

Abstract
Mutations in spliceosome components have been implicated in carcinogenesis of various types of cancer. One of the most frequently found is U2AF1 S34F missense mutation. Functional analyses of this mutation have been largely limited to hematological malignancies although the mutation is also frequently seen in other cancer types including lung adenocarcinoma (LUAD). We examined the impact of knockdown (KD) of wild type (wt) U2AF1 and ectopic expression of two splice variant S34F mutant proteins in terms of alternative splicing (AS) pattern and cell cycle progression in A549 lung cancer cells. We demonstrate that induction of distinct AS events and disruption of mitosis at distinct sub-stages result from KD and ectopic expression of the mutant proteins. Importantly, when compared with the splicing pattern seen in LUAD patients with U2AF1 S34F mutation, ectopic expression of S34F mutants but not KD was shown to result in common AS events in several genes involved in cell cycle progression. Our study thus points to an active role of U2AF1 S34F mutant protein in inducing cell cycle dysregulation and mitotic stress. In addition, alternatively spliced genes which we describe here may represent novel potential markers of lung cancer development.
Keywords: alternative splicing, lung adenocarcinoma, mitotic stress, S34F, U2AF1
INTRODUCTION
U2AF1, a subunit of the U2AF dimer which recognizes the 3′ splice site, has been found to be frequently mutated in several types of cancer including acute myeloid leukemia (AML) and lung adenocarcinoma (LUAD) (Brooks et al., 2014). The mutations are heterozygous and missense in nature and occur mostly at S34 and to a less extent at Q157 positions (Graubert et al., 2012; Makishima et al., 2012; Przychodzen et al., 2013; Yoshida et al., 2011). The missense mutations at these two amino acid positions are proposed to have an initiating effect based on its presence in myelodysplastic syndromes (MDS) which can later evolve into AML. (Graubert et al., 2012; Makishima et al., 2012; Yoshida et al., 2011). The recurrence of the mutation at specific amino acids strongly suggests a gain of oncogenic activity, but the exact mode of action during carcinogenesis remains unknown.
Changes in splicing pattern have been profiled and examined using transcriptome data mostly from myeloid neoplasms including MDS and AML patient samples although limited efforts have been made with LUAD as well (Brooks et al., 2014; Ilagan et al., 2015; Przychodzen et al., 2013). Splicing patterns have also been examined using ectopic expression of U2AF1 mutants in various cell lines and transgenic murine models (Brooks et al., 2014; Ilagan et al., 2015; Shirai et al., 2015; Yoshida et al., 2011). Multiple cellular pathways have been implicated, but results vary among the studies. Thus far, the exact cancer-associated splicing changes induced by the mutation have not been pinpointed.
Other related issues also merit further examinations. The first is whether the frequently seen S34F or Q157P missense mutations in U2AF1 represents not just a gain of oncogenic function but partly a loss-of-function mutation or a dominant negative mutation. Of interest, it has been reported that knockdown (KD) of U2AF1 leads to alternative splicing (AS) events of its own including that of a cell cycle regulator Cdc25 (Fu et al., 2011; Pacheco et al., 2006). The second is the apparent lack in the part of the mutant U2AF1 of cell growth promoting activity as an oncogene. In fact, cell growth is retarded at least initially by KD and by ectopic expression of U2AF1 mutants (Pacheco et al., 2006; Yoshida et al., 2011).
Here, we report characterization of changes in splicing pattern and cell growth brought by KD of U2AF1 and by expression of S34F mutants using a single in vitro system. We demonstrate that KD and mutant expression lead to distinct AS patterns in A549 lung cancer cells. Through integration with AS data from LUAD patients with U2AF1 S34F mutation, AS events in a set of genes involved in cell cycle progression and mitosis are proposed as potential contributors to carcinogenesis. We also show that KD and mutant expression retard mitotic progression at distinct sub-stages and therefore via distinct mechanisms. In sum, our data provide novel mechanistic insights into the role of U2AF1 S34F mutation in carcinogenesis and a list of AS events that may serve as novel biomarkers of LUAD development.
MATERIALS AND METHODS
Cell culture
A549 lung carcinoma cells were obtained from the American Type Culture Collection (ATCC, USA) and were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (Hyclone, USA).
Knockdown and ectopic expression of U2AF1 variants
For KD, we used a synthetic RNA duplexes (U2AF1 siRNA#1, 5′-CUAGAAAGUGUUGUAGUUGAUUG-3′; GE Dharmacon, USA) specifically targeting human U2AF1 3′UTR region. To generate retroviruses expressing the two wild type (wt) splice variants of U2AF1, the coding regions were PCR-amplified and subcloned into the LZRS retroviral vector with V5 and three FLAG epitopes at the C-terminus (Jung et al., 2015; Kim et al., 2003). Site-directed mutagenesis was used to create the S34F mutants. Cells were transfected with 40 nM siRNAs for U2AF1 or scramble control using Lipofectamine RNAiMAX (Invitrogen, USA). A549 were infected with viruses 6 h after siRNA treatment and cultured for 48 h for RNA-seq analyses, RT-PCR and western blotting. Immunofluorescence and flow cytometric analyses were typically carried out 72 and 96 h respectively after viral infections in the absence of pre-treatment with siRNAs. For long-term growth upto 15 days, cell proliferation rate was determined using EZ-Cytox Cell Viability Assay Kit (DoGen, Korea). Clonal expansion of mutant-expressing cells was shown after 15 days of culture in 35 mm plates. Cells were stained with 0.1% Coomassie Blue in 45% methanol and 10% acetic acid solution for visualization.
RNA-seq data acquisition and analyses
Total RNA for RNA-seq was isolated using RNeasy Mini Kit according to the manufacturer’s protocol (Qiagen, Germany). The quality of RNA was assessed by Agilent 2100 Bioanalyzer (Agilent Technologies, The Netherlands) with an RNA Integrity Number value greater than 8. mRNA sequencing libraries were prepared according to the manufacturer’s instructions using the Illumina Truseq RNA Prep kit v2. The quality of the amplified libraries was verified by capillary electrophoresis (Bioanalyzer, Agilent). Sequencing of pooled libraries were performed on the HiSeq 2000 sequencing system (Illumina) with paired-end reads of 100 bp length. The duplicate RNA-Seq data set to confirm the effect of U2AF1 mutation on splicing have been deposited in Gene Expression Omnibus (GEO) database (GSE66553). First, low quality reads were trimmed, and alignment using Tophat version 2.0.12 to the reference genome GRCh37 was carried out. For AS event quantification, the gene model Ensembl release 75 was used for all required applications. Differential AS events were analyzed using MATS version 3.0.8. WT U2AF1-induced AS events were compared with AS events from S34F mutation or KD, and AS events (605 events) within cutoff FDR < 0.01 were chosen for each comparison. From The Cancer Genome Atlas (TCGA) dataset, transcriptome data of eight LUAD patients with U2AF1 S34F mutation (TCGA-49-4488, TCGA-49-4505, TCGA-49-6744, TCGA-50-5941, TCGA-55-7727, TCGA-55-7903, TCGA-64-1680, TCGA-78-7145) and three patients without the mutation (TCGA-44-6148, TCGA-55-6980, TCGA-55-6984) were obtained. Differential AS event analyses was carried out using MATS 3.0.8 with cutoff p-value < 0.05. Finally, TCGA differential AS events were extracted to intersect with results from our analyses with A549 cells. Determination of biological functions using gene set enrichment analysis for these AS event genes was carried out using DAVID. Further details for all procedures are available upon request.
Conventional RT-PCR analysis
Total RNA was extracted using Trizol (Invitrogen), and cDNA was synthesized by using GoScript™ reverse transcriptase (Promega, USA) according to the manufacturer’s instructions. For validation of differential AS events between control and U2AF1 KD and between wt U2AF1 and S34F mt U2AF1, cDNA was amplified using Platinum Taq polymerase (Invitrogen). The gene-specific oligonucleotide primers used for RT-PCR are listed in Supplementary Table S1. Each cDNA was amplified in a 35-cycle reaction at the annealing temperature of 60°C. ACTB gene was used as an endogenous reference. PCR products were analyzed on agarose gels.
Immunoblot analysis
The transduced cells were subjected to immunoblot analyses with antibodies against U2AF1 (Novus Biologicals, USA) and α-tubulin (AbFrontier, Korea).
Flow cytometry
Flow cytometric analyses were carried out using a BD LSR-Fortessa cell analyzer (BD Biosciences, USA). Typically, the siRNA-treated or virus-transduced cells were cultured for 96 hours, fixed with 70% ethanol, stained with 50 μg/ml propidium iodide (Sigma, USA) and then examined for cell cycle progression.
Immunocytochemistry
After the siRNA-treated or virus-transduced cells were cultured for 72 h, chromosomes were stained with 4′6-diamidino-2-phenylindole (DAPI; Sigma) and examined by epifluorescence microscopy. For immunocytochemistry, antibodies against α-tubulin (Sigma) and CENP-A (Abcam, USA) were used.
RESULTS
For the functional analysis of U2AF1 S34F mutation, we chose A549 cell line derived from LUAD of a Caucasian male which was readily transfected with siRNAs and transduced with retroviruses. Endogenous wt U2AF1 was down-regulated with a specific siRNA (siRNA#1), and ectopic expression of the two splice variants of wt and of S34F U2AF1 was accomplished by infection with retroviruses (Fig. 1A; for the structure and RefSeq ID of variants a and b, see Supplementary Fig. S1). Transcriptome data from RNA-seq were analyzed in a pair-wise fashion (i.e. control siRNA vs U2AF1 siRNA; wt U2AF1 variant b vs mutant U2AF1 variant b; wt U2AF1 variant a vs mutant U2AF1 variant a) to identify changes in AS pattern. An extensive overlap was evident between AS events induced by the two mutant variants while the overlap between AS events induced by KD and by mutant expression was limited (Fig. 1B; a complete list and details of AS events in the Supplementary File S1). Multiple AS events from transcriptome mapping were re-examined by RT-PCR, and virtually all tested AS events confirmed the transcriptome data (Figs. 1C–1F; Supplementary Fig. S2). Of note, KD with another siRNA#2 was carried out, and AS was examined for EHMT2, ASXL2, and CDC25A which are three genes that show AS upon KD with the siRNA#1. Qualitatively similar results were obtained for all three genes (Supplementary Fig. S3) supporting that significant AS from KD are not likely from off-target effects. Interestingly, even the AS events computationally assigned uniquely to either of the two mutant expression groups were in fact commonly seen albeit often with quantitative differences (Figs. 1D and 1E). We thus merged the two sets for subsequent clustering and Gene Ontology (GO) analyses.
Fig. 1. KD and mutant expression lead to distinct AS events.
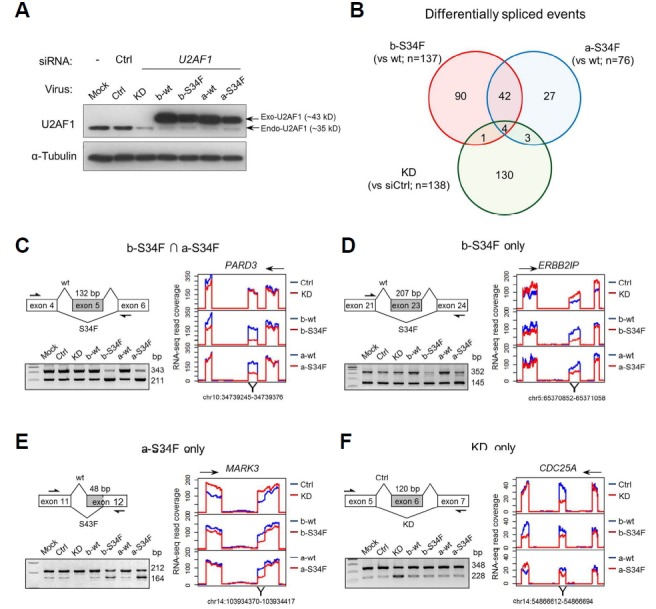
(A) Immunoblot showing KD of endogenous U2AF1 and expression of exogenous wt and S34F mutant U2AF1. Ctrl indicates control empty vector virus infection. (B) Venn diagram of genes showing AS upon KD or mutant expression. Note the limited overlap between KD and mutant expression in contrast to expression of the two mutants. (C–F) Representative cases of AS from the four indicated gene set categories. The alternatively spliced exon and surrounding exons are diagrammed. Arrows indicate oligonucleotide primers used for RT-PCR shown below. To the right are RNA-seq read coverage of the diagrammed exons. Directions of transcription are indicated above. Note the similarity in AS induced by the two U2AF1 splice variants.
We examined the nature of AS by examining 3′ splice sites of introns where U2AF1 binds. AS can occur in various patterns, but most of the AS induced from mutant expression and KD came in the forms of alternative 3′ spice site (A3SS) and skipped exon (SE) on which we focused for subsequent analyses (Fig. 2A). Both A3SS and SE can result in either exclusion or inclusion of exons, in whole or in part, resulting in selection of alternative 3′ splice sites by mutant expression or KD compared to wt U2AF1 expression (Fig. 2B). A clear preference of S34F mutants was readily revealed: −3 nucleotide from 3′ exon was overwhelmingly T in the case of wt U2AF1 while C/A was the choice in the case of mutant U2AF1 (Fig. 2C). This is similar to the effect seen in hematological malignancies (Ilagan et al., 2015; Przychodzen et al., 2013). In contrast, no significant change of 3′ spice site preference was seen by KD (Fig. 2C).
Fig. 2. Sequence preferences at altered 3′ splice sites associated with U2AF1 S34F mutant expression or KD.
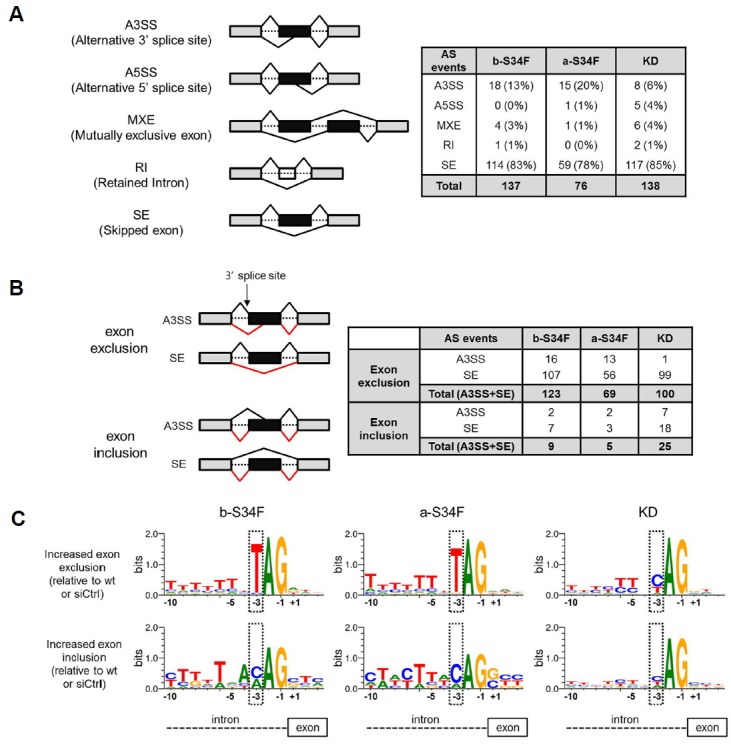
(A) On the left side, various types of alternative splicing (AS) events are schematically shown. Exons and introns are represented as blocks and dotted lines respectively. On the right side, the counts of AS events associated with U2AF1 S34F expression or KD are tabluated (FDR < 0.01). (B) On the left side, exon exclusion and inclusion are shown schematically for SE and A3SS. Wild type splicing is indicated by black lines above the genes and AS induced by mutant expression or KD are shown in red below. Tabluation shows that most AS events induced by mutant expression or KD are in fact exon exclusion. (C) The sequences near the 3′ splicing site are shown. Note that nucleotide preference at the −3 position is changed from T to C/A upon mutant expression. Sequence logos are obtained using WebLogo (http://weblogo/berkeley.edu/loco/cgi).
In order to further prioritize the AS events in terms of clinical relevance, we sought to integrate our transcriptome data with those from LUAD patients with U2AF1 S34F mutation. We obtained transcriptome of 8 LUAD patients from The Cancer Genome Atlas (TCGA) dataset and were able to enlist 5,418 comprehensive tumor-specific AS events (p < 0.05). Out of 167 AS events induced by U2AF1 S34F mutant expression (FDR < 0.01), 47 were also seen in LUAD patients while 28 common events were seen between the AS sets of KD and the patients (Fig. 3A; a complete list and details of AS events in the Supplementary File S2). We performed gene set over-representation analyses using each of the two intersecting gene sets (47 from mutant expression and 28 from KD). This has led to significant Gene Ontology (GO) terms only from the intersection gene set from S34F mutant expression and LUAD patients (Fig. 3B). Remarkably, all GO terms except one were associated with cell cycle and mitosis. We carried out RT-PCR for the seven genes associated with these GO terms and confirmed the induction of AS or of change in the expression level of specific variants (Figs. 1C and 1D and Supplementary Fig. S2).
Fig. 3. Integrated profiling of alternative splicing with LUAD patient data and GO analysis.
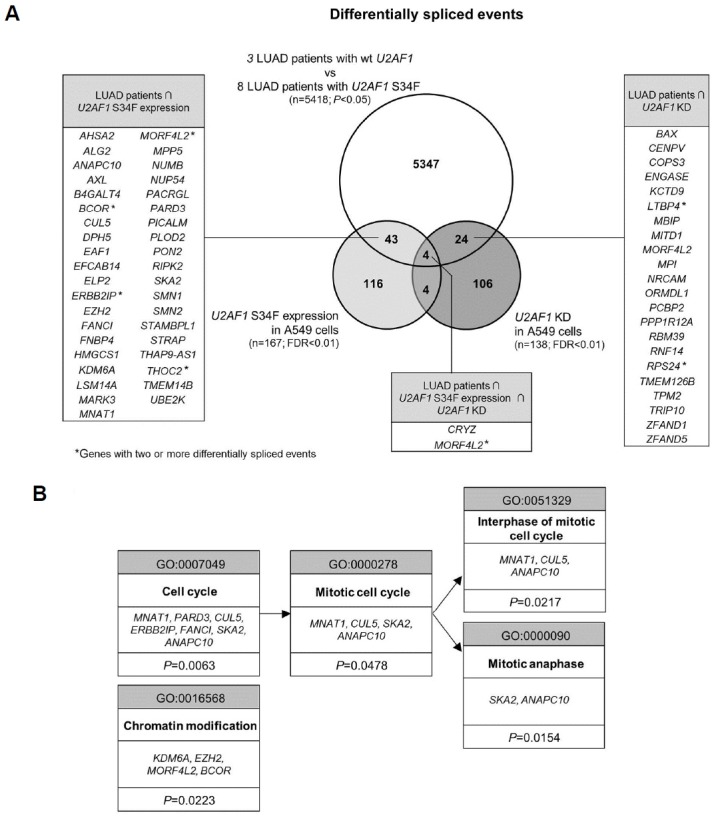
(A) Venn diagram of genes that have undergone AS from KD, from mutant expression and in LUAD cases with S34F mutation. Gene lists are presented for intersection gene sets. (B) GO terms and genes from gene set enrichment analysis with DAVID on 47 AS events found in the intersection of LUAD patient set and mutant expression set. Note that no enriched term was found for genes from intersection of LUAD patient set and KD set.
It has been reported that both KD and mutant expression lead to cell cycle arrest (Pacheco et al., 2006; Yoshida et al., 2011). Consistently, we also noticed G2/M arrest upon KD or expression of either of the mutant variants (Fig. 4A). It has also been shown that KD leads to accumulation of cells in prometaphase during mitosis (Pacheco et al., 2006). Given these facts, that AS events induced by KD and mutant expression are distinct and that mutant expression but not KD induced AS events in genes involved in mitotic cell cycle progression in common with LUAD cases led us to hypothesize that KD and mutant expression affect mitosis in distinct fashions. We thus examined the substage distribution of mitotic cells. As has been reported, we found that KD stalled mitosis at the prometaphase step (Figs. 4B–4D). Over 70% of the cells with condensed chromosomes did not show separation of centrosomes. Interestingly, this was not the case with mutant expression: the mitotic arrest occurred at the metaphase (Figs. 4B–4D). Among the cells with clear separation of centrosomes to opposite ends, significant fractions had mis-aligned chromosomes located outside the mid-plane. This was the case with expression of either of the mutant splice variants. The results indicate that consistent with induction of distinct AS profiles, KD and mutant expression have distinct effects on cellular physiology particularly in terms of mechanisms of mitotic stress.
Fig. 4. KD and mutant expression lead to mitotic stress at distinct stages.
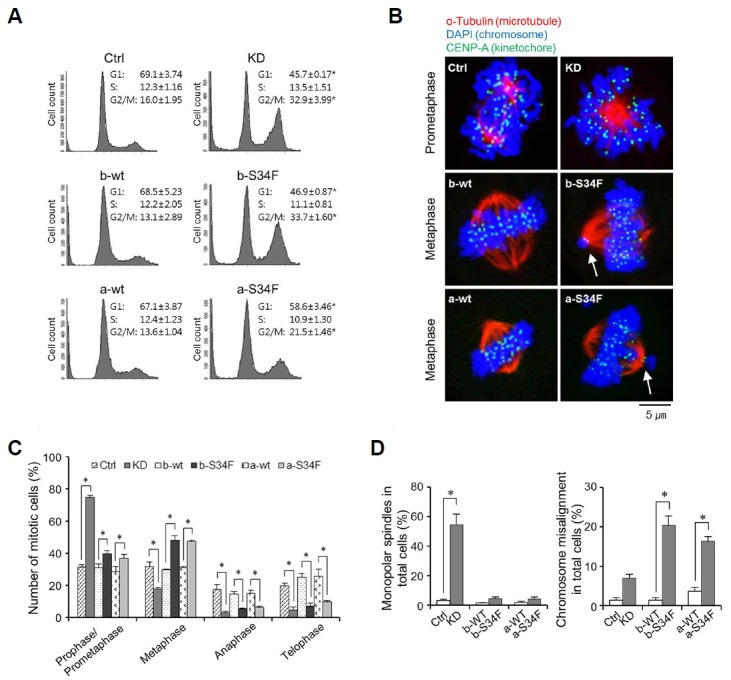
(A) Flow cytometric analyses of cell cycle progression. Note that both KD and mutant expression induce G2/M arrest. Results are average ± standard error of the mean (SEM) of three independent assays. (*) represents P-value of < 0.05 compared to control condition. (B) Representative images of cells after KD and mutant expression in the affected sub-stages. Note that after KD, centrosomes are not separating even though chromosomes are condensed. Also note that not all chromosomes (white arrows) are aligned in the center after mutant expression during metaphase. (B, C) Mitotic sub-stage distribution of cells after KD and mutant expression. Cells with condensed chromosomes with microtubules extending from centrosome are grouped together as prophase/prometaphase cells. Cells with centrosomes in opposite ends with chromosomes aligned in the middle are considered metaphase cells. Cells showing mitotic spindle-mediated chromosomal migration are considered anaphase cells. Note that KD results in accumulation of cells in prophase/prometaphase and decrease in fractions of cells in all subsequent stages while mutant expression increases the fraction of cells in metaphase and decreases those in anaphase and telophase. Results are average ± SEM of three independent assays. (*) represents P-value of < 0.05 compared to control condition. (D) Fractions of cells with monopolar spindles after KD and of cells with misaligned chromosomes after mutant expression (b and a) are shown. Results are average ± SEM of three independent assays. (*) represents P-value of < 0.05 compared to control condition.
Despite initially stunted cell growth, stable clonal cell population eventually emerged from mutant expressing cells but not from KD cells that divided at a rate comparable to control empty vector virus-infected cells (Fig. 5A). The surviving mutant expressing cells grew as clones suggesting that they descended from individual cells after gaining additional mutations or altered gene expression patterns (Fig. 5B). Such rebound proliferation phenomenon has also been reported recently in Ba/F3 cells in which U2AF1 S34 mutant was stably overexpressed (Park et al., 2016). We also confirmed that expression of the ectopic U2AF1 S34F mutant was maintained in over 80% of these cells by immunocytochemical staining (data not shown).
Fig. 5. Rebound proliferation of U2AF1 S34 mutant-expressing A549 cells.
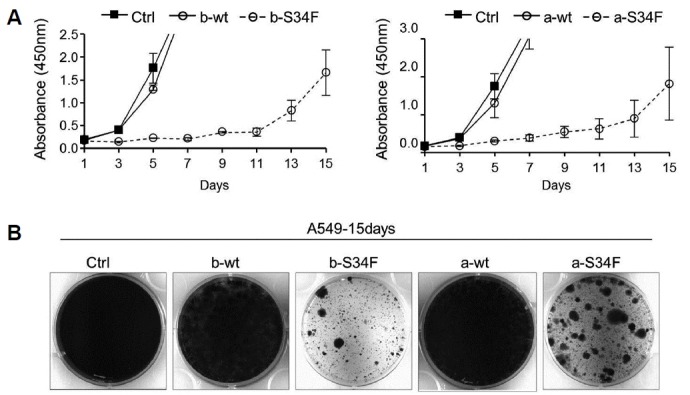
(A) 2 days after infection, the trypsinized cells were plated in 96-well plate, and cell proliferation was examined for upto 15 days. Note that U2AF1 S34 mutant-expressing cells grew in a robust manner after initially stunted cell division. (B) U2AF1 S34 mutant-expressing cells grew via clonal expansion. Virus-transduced cells were cultured for 15 days in 35mm plates. Note the clonal expansion of S34F mutant -expressing cells after presumptive apoptosis of most of the cells.
DISCUSSION
In this study, we demonstrate using a single lung cancer cell line system that KD of wt U2AF1 and expression of S34F mutants lead to distinct AS events and distinct effects on cell cycle progression. We show for the first time that although both KD and mutant expression lead to G2/M cell cycle arrest as has been reported previously they do so via inhibiting different steps within mitosis. In addition, we define a group of genes involved in mitosis which undergo identical AS in S34F mutant expressing cells and in LUAD patients with S34F mutation. A clear limitation to our screen is the use of over-expression of mutant genes in combination with KD of the WT U2AF1. The mutation in patients is heterozygous in nature, and this implies that the ratios of wild type and mutant proteins utilized in this study are not likely to reflective of those in patient cancer cells. Our goal was to accentuate AS events induced by mutants for facilitated identification at the risk of distorting the cellular physiology. It was thus important in the end to limit the candidate AS events of importance to those also seen in LUAD patients. It should be pointed out that siRNA treatment was not applied in combination with mutant expression in subsequent experiments examining the mitotic stress.
That KD and S34F mutant expression have distinct effects in 3′ splice site selection and in cell cycle arrest clearly indicates that U2AF1 S34F mutation represents a gain of activity whether oncogenic or not. We cannot rule out the possibility that AS resulting from partial loss of wt U2AF1 activity also contributes to carcinogenesis. Still, the recurrence of this missense mutation in various cancer types and the induction of AS in common mitotic genes with LUAD tissues strongly suggest that S34F mutation is oncogenic in nature.
One effect resulting from either KD or mutant expression is the mitotic stress. In fact, it appears that expression of mutant does not just stall mitosis but rather induce dysregulated chromosomal arrangements (Fig. 4). It has been proposed that oncogenes can often induce mitotic stress and subsequent chromosomal instability (CIN) (Duijf and Benezra, 2013; Roschke and Rozenblum, 2013). It is interesting to note that U2AF1 mutation positive cancers often show cytogenetic abnormalities including trisomy 8, trisomy 21, monosomy 7, and partial deletion of chromosome 20 in MDS and AML (Damm et al., 2012; Makishima et al., 2012; Qian et al., 2012). It is thus a possible scenario that the mitotic stress seen with the mutant U2AF1 expression originates from AS of a set of mitosis regulator genes and leads to CIN and aneuploidy. In sum, although inhibition of cell growth and apoptosis are initially seen due to the stress from U2AF1 S34F expression, a genetic heterogeneity could be generated and a more malignant progenitor cells with additional mutations could ultimately emerge (Duijf and Benezra, 2013; Roschke and Rozenblum, 2013). It may thus be interesting to examine the clones from surviving cells after S34F mutants for further mutations and/or chromosomal aberrations.
Among the critical remaining questions is which of the AS events has the carcinogenic activity albeit possibly indirectly. Strictly speaking, we cannot rule out any of them including KD-induced AS events as potential contributors until each AS event is fully examined for its function. Plus, although our study was designed to test the differential activity of mutants of the two naturally occurring splice variants of U2AF1, we were unable to distinguish their function in terms of AS or mitotic stress. It could be that subtle quantitative differences in AS may turn out to be biologically meaningful. Another possibility worthy of consideration is that more than one AS events contribute to cell cycle stress and set the table for a more definitive driving event, be it another oncogenic mutation or aneuploidic cell division. Accumulation of additional patient data down the road should give more informative AS profiles and narrow down the AS candidates that are mechanistically involved in carcinogenesis.
Supplementary data
ACKNOWLEDGMENTS
This research was supported by funding from the Ministry of Science and ICT via National Research Foundation, Republic of Korea (NRF-2013M3C7A1056562 to J. Kim; NRF-2015 K1A4A3047851 to S. Lee; NRF-2016R1A6A3A11932702 to Y. Jung).
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Brooks A.N., Choi P.S., de Waal L., Sharifnia T., Imielinski M., Saksena G., Pedamallu C.S., Sivachenko A., Rosenberg M., Chmielecki J., et al. A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PLoS One. 2014;9:e87361. doi: 10.1371/journal.pone.0087361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damm F., Kosmider O., Gelsi-Boyer V., Renneville A., Carbuccia N., Hidalgo-Curtis C., Della Valle V., Couronne L., Scourzic L., Chesnais V., et al. Mutations affecting mRNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes. Blood. 2012;119:3211–3218. doi: 10.1182/blood-2011-12-400994. [DOI] [PubMed] [Google Scholar]
- Duijf P.H., Benezra R. The cancer biology of whole-chromosome instability. Oncogene. 2013;32:4727–4736. doi: 10.1038/onc.2012.616. [DOI] [PubMed] [Google Scholar]
- Fu Y., Masuda A., Ito M., Shinmi J., Ohno K. AG-dependent 3-splice sites are predisposed to aberrant splicing due to a mutation at the first nucleotide of an exon. Nucleic Acids Res. 2011;39:4396–4404. doi: 10.1093/nar/gkr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal P.A., Coin L., Marshall M., Down T., Hubbard T., Wooster R., Rahman N., Stratton M.R. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graubert T.A., Shen D., Ding L., Okeyo-Owuor T., Lunn C.L., Shao J., Krysiak K., Harris C.C., Koboldt D.C., Larson D.E., et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. 2012;44:53–57. doi: 10.1038/ng.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilagan J.O., Ramakrishnan A., Hayes B., Murphy M.E., Zebari A.S., Bradley P., Bradley R.K. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015;25:14–26. doi: 10.1101/gr.181016.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski M., Berger A.H., Hammerman P.S., Hernandez B., Pugh T.J., Hodis E., Cho J., Suh J., Capelletti M., Sivachenko A., et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Y., Jun Y., Lee H.Y., Kim S., Jung Y., Keum J., Lee Y.S., Cho Y.B., Lee S., Kim J. Characterization of SLC22A18 as a tumor suppressor and novel biomarker in colorectal cancer. Oncotarget. 2015;6:25368–25380. doi: 10.18632/oncotarget.4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Lo L., Dormand E., Anderson D.J. SOX10 maintains multipotency and inhibits neuronal differentiation of neural crest stem cells. Neuron. 2003;38:17–31. doi: 10.1016/s0896-6273(03)00163-6. [DOI] [PubMed] [Google Scholar]
- Makishima H., Visconte V., Sakaguchi H., Jankowska A.M., Abu Kar S., Jerez A., Przychodzen B., Bupathi M., Guinta K., Afable M.G., et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012;119:3203–3210. doi: 10.1182/blood-2011-12-399774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Network, T.C.G.A.R. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco T.R., Moita L.F., Gomes A.Q., Hacohen N., Carmo-Fonseca M. RNA interference knockdown of hU2AF35 impairs cell cycle progression and modulates alternative splicing of Cdc25 transcripts. Mol Biol Cell. 2006;17:4187–4199. doi: 10.1091/mbc.E06-01-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.M., Ou J., Chamberlain L., Simone T.M., Yang H., Virbasius C.M., Ali A.M., Zhu L.J., Mukherjee S., Raza A., et al. U2AF35(S34F) Promotes transformation by directing aberrant ATG7 Pre-mRNA 3′ end formation. Mol Cell. 2016;62:479–490. doi: 10.1016/j.molcel.2016.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przychodzen B., Jerez A., Guinta K., Sekeres M.A., Padgett R., Maciejewski J.P., Makishima H. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood. 2013;122:999–1006. doi: 10.1182/blood-2013-01-480970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J., Yao D.M., Lin J., Qian W., Wang C.Z., Chai H.Y., Yang J., Li Y., Deng Z.Q., Ma J.C., et al. U2AF1 mutations in Chinese patients with acute myeloid leukemia and myelodysplastic syndrome. PLoS One. 2012;7:e45760. doi: 10.1371/journal.pone.0045760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roschke A.V., Rozenblum E. Multi-layered cancer chromosomal instability phenotype. Front Oncol. 2013;3:302. doi: 10.3389/fonc.2013.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai C.L., Ley J.N., White B.S., Kim S., Tibbitts J., Shao J., Ndonwi M., Wadugu B., Duncavage E.J., Okeyo-Owuor T., et al. Mutant U2AF1 expression alters hmatopoiesis and pre-mRNA splicing in vivo. Cancer Cell. 2015;27:631–643. doi: 10.1016/j.ccell.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thol F., Kade S., Schlarmann C., Loffeld P., Morgan M., Krauter J., Wlodarski M.W., Kolking B., Wichmann M., Gorlich K., et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood. 2012;119:3578–3584. doi: 10.1182/blood-2011-12-399337. [DOI] [PubMed] [Google Scholar]
- Yoshida K., Sanada M., Shiraishi Y., Nowak D., Nagata Y., Yamamoto R., Sato Y., Sato-Otsubo A., Kon A., Nagasaki M., et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.