 10.1074/jbc.M114.553297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ding TT, Lee SJ, Rochet JC, Lansbury PT. Annular α-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry. 2002;41:10209–10217. doi: 10.1021/bi020139h. [DOI] [PubMed] [Google Scholar]
- 32.Gosavi N, Lee HJ, Lee JS, Patel S, Lee SJ. Golgi fragmentation occurs in the cells with prefibrillar α-synuclein aggregates and precedes the formation of fibrillar inclusion. J Biol Chem. 2002;277:48984–48992. doi: 10.1074/jbc.M208194200. [DOI] [PubMed] [Google Scholar]
- 33.Fujita Y, Ohama E, Takatama M, Al-Sarraj S, Okamoto K. Fragmentation of Golgi apparatus of nigral neurons with α-synuclein-positive inclusions in patients with Parkinson’s disease. Acta Neuropathologica. 2006;112:261–265. doi: 10.1007/s00401-006-0114-4. [DOI] [PubMed] [Google Scholar]
- 34.Nakamura K, Nemani VM, Azarbal F, Skibinski G, Levy JM, Egami K, Munishkina L, Zhang J, Gardner B, Wakabayashi J, Sesaki H, Cheng YF, Finkbeiner S, Nussbaum RL, Masliah E, Edwards RH. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein α-synuclein. J Biol Chem. 2011;286:20710–20726. doi: 10.1074/jbc.M110.213538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Freeman D, Cedillos R, Choyke S, Lukic Z, McGuire K, Marvin S, Burrage AM, Sudholt S, Rana A, O’Connor C, Wiethoff CM, Campbell EM. α-Synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. Plos One. 2013;8 doi: 10.1371/journal.pone.0062143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meredith GE, Totterdell S, Petroske E, Cruz KS, Callison RC, Lau YS. Lysosomal malfunction accompanies α-synuclein aggregation in a progressive mouse model of Parkinson’s disease. Brain Res. 2002;956:156–165. doi: 10.1016/s0006-8993(02)03514-x. [DOI] [PubMed] [Google Scholar]
- 37.Butterfield SM, Lashuel HA. Amyloidogenic protein membrane interactions: Mechanistic insight from model systems. Angew Chem Int Ed. 2010;49:5628–5654. doi: 10.1002/anie.200906670. [DOI] [PubMed] [Google Scholar]
- 38.Beyer K. Mechanistic aspects of Parkinson’s disease: α-synuclein and the biomembrane. Cell Biochem Biophys. 2007;47:285–299. doi: 10.1007/s12013-007-0014-9. [DOI] [PubMed] [Google Scholar]
- 39.Dikiy I, Eliezer D. Folding and misfolding of α-synuclein on membranes. Biochim Biophys Acta Biomembr. 2012;1818:1013–1018. doi: 10.1016/j.bbamem.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pfefferkorn CM, Jiang Z, Lee JC. Biophysics of α-synuclein membrane interactions. Biochim Biophys Acta Biomembr. 2012;1818:162–171. doi: 10.1016/j.bbamem.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Middleton ER, Rhoades E. Effects of curvature and composition on α-synuclein binding to lipid vesicles. Biophys J. 2010;99:2279–2288. doi: 10.1016/j.bpj.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 43.Pfefferkorn CM, Heinrich F, Sodt AJ, Maltsev AS, Pastor RW, Lee JC. Depth of α-synuclein in a bilayer determined by fluorescence, neutron reflectometry, and computation. Biophys J. 2012;102:613–621. doi: 10.1016/j.bpj.2011.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang Z, Hess SK, Heinrich F, Lee JC. Molecular details of α-synuclein membrane association revealed by neutrons and photons. J Phys Chem B. 2015;119:4812–4823. doi: 10.1021/jp512499r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jiang Z, de Messieres M, Lee JC. Membrane remodeling by α-synuclein and effects on amyloid formation. J Am Chem Soc. 2013;135:15970–15973. doi: 10.1021/ja405993r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Varkey J, Mizuno N, Hegde BG, Cheng NQ, Steven AC, Langen R. α-Synuclein oligomers with broken helical conformation form lipoprotein nanoparticles. J Biol Chem. 2013;288:17620–17630. doi: 10.1074/jbc.M113.476697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eichmann C, Campioni S, Kowal J, Maslennikov I, Gerez J, Liu XX, Verasdonck J, Nespovitaya N, Choe S, Meier BH, Picotti P, Rizo J, Stahlberg H, Riek R. Preparation and characterization of stable α-synuclein lipoprotein particles. J Biol Chem. 2016;291:8516–8527. doi: 10.1074/jbc.M115.707968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eichmann C, Kumari P, Riek R. High-density lipoprotein-like particle formation of synuclein variants. FEBS Lett. 2017;591:304–311. doi: 10.1002/1873-3468.12543. [DOI] [PubMed] [Google Scholar]
- 49.Varkey J, Isas JM, Mizuno N, Jensen MB, Bhatia VK, Jao CC, Petrlova J, Voss JC, Stamou DG, Steven AC, Langen R. Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J Biol Chem. 2010;285:32486–32493. doi: 10.1074/jbc.M110.139576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mizuno N, Varkey J, Kegulian NC, Hegde BG, Cheng NQ, Langen R, Steven AC. Remodeling of lipid vesicles into cylindrical micelles by α-synuclein in an extended α-helical conformation. J Biol Chem. 2012;287:29301–29311. doi: 10.1074/jbc.M112.365817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Westphal CH, Chandra SS. Monomeric synucleins generate membrane curvature. J Biol Chem. 2013;288:1829–1840. doi: 10.1074/jbc.M112.418871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pfefferkorn CM, Lee JC. Tryptophan probes at the α-synuclein and membrane interface. J Phys Chem B. 2010;114:4615–4622. doi: 10.1021/jp908092e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Braun AR, Lacy MM, Ducas VC, Rhoades E, Sachs JN. α-Synuclein-induced membrane remodeling is driven by binding affinity, partition depth, and interleaflet rrder asymmetry. J Am Chem Soc. 2014;136:9962–9972. doi: 10.1021/ja5016958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zimmerberg J, Kozlov MM. How proteins produce cellular membrane curvature. Nat Rev Mol Cell Biol. 2006;7:9–19. doi: 10.1038/nrm1784. [DOI] [PubMed] [Google Scholar]
- 55.Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, Urlaub H, Schenck S, Brugger B, Ringler P, Muller SA, Rammner B, Grater F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmuller H, Heuser J, Wieland F, Jahn R. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 56.Rappley I, Myers DS, Milne SB, Ivanova PT, LaVoie MJ, Brown HA, Selkoe DJ. Lipidomic profiling in mouse brain reveals differences between ages and genders, with smaller changes associated with α-synuclein genotype. J Neurochem. 2009;111:15–25. doi: 10.1111/j.1471-4159.2009.06290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Giusto NM, Salvador GA, Castagnet PI, Pasquare SJ, de Boschero MGI. Age-associated changes in central nervous system glycerolipid composition and metabolism. Neurochem Res. 2002;27:1513–1523. doi: 10.1023/a:1021604623208. [DOI] [PubMed] [Google Scholar]
- 58.Sani MA, Gehman JD, Separovic F. Lipid matrix plays a role in Aβ fibril kinetics and morphology. FEBS Letters. 2011;585:749–754. doi: 10.1016/j.febslet.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 59.Korshavn KJ, Satriano C, Lin YX, Zhang RC, Dulchavsky M, Bhunia A, Ivanova MI, Lee YH, La Rosa C, Lim MH, Ramamoorthy A. Reduced lipid bilayer thickness regulates the aggregation and cytotoxicity of amyloid-β. J Biol Chem. 2017;292:4638–4650. doi: 10.1074/jbc.M116.764092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brender JR, Salamekh S, Ramamoorthy A. Membrane disruption and early events in the aggregation of the diabetes related peptide IAPP from a molecular perspective. Acc Chem Res. 2012;45:454–462. doi: 10.1021/ar200189b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brender JR, Heyl DL, Samisetti S, Kotler SA, Osborne JM, Pesaru RR, Ramamoorthy A. Membrane disordering is not sufficient for membrane permeabilization by islet amyloid polypeptide: studies of IAPP(20-29) fragments. Phys Chem Chem Phys. 2013;15:8908–8915. doi: 10.1039/c3cp44696d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brender JR, Durr UHN, Heyl D, Budarapu MB, Ramamoorthy A. Membrane fragmentation by an amyloidogenic fragment of human Islet amyloid polypeptide detected by solid-state NMR spectroscopy of membrane nanotubes. Biochim Biophys Acta-Biomembr. 2007;1768:2026–2029. doi: 10.1016/j.bbamem.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kegulian NC, Sankhagowit S, Apostolidou M, Jayasinghe SA, Malmstadt N, Butler PC, Langen R. Membrane curvature-sensing and curvature-inducing activity of Islet amyloid polypeptide and its implications for membrane disruption. J Biol Chem. 2015;290:25782–25793. doi: 10.1074/jbc.M115.659797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Perni M, Galvagnion C, Maltsev A, Meisl G, Muller MBD, Challa PK, Kirkegaard JB, Flagmeier P, Cohen SIA, Cascella R, Chen SW, Limboker R, Sormanni P, Heller GT, Aprile FA, Cremades N, Cecchi C, Chiti F, Nollen EAA, Knowles TPJ, Vendruscolo M, Bax A, Zasloff M, Dobson CM. A natural product inhibits the initiation of α-synuclein aggregation and suppresses its toxicity. Proc Natl Acad Sci U S A. 2017;114:E1009–E1017. doi: 10.1073/pnas.1610586114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen M, Margittai M, Chen J, Langen R. Investigation of α-synuclein fibril structure by site-directed spin labeling. J Biol Chem. 2007;282:24970–24979. doi: 10.1074/jbc.M700368200. [DOI] [PubMed] [Google Scholar]
- 66.Vilar M, Chou HT, Luhrs T, Maji SK, Riek-Loher D, Verel R, Manning G, Stahlberg H, Riek R. The fold of α-synuclein fibrils. Proc Natl Acad Sci U S A. 2008;105:8637–8642. doi: 10.1073/pnas.0712179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Meier BH, Bockmann A. The structure of fibrils from ‘misfolded’ proteins. Curr Opin Struct Biol. 2015;30:43–49. doi: 10.1016/j.sbi.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 68.McGlinchey RP, Dominah GA, Lee JC. Taking a bite out of amyloid: mechanistic insights into α-synuclein degradation by cathepsin L. Biochemistry. 2017;56:3881–3884. doi: 10.1021/acs.biochem.7b00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hellstrand E, Nowacka A, Topgaard D, Linse S, Sparr E. Membrane lipid co-aggregation with α-synuclein fibrils. Plos One. 2013;8:10. doi: 10.1371/journal.pone.0077235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.