

Abstract
Adiponectin, a hormone produced by adipose tissue, is very abundant in plasma, and its anti- and pro-inflammatory effects are reported. However, the mechanisms of these pro- and anti-inflammatory effects are not fully defined. Herein, we evaluated the dual inflammatory response mechanism of adiponectin in macrophages. Short-term globular adiponectin (gAd) treatment induced IκBα degradation, NF-κB nuclear translocation, and TNF-α production in RAW 264.7 cells. Polymyxin B pretreatment did not block gAd-induced IκBα degradation, and heated gAd was unable to degrade IκBα, suggesting that the effects of gAd were not due to endotoxin contamination. gAd activated IKK and Akt, and inhibition of either IKK or Akt by dominant-negative IKKβ (DN-IKKβ) or DN-Akt overexpression blocked gAd-induced IκBα degradation, suggesting that short-term incubation with gAd mediates inflammatory responses by activating the IκB/NF-κB and PI3K/Akt pathways. Contrastingly, long-term stimulation with gAd induced, upon subsequent stimulation, tolerance to gAd, lipopolysaccharide, and CpG-oligodeoxynucleotide, which is associated with gAd-induced downregulation of IL-receptor-associated kinase-1 (IRAK-1) due to IRAK-1 transcriptional repression. Conclusively, our findings demonstrate that the pro- and anti-inflammatory responses to gAd in innate immune cells are time-dependent, and mediated by the activation of the IκB/NF-κB pathway, and IRAK-1 downregulation, respectively.
Keywords: globular adiponection, NF-κB, IRAK-1
INTRODUCTION
Adiponectin is one of the adipokines mainly produced by the adipose tissue (Tilg and Moschen, 2006). It has been reported that adiponectin has insulin-sensitizing (Berg et al., 2001; Fruebis et al., 2001; Yamauchi et al., 2001), atherogenic (Kubota et al., 2002; Okamoto et al., 2002), and anti-cancer effects (Dalamaga et al., 2012), and decreased adiponectin levels are associated with obesity (Arita et al., 1999), insulin resistance (Hulthe et al., 2003), type 2 diabetes (Lindsay et al., 2002), and cardiovascular diseases (Matsuzawa et al., 2004; Schlegel, 2004).
In immune system, it has been widely recognized that adiponectin has anti-inflammatory properties including attenuation of nuclear factor-kappaB (NF-κB) activity (Yamaguchi et al., 2005) and reduced TNF-α production (Maeda et al., 2002). However, there are several studies reporting the pro-inflammatory activities of adiponectin (Haugen and Drevon, 2007; Park et al., 2007; Tsatsanis et al., 2005). In fact, plasma adiponectin levels are elevated during inflammatory disorders such as pre-eclampsia (Haugen et al., 2006; Hendler et al., 2005) and arthritis (Otero et al., 2006; Senolt et al., 2006). Furthermore, patients with chronic obstructive pulmonary disease, wherein inflammation is regarded to play pathological roles, exhibit elevated plasma adiponectin levels (Chan et al., 2010), and elevated adiponectin levels are associated with disease exacerbation (Kirdar et al., 2009) and respiratory-related death (Yoon et al., 2012). These findings suggest that adiponectin may have dual anti- and pro-inflammatory properties. However, the detailed mechanism underlying the potential dual anti- and pro-inflammatory activities of adiponectin has not yet been fully understood.
In this study, we investigated whether adiponectin has dual anti- and pro-inflammatory effects as well as their plausible mechanisms in innate immune cells.
MATERIALS AND METHODS
Cell culture
RAW 264.7 (murine microphage) cells were maintained in Dulbecco’s modified eagle medium (DMEM) (Thermo Fisher Scientific, USA) with 10% FBS and 1% penicillin/streptomycin at 37°C under 5% CO2 and 95% air.
Reagents
Recombinant human globular adiponectin (gAd) was purchased from BioVision (USA). A stock solution of gAd was prepared in distilled water containing 50 mg/ml BSA to a concentration of 1 mg/ml and aliquots were stored at −20°C until needed. MG132 was obtained from Calbiochem (Germany). PS-341 and SC-514 were supplied by Selleck Chemicals (USA). Lipopolysaccharide (LPS) and polymyxin B (PMB) were obtained from Sigma-Aldrich (USA). CpG-oligodeoxynucleotide (CpG-ODN) was supplied by InvivoGen (USA). Control and IRAK-1 siRNAs were purchased from Santa Cruz Biotechnology Inc. (USA).
Cytoplasmic and nuclear extraction
Cells were washed with cold PBS and allowed to equilibrate in 1X cytoplasmic extraction buffer (CEB containing 10 mM Tris-HCl (pH 7.8), 10 mM KCl, 1.5 mM EDTA, 0.5 mM DTT, 1 mM Na2VO4, and 2 mM Levamisole) on ice for 5 min. The cells were lysed with cytoplasmic lysis buffer (CEB containing 0.4 % NP-40 and a protease inhibitor cocktail), and scraped gently. Following centrifugation at 3,500 rpm at 4°C for 5 min, the supernatants (cytoplasmic extracts) were collected. The nuclear pellets were rinsed with nuclear extraction buffer (NEB containing 20 mM Tris-HCl (pH 7.8), 150 mM NaCl, 50 mM KCl, 1.5 mM EDTA, 5 mM DTT, 1 mM Na2VO4, and 2 mM levamisole), and resuspended in nuclear lysis buffer (NEB/0.4% NP-40/protease inhibitor cocktail). The suspension was subjected to centrifugation at 13,000 rpm at 4°C for 10 min. The supernatants (nuclear extracts) were either used immediately or stored at −70°C.
Immunoblotting analysis and Antibodies
Total cellular extracts were prepared using 1X Cell Lysis Buffer (Cell Signaling Technology, USA) supplemented with 1 mM PMSF. Protein concentration was determined using the Bradford assay. Cell extracts were subjected to sodium do-decyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and gels were transferred to Hybond ECL nitrocellulose membranes (GE Heathcare, Germany) for 100 min at 90 V. The membranes were blocked with 5% skim milk in 1X tris-buffered saline (TBS) containing 0.1% Tween 20 (TBS-T) for 1 h at room temperature. The following antibodies were used for protein detection: anti-IκBα, anti-p65, anti-IKKα, anti-IKKβ, anti-MyD88, anti-IRAK1, anti-TRAF6, anti-TAK1, anti-actin antibodies (Santa Cruz Biotechnology Inc.), anti-phosphorylated TAK1 (p-TAK1) at Ser 412, anti-p-IKKα/β at Ser176/180, anti-p-IκBα at Ser32, and anti-p-Akt at Ser473 antibodies (Cell Signaling Technology, USA), and anti-p-IRAK-1 at Thr209 (Abcam, USA). After successful washes, the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h. Blots were developed using West Pico Western blot detection kit (Thermo Fisher Scientific).
In vitro kinase assay
RAW 264.7 cells were stimulated with gAd and LPS. The IKK complex in total cell extracts was immunoprecipitated using anti-IKKα antibody. Protein A/G UltraLink Resin (Thermo Fisher Scientific) was added to each tube, and rotated at 4°C for 1 h. The beads were washed sequentially in 1X cell lysis buffer, wash buffer (20 mM Tris-HCl (pH 7.4), 0.1 % NP-40), and 1X kinase buffer (Cell Signaling Technology, USA). The immunoprecipitates were incubated at 30°C for 30 min in a kinase buffer containing 0.5 μg recombinant IκBα and 0.2 mM of ATP. The kinase reaction products were subjected to SDS-PAGE, then, transferred to a nitrocellulose membrane, and analyzed by immunoblotting.
Quantitative real time-PCR
Total RNA from RAW 264.7 cells was isolated using RNeasy kit (Qiagen, Germany). cDNA was synthesized from 1μg of total RNA using Reverse Transcription System (Promega, USA). Power SYBR Green PCR Master Mix (Applied Biosystems, USA) was used for amplification of TNF-α, IRAK-1 and GAPDH. The primers used were moues TNF-α (fwd:5′-CAC AGA AAG CAT GAT CCG CGA CGT-3′, rev:5′-CGG CAG AGA GGA GGT TGA CTT TCT-3′), mouse IRAK-1 (fwd: 5′-CAG AAC CAC CAC AGA TCA TCA TC-3′, rev: 5′-GGC TAT CCA AGA CCC CTT CTT C-3′) and mouse GAPDH (fwd: 5′-TCC CTC AAG ATT GTC AGC AAT G-3′, rev: 5′-AGA TCC ACA ACG GAT ACA TTG G-3′).
Transfection of plasmid vectors and siRNAs
Transfection with plasmid vectors or siRNAs was performed using a Neon Transfection System (Thermo Fisher Scientific) according to the manufacturer’s specifications. After 48 h, cells were used in experiments.
Multiplex bead assay
TNF-α and IL-6 levels in culture supernatants were determined using a commercially available Bio-Plex Pro™ Cytokine Assay Kit (Bio-Rad, USA) according to the manufacturer’s instructions.
Statistical analysis
Data was subjected to Student’s t-test to determine statistical significance, and a P value of < 0.05 was considered significant.
RESULTS
Pro-inflammatory effect of gAd is mediated through IκB/NF-κB pathway
Since the inhibitory protein, IκBα, sequesters NF-κB in an inactive form in the cytoplasm, the phosphorylation and subsequent degradation of IκBα is prerequisite for the activation of NF-κB. To investigate if gAd activates the IκB/NF-κB pathway, we evaluated the effect of gAd on IκBα expression. RAW 264.7 macrophage cells were treated with gAd (1, 3, 5, and 10 μg/ml) and lipopolysaccharide (LPS, 1 μg/ml) for 30 min. LPS was used as a positive control to induce IκBα degradation. gAd markedly induced the degradation of IκBα in a dose-dependent manner (Fig. 1A, upper panel). Next, we tested the time-course effect of gAd on IκBα phosphorylation and degradation. Cells were treated with gAd (5 μg/ml) for 5, 10, 20, 30, and 60 min. Phosphorylation of IκBα started to increase 5 min after gAd stimulation. gAd induced maximum degradation of IκBα at 30 min and gAd-induced IκBα degradation was recovered to baseline (Fig. 1A, lower panel). This indicates that the gAd-induced IκBα degradation is reversible. To exclude the possibility that the effect of gAd on IκBα expression could be due to endotoxin contamination in the recombinant protein, we pretreated cells with polymyxin B (PMB, endotoxin inactivator) prior to gAd stimulation and treated cells with heated gAd. The PMB treatment did not block gAd-induced IκBα phosphorylation and degradation (Fig. 1B, upper panel). Moreover, the treatment with heated gAd did not induce phosphorylation and degradation of IκBα (Fig. 1B, lower panel). These results suggest that gAd-mediated IκBα phosphorylation and degradation are not due to contamination by endotoxin.
Fig. 1. gAd activates NF-κB and induces pro-inflammatory cytokine secretion.
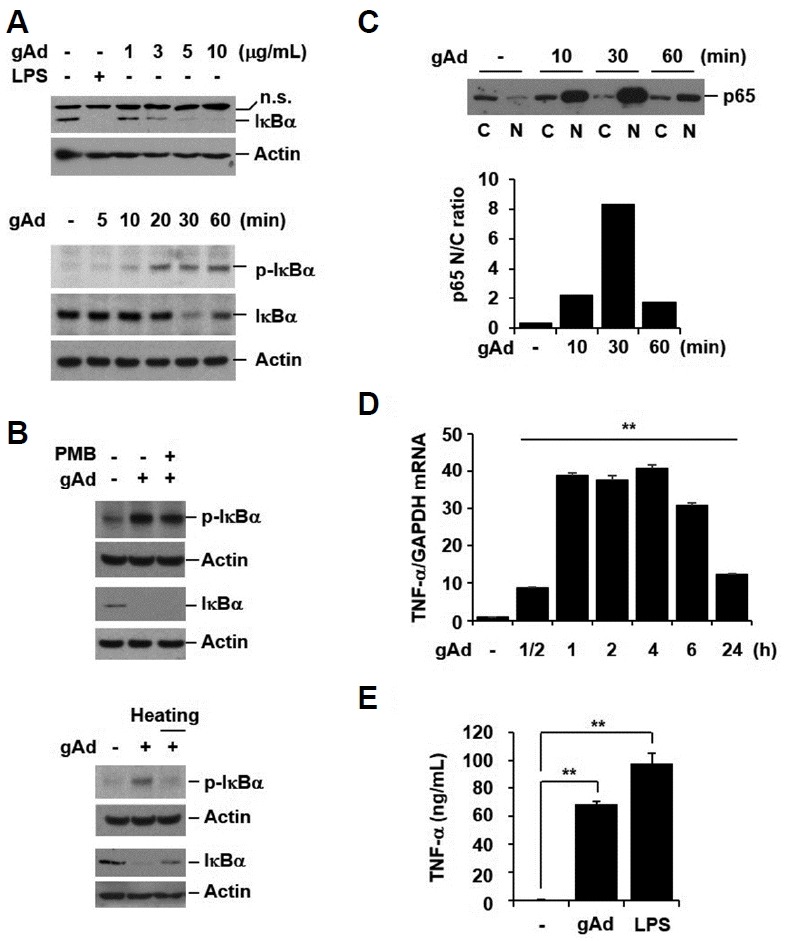
(A) RAW 264.7 cells were incubated with gAd (1, 3, 5, 10 μg/ml) and LPS (1 μg/ml) for 30 min. Cells were stimulated with gAd (5 μg/ml) for the indicated times. (B) RAW 264.7 cells were pretreated with polymyxin B (PMB, 10 μg/ml) for 1 h, and then stimulated with gAd (5 μg/ml) for 30 min in the presence or absence of PMB. Cells were treated with gAd and heated gAd (5 μg/ml) for 30 min. Total cellular extracts were subjected to Western blot analysis for p-IκBα, IκBα and actin. (C) Cells were stimulated with gAd (5 μg/ml) for the indicated times. Cytoplasmic (C) and nuclear (N) proteins were extracted. Equal amounts of protein were subjected to Western blot analysis for p65. Ratio of nuclear to cytoplasmic (N/C) p65 densitometry values (lower panel of (C)). (D) RAW 264.7 cells were stimulated with gAd (5 μg/ml) for the indicated times. Total RNA was isolated and quantitative real-time PCR for TNF-α and GAPDH was performed. Data represent the mean ± SD of triplicates. **P < 0.05 (E) Cells were treated with gAd (5 μg/ml) and LPS (1 μg/ml) for 24 h. TNF-α concentration in culture medium was measured by multiplex bead assay. Data represent the mean ± SD of triplicates. **P < 0.05 Results are representative of three separate experiments. n.s., non-specific.
Activation of NF-κB is associated with nuclear translocation of the p65 component. We treated RAW 264.7 cells with gAd (5 μg/ml) for 10, 30, and 60 min, and extracted cytoplasmic (C) and nuclear (N) proteins. The expression of p65 was analyzed by Western blotting. gAd induced nuclear translocation of p65 (Fig. 1C). Moreover, gAd increased the mRNA expression and release of NF-κB-regulated pro-inflammatory cytokine, TNF-α (Figs. 1D and 1E). Collectively, these findings indicate that gAd activates the IκB/NF-κB pathway, which subsequently leads to proinflammatory cytokine production.
gAd-induced IκBα degradation is caused by IKK and Akt activation
To determine the mechanism of gAd-mediated IκBα degradation, we investigated the effect of gAd on IκB kinase (IKK) activity. RAW 264.7 cells were incubated with gAd and LPS for 10 and 20 min, respectively. The IKK complex was immunoprecipitated with anti-IKKα antibodies. The in vitro kinase activity assay was performed as described in the Materials and Methods section. gAd as well as LPS activated IKK (Fig. 2A). When IKK activity was blocked by IKKβ-specific inhibitor (SC-514, 100 μM) pretreatment, gAd-induced phosphorylation and degradation of IκBα were suppressed (Fig. 2B). To further confirm that IKK activity is required for gAd-induced IκBα phosphorylation and degradation, IKK activity was blocked by the overexpression of dominant-negative IKKβ (DN-IKKβ). In cells overexpressing DN-IKKβ, gAd-induced phosphorylation and degradation of IκBα were suppressed (Fig. 2C). These results suggest that IKK activation is responsible for gAd-induced IκBα degradation.
Fig. 2. gAd-induced IκBα degradation is mediated by IKK and Akt activation.
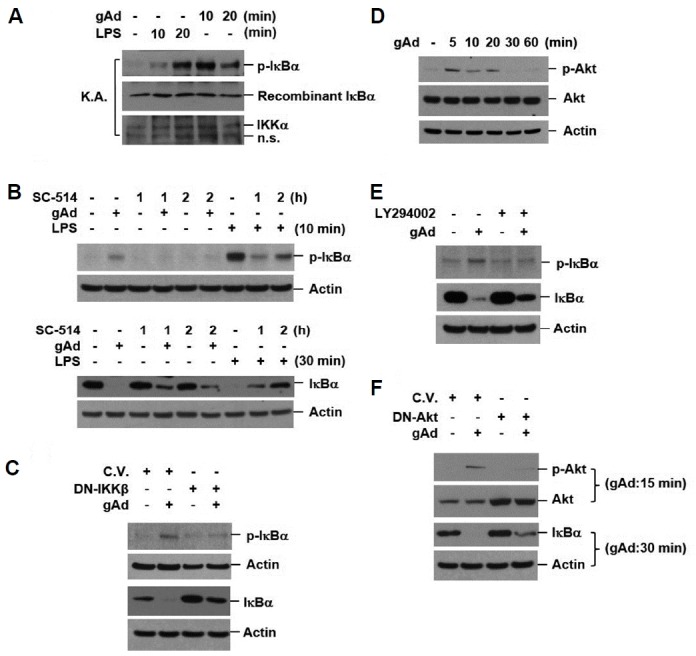
(A) RAW 264.7 cells were treated with gAd (5 μg/ml) and LPS (1 μg/ml) for the indicated times. The IKK complex was immunoprecipitated using anti-IKKα antibodies. The kinase activity of IKK was assayed as described in the materials and methods section. (B) RAW 264.7 cells were preincubated with SC-514 (an IKKβ specific inhibitor, 100 μM) and then, stimulated with gAd (5 μg/ml) for 30 min and LPS (1 μg/ml) for 10 or 30 min in the presence or absence of SC-514. (C) Cells were transiently transfected with DN-IKKβ (K44A) and control plasmid vector (C.V.). At 48 h after transfection, cells were incubated with gAd (5 μg/ml) for 30 min. (D) Cells were treated with gAd (5 μg/ml) for the indicated times. (E) RAW 264.7 cells were pretreated with LY294002 (40 μM) for 2 h and then stimulated with gAd (5 μg/ml) for 30 min. (F) Cells were transfected with DN-Akt and control plasmid vector. At 48 h after transfection, cells were incubated with gAd (5 μg/ml) for 15 or 30 min. Total cellular extracts were subjected to Western blot analysis for p-IκBα, IKKα, IκBα, p-Akt, Akt, and actin. Results are representative of three separate experiments. K.A., kinase activity assay.
Next, we analyzed the ability of gAd to activate the phosphatidylinositol 3-kinase (PI3K)/Akt pathway, because previous studies suggested that PI3K/Akt modulates the NF-κB signaling pathway (Zhao et al., 2008), and gAd activates Akt in monocytes (Sattar and Sattar, 2012). Treatment with gAd induced phosphorylation of Akt on serine 473 (Fig. 2D) and blocking Akt activation by treatment with LY294002 (PI3K inhibitor) and overexpression of dominant-negative Akt (DN-Akt) inhibited gAd-induced IκBα phosphorylation and degradation (Figs. 2E and 2F), suggesting that the activation of the PI3K/Akt pathway is involved in gAd-induced IκBα degradation. Taken together, these results indicate that the activation of either Akt or IKK is involved in the gAd-mediated activation of the IκB/NF-κB pathway.
Anti-inflammatory effect of gAd results from induction of immune tolerance
Continued exposure to gAd negatively regulates macrophage response to Toll-like receptor (TLR) 4 ligand LPS or TLR3 ligand poly(I:C) (Tsatsanis et al., 2005; 2006; Zacharioudaki et al., 2009). To confirm the anti-inflammatory properties of gAd, we treated RAW 264.7 cells with gAd for 4 and 24 h, and then re-stimulated the cells with gAd, LPS, and CpG-ODN. IκBα expression was examined by Western blot analysis and pro-inflammatory cytokine concentration in culture supernatants was assessed by multiplex bead assay 20 h after stimulation. Although phosphorylation of IκBα by gAd, LPS, or CpG-ODN was not suppressed by gAd pre-treatment, IκBα degradation was blocked in gAd-pretreated cells (Figs. 3A and 3B). Both TNF-α and IL-6 productions were reduced by pre-exposure with gAd (Fig. 3C). These results indicate that gAd induces a resistance to IκBα degradation and pro-inflammatory cytokine production to subsequent challenge with gAd and TLR ligands.
Fig. 3. gAd pretreatment suppresses inflammatory response to gAd, LPS, and CpG-ODN.
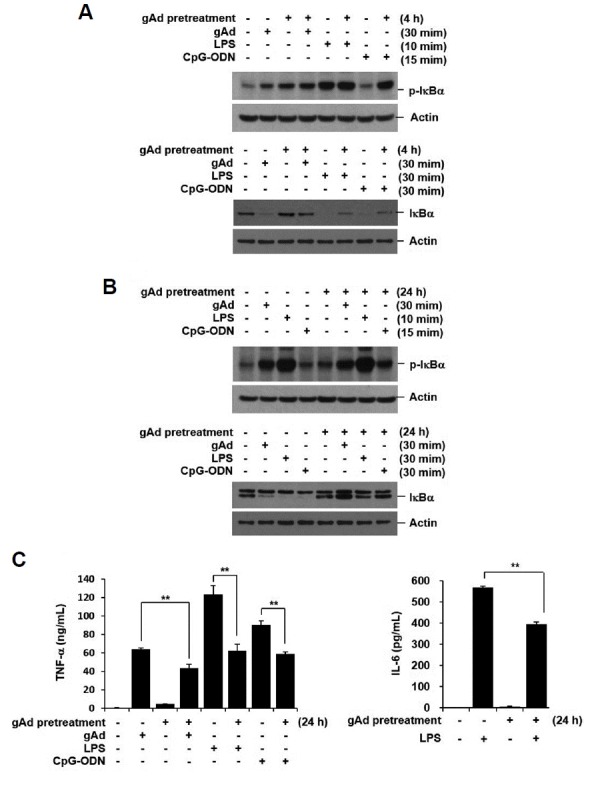
(A, B) RAW 264.7 cells were preincubated with gAd (5 μg/ml) for 4 h or 24 h, and then, washed several times with fresh medium. The cells were stimulated with gAd (5 μg/ml), LPS (1 μg/ml), and CpG-ODN (1 μM) for the indicated times. Total cellular extracts were subjected to Western blot analysis for p-IκBα, IκBα, and actin. (C) After preincubation with gAd for 24 h, RAW 264.7 cells were washed, and then stimulated with gAd, LPS and CpG-ODN for 20 h. TNF-α and IL-6 concentrations in the culture supernatant were measured by multiplex bead assay. Data represent the mean ± SD of triplicates. **P < 0.05 Results are representative of three separate experiments.
Tolerance induction by long-term incubation with gAd is associated with transcriptional repression of IRAK-1
To determine the mechanism responsible for tolerance to TLR signaling by long-term incubation with gAd, we investigated the effect of gAd on the expression of the upstream regulators of IκBα. Long-term treatment with gAd increased the expression of myeloid differentiation factor 88 (MyD88), tumor necrosis factor receptor (TNFR)-associated factor 6 (TRAF6), and TGF-β-activated kinase 1 (TAK1)(Fig. 4A). Interestingly, IRAK-1 protein expression was markedly down-regulated by gAd (Fig. 4A). The IRAK-1 downregulation could be due to post-translational modifications, since it has been shown to be degraded via the proteasome following its phosphorylation (Yamin and Miller, 1997). To evaluate if gAd-mediated IRAK-1 downregulation is due to proteasomal degradation, we pretreated cells with proteasome inhibitors (MG132 or PS-341). The proteasomal inhibitors did not prevent gAd-induced IRAK-1 downregulation (Fig. 4B). Thus, post-translational modification of IRAK-1 by gAd is less likely to occur. We next examined the mRNA levels of IRAK-1 after gAd stimulation by quantitative real-time PCR analysis. IRAK-1 mRNA was constitutively expressed in RAW 264.7 cells and its expression was significantly decreased by gAd (Fig. 4C). To determine if downregulation of IRAK-1 is at least partially responsible for tolerance to gAd and TLR signaling, we transiently transfected cells with control and IRAK-1 siRNAs. At 48 h after transfection, cells were treated with gAd, LPS, and CpG-ODN for 24 h. Knock-down of IRAK-1 significantly suppressed TNF-α and IL-6 production in gAd, LPS, and CpG-ODN-treated cells (Figs. 4D and 4E). This indicates that tolerance induction by long-term incubation with gAd might be associated with transcriptional repression of IRAK-1.
Fig. 4. gAd downregulates IRAK-1 mRNA and protein expression.
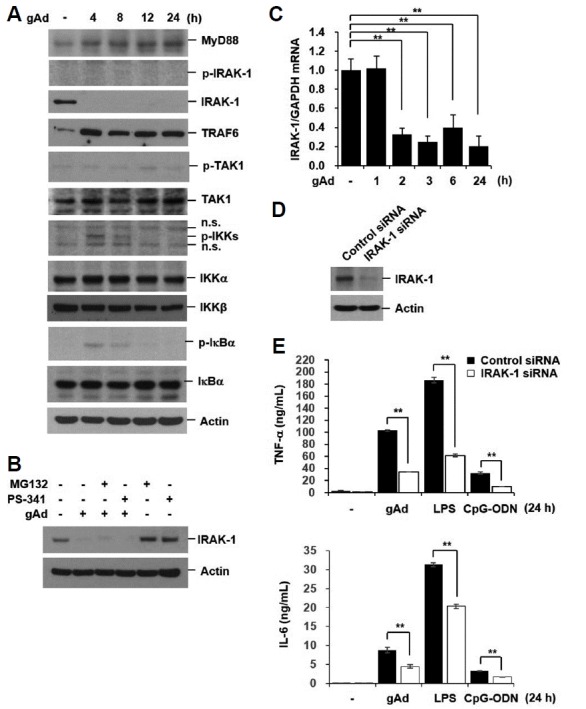
(A) RAW 264.7 cells were incubated with gAd (5 μg/ml) for the indicated times. (B) Cells were pretreated with MG132 (10 μM) and PS-341 (200 nM) for 1 h, and then stimulated with gAd (5 μg/ml) for 4 h in the presence or absence of MG132 and PS-341. Total cellular extracts were subjected to Western blot analysis for MyD88, p-IRAK-1, IRAK-1, TRAF6, p-TAK1, TAK1, p-IKKα/β, IKKα, IKKβ, p-IκBα, IκBα, and actin. (C) Cells were treated with gAd (5 μg/ml) for the indicated times. Quantitative real-time PCR for IRAK-1 and GAPDH was performed. Data represent the mean ± SD of triplicates. **P < 0.05 (D-E) RAW 264.7 cells were transiently transfected with control or IRAK-1 siRNAs. At 48 h after transfection, cells were treated with gAd (5 μg/ml), LPS (1 μg/ml), or CpG-ODN (1 μM) for 24 h. Total cellular extracts were subjected to Western blot analysis for IRAK-1 and Actin. TNF-α and IL-6 concentrations in culture medium were measured by multiplex bead assay. Data represent the mean ± SD of triplicates. **P < 0.05 Results are representative of three separate experiments.
DISCUSSION
Adiponectin is the most abundant hormone in the plasma secreted by the adipose tissue. Adiponectin exists in its full length and forms homomultimers (Tanianskii and Denisenko, 2012). Proteolytic cleavage of the full-length adiponectin by macrophage-derived elastase yields a smaller product, gAd, which is also present in the circulation (Waki et al., 2005), and has several biological effects.
The anti-inflammatory effects of adiponectin in innate immune cells are well-known. The molecular mechanisms of these effects include increased expressions of interleukin-10 (IL-10) (Kumada et al., 2004; Park et al., 2007; Wolf et al., 2004), peroxisome proliferator-activated receptor γ2 (PPARγ2) (Ajuwon and Spurlock, 2005), and IRAK-M (Zacharioudaki et al., 2009). Active ERK-dependent early growth response protein 1 (Egr-1) expression and NF-κB activation by gAd increase TNF-α production, which, enhances IL-10 release, in turn, mediating desensitization to secondary LPS stimulation (Huang et al., 2008). Enhanced activity of PPARγ2 (Ajuwon and Spurlock, 2005) suppresses the transactivation ability of NF-κB. IRAK-M mediates LPS tolerance by inhibiting the association of IRAK-1 and IRAK-4 with TRAF6 (Zacharioudaki et al., 2009). However, the mechanisms for the inhibitory effects of adiponectin are not fully understood. Moreover, several recent reports demonstrate that short-term stimulation with adiponectin elicits a pro-inflammatory activity (Tsatsanis et al., 2005) via ERK and NF-κB activation (Park et al., 2007). Thus, adiponectin appears to have dual (anti- and pro-) inflammatory properties.
In this study, we also found that gAd treatment for a relatively short time activates NF-κB via the degradation of its inhibitor, IκBα, resulting in pro-inflammatory cytokine production. One report suggested that the action of adiponectin in human monocyte-derived macrophages may be mediated by endotoxin contamination during gAd preparation (Turner et al., 2009). To rule out this possibility, we pretreated cells with PMB, an antibiotic which binds and inactivates LPS, and then evaluated the effects of gAd on IκBα degradation. PMB had no effect on gAd-induced IκBα phosphorylation and degradation. It is further supported by the fact that heated gAd was unable to degrade IκBα because heat treatment inactivates gAd, but not LPS. IκBα degradation by gAd resulted in the release and nuclear translocation of NF-κB subunit, leading to the expression of pro-inflammatory genes such as TNF-α.
Phosphorylation of two serine residues (serine 32 and 36) of IκB by active IKK is required for its subsequent polyubiquitination and degradation via the proteasome. Our findings showed that gAd activated IKK, which was involved in gAd-induced phosphorylation and degradation of IκBα. Moreover, we found that gAd induced Akt activation, and the blockade of Akt activation suppressed gAd-induced phosphorylation and degradation of IκBα. Collectively, these findings suggest that active IKK and Akt mediate gAd-induced phosphorylation and degradation of IκBα, resulting in NF-κB activation and pro-inflammatory cytokine production. These results are inconsistent with the effects of adiponectin in IL-18-treated endothelial cells. IL-18-treated endothelial cells underwent apoptosis, which was blocked by adiponectin treatment. The anti-apoptotic effects of adiponectin are mediated by the inhibition of IKK/NF-κB/PTEN signaling. Treatment of endothelial cells with adiponectin reverses IL-18-induced IKK activity (Chandrasekar et al., 2008). The inhibitory effects of gAd on NF-κB pathway might be cell type-specific.
The TLR family, in humans, consists of 10 members (TLR1–TLR10), which are involved in two distinct signaling pathways, the MyD88-dependent and MyD88-independent pathway. Prolonged stimulation with adiponectin has been reported to suppress TLR signaling involving TLR3 and TLR4 (Tsatsanis et al., 2005). One report suggests that adiponectin suppresses the TLR4/MyD88-independent pathway via the heme oxygenase-1 (HO-1)-mediated reduction of TLR4 expression (Mandal et al., 2010). In our study, we found that continued stimulation with gAd suppressed the production of pro-inflammatory cytokines such as TNF-α and IL-6 to subsequent challenge with gAd or TLR ligands. While TLR4 signaling utilizes both the MyD88-dependent and MyD88-independent pathways (Takeda and Akira, 2004), TLR9 signaling utilizes only the MyD88-dependent pathway. Thus, these findings suggest that gAd might target upstream mediators (MyD88, IRAK, TRAF6, TAK1, and IKKs) of IκBα in the MyD88-dependent pathway. Among these mediators, we found that the gAd treatment decreased IRAK-1 expression. IRAK-1 is a serine/threonine kinase, which plays critical roles in TLR signaling (Takeda and Akira, 2004) and the activation of NF-κB (Liu et al., 2008). IRAK-1 deficiency is known to mediate inefficient IκBα degradation and decreased activation of NF-κB. As IRAK-1 is responsible for IKK phosphorylation and gAd depleted IRAK-1 in our experiments, we first speculated that tolerance induction by gAd might be mediated by blocking TLR stimulation-induced IκBα phosphorylation and its degradation. Unexpectedly, IκBα was phosphorylated by gAd and TLR stimulation in gAd-pretreated cells, suggesting that IκBα phosphorylation (at Ser32) does not efficiently lead to IκBα degradation in gAd pretreated cells in which IRAK-1 is down-regulated. These results raise the possibility that tolerance induction in IRAK-1 down-regulated cells by gAd occurs independently of IκBα phosphorylation. In support of this explanation, IκBα degradation is significantly less and NF-κB activation is defective in response to TLR ligands and IL-1 stimulation in IRAK-1 deficient cells including macrophages and fibroblast (Chandra et al., 2013; Kanakaraj et al., 1998; Kawagoe et al., 2008; Thomas et al., 1999; Zhou et al., 2013). We also demonstrated that knock-down of IRAK-1 significantly suppressed pro-inflammatory cytokine production in gAd, LPS, and CpG-ODN-treated cells. Thus, the reduction in IRAK-1 expression by gAd is associated with the tolerance.
In response to stimuli such as LPS, IRAK undergoes proteasomal degradation following its phosphorylation (Yamin and Miller, 1997). In this study, it was shown that inhibition of proteasomal activity by pretreating its specific inhibitors (MG132 or PS-341) did not block gAd-induced IRAK-1 downregulation, which suggests that the gAd-mediated reduction in IRAK-1 expression is not due to proteasomal degradation. Bacterial lipoprotein (BLP)-induced self-tolerance and cross-tolerance to LPS were reported to be associated with reduced IRAK-1 expression (Li et al., 2006). However, IRAK-1 mRNA expression remained unchanged in BLP-tolerant cells, which suggests that BLP self-tolerance and cross-tolerance to LPS occur at the post-transcriptional level. Interestingly, we found that gAd treatment significantly decreased IRAK-1 mRNA expression in a time-dependent manner. This was not due to the effect of gAd on cell viability since gAd treatment did not change cell viability, but rather slightly increased proliferation rate (data not shown). These findings demonstrate that the mechanism of gAd-induced tolerance to TLR is different from that of BLP or LPS-induced tolerance.
In summary, our study shows that gAd exerts a pro-inflammatory response via the IKK- and Akt-dependent activation of NF-κB at an early phase after stimulation, and continued exposure with gAd induces tolerance to TLR signaling via the reduction of IRAK-1 mRNA and protein expression.
ACKNOWLEDGMENTS
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
REFERENCES
- Ajuwon K.M., Spurlock M.E. Adiponectin inhibits LPS-induced NF-kappaB activation and IL-6 production and increases PPARgamma2 expression in adipocytes. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1220–1225. doi: 10.1152/ajpregu.00397.2004. [DOI] [PubMed] [Google Scholar]
- Arita Y., Kihara S., Ouchi N., Takahashi M., Maeda K., Miyagawa J., Hotta K., Shimomura I., Nakamura T., Miyaoka K., et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257:79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- Berg A.H., Combs T.P., Du X., Brownlee M., Scherer P.E. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med. 2001;7:947–953. doi: 10.1038/90992. [DOI] [PubMed] [Google Scholar]
- Chan K.H., Yeung S.C., Yao T.J., Ip M.S., Cheung A.H., Chan-Yeung M.M., Mak J.C. Society CSGotHKT. Elevated plasma adiponectin levels in patients with chronic obstructive pulmonary disease. Int J Tuberc Lung Dis. 2010;14:1193–1200. [PubMed] [Google Scholar]
- Chandra R., Federici S., Bishwas T., Németh Z.H., Deitch E.A., Thomas J.A., Spolarics Z. IRAK1-dependent signaling mediates mortality in polymicrobial sepsis. Inflammation. 2013;36:1503–1512. doi: 10.1007/s10753-013-9692-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekar B., Boylston W.H., Venkatachalam K., Webster N.J., Prabhu S.D., Valente A.J. Adiponectin blocks interleukin-18-mediated endothelial cell death via APPL1-dependent AMP-activated protein kinase (AMPK). activation and IKK/NF-kappaB/PTEN suppression. J Biol Chem. 2008;283:24889–24898. doi: 10.1074/jbc.M804236200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalamaga M., Diakopoulos K.N., Mantzoros C.S. The role of adiponectin in cancer: a review of current evidence. Endocrine Rev. 2012;33:547–594. doi: 10.1210/er.2011-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruebis J., Tsao T.S., Javorschi S., Ebbets-Reed D., Erickson M.R., Yen F.T., Bihain B.E., Lodish H.F. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci USA. 2001;98:2005–2010. doi: 10.1073/pnas.041591798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugen F., Drevon C.A. Activation of nuclear factor-kappaB by high molecular weight and globular adiponectin. Endocrinology. 2007;148:5478–5486. doi: 10.1210/en.2007-0370. [DOI] [PubMed] [Google Scholar]
- Haugen F., Ranheim T., Harsem N.K., Lips E., Staff A.C., Drevon C.A. Increased plasma levels of adipokines in preeclampsia: relationship to placenta and adipose tissue gene expression. Am J Physiol Endocrinol Metab. 2006;290:E326–333. doi: 10.1152/ajpendo.00020.2005. [DOI] [PubMed] [Google Scholar]
- Hendler I., Blackwell S.C., Mehta S.H., Whitty J.E., Russell E., Sorokin Y., Cotton D.B. The levels of leptin, adiponectin, and resistin in normal weight, overweight, and obese pregnant women with and without preeclampsia. Am J Obstet Gynecol. 2005;193:979–983. doi: 10.1016/j.ajog.2005.06.041. [DOI] [PubMed] [Google Scholar]
- Huang H., Park P.H., McMullen M.R., Nagy L.E. Mechanisms for the anti-inflammatory effects of adiponectin in macrophages. J Gastroenterol Hepatol. 2008;23 Suppl 1:S50–53. doi: 10.1111/j.1440-1746.2007.05284.x. [DOI] [PubMed] [Google Scholar]
- Hulthe J., Hulten L.M., Fagerberg B. Low adipocyte-derived plasma protein adiponectin concentrations are associated with the metabolic syndrome and small dense low-density lipoprotein particles: atherosclerosis and insulin resistance study. Metabolism. 2003;52:1612–1614. doi: 10.1016/s0026-0495(03)00313-5. [DOI] [PubMed] [Google Scholar]
- Kanakaraj P., Schafer P.H., Cavender D.E., Wu Y., Ngo K., Grealish P.F., Wadsworth S.A., Peterson P.A., Siekierka J.J., Harris C.A., et al. Interleukin (IL).-1 receptor-associated kinase (IRAK). requirement for optimal induction of multiple IL-1 signaling pathways and IL-6 production. J Exp Med. 1998;187:2073–2079. doi: 10.1084/jem.187.12.2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawagoe T., Sato S., Matsushita K., Kato H., Matsui K., Kumagai Y., Saitoh T., Kawai T., Takeuchi O., Akira S. Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat Immunol. 2008;9:684–691. doi: 10.1038/ni.1606. [DOI] [PubMed] [Google Scholar]
- Kirdar S., Serter M., Ceylan E., Sener A.G., Kavak T., Karadag F. Adiponectin as a biomarker of systemic inflammatory response in smoker patients with stable and exacerbation phases of chronic obstructive pulmonary disease. Scand J Clin Lab Invest. 2009;69:219–224. doi: 10.1080/00365510802474400. [DOI] [PubMed] [Google Scholar]
- Kubota N., Terauchi Y., Yamauchi T., Kubota T., Moroi M., Matsui J., Eto K., Yamashita T., Kamon J., Satoh H., et al. Disruption of adiponectin causes insulin resistance and neointimal formation. J Biol Chem. 2002;277:25863–25866. doi: 10.1074/jbc.C200251200. [DOI] [PubMed] [Google Scholar]
- Kumada M., Kihara S., Ouchi N., Kobayashi H., Okamoto Y., Ohashi K., Maeda K., Nagaretani H., Kishida K., Maeda N., et al. Adiponectin specifically increased tissue inhibitor of metalloproteinase-1 through interleukin-10 expression in human macrophages. Circulation. 2004;109:2046–2049. doi: 10.1161/01.CIR.0000127953.98131.ED. [DOI] [PubMed] [Google Scholar]
- Li C.H., Wang J.H., Redmond H.P. Bacterial lipoprotein-induced self-tolerance and cross-tolerance to LPS are associated with reduced IRAK-1 expression and MyD88-IRAK complex formation. J Leukoc Biol. 2006;79:867–875. doi: 10.1189/jlb.0905505. [DOI] [PubMed] [Google Scholar]
- Lindsay R.S., Funahashi T., Hanson R.L., Matsuzawa Y., Tanaka S., Tataranni P.A., Knowler W.C., Krakoff J. Adiponectin and development of type 2 diabetes in the Pima Indian population. Lancet. 2002;360:57–58. doi: 10.1016/S0140-6736(02)09335-2. [DOI] [PubMed] [Google Scholar]
- Liu G., Park Y.J., Abraham E. Interleukin-1 receptor-associated kinase (IRAK). -1-mediated NF-kappaB activation requires cytosolic and nuclear activity. FASEB J. 2008;22:2285–2296. doi: 10.1096/fj.07-101816. [DOI] [PubMed] [Google Scholar]
- Maeda N., Shimomura I., Kishida K., Nishizawa H., Matsuda M., Nagaretani H., Furuyama N., Kondo H., Takahashi M., Arita Y., et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med. 2002;8:731–737. doi: 10.1038/nm724. [DOI] [PubMed] [Google Scholar]
- Mandal P., Roychowdhury S., Park P.H., Pratt B.T., Roger T., Nagy L.E. Adiponectin and heme oxygenase-1 suppress TLR4/MyD88-independent signaling in rat Kupffer cells and in mice after chronic ethanol exposure. J Immunol. 2010;185:4928–4937. doi: 10.4049/jimmunol.1002060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa Y., Funahashi T., Kihara S., Shimomura I. Adiponectin and metabolic syndrome. Arterioscler Thromb Vasc Biol. 2004;24:29–33. doi: 10.1161/01.ATV.0000099786.99623.EF. [DOI] [PubMed] [Google Scholar]
- Okamoto Y., Kihara S., Ouchi N., Nishida M., Arita Y., Kumada M., Ohashi K., Sakai N., Shimomura I., Kobayashi H., et al. Adiponectin reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2002;106:2767–2770. doi: 10.1161/01.cir.0000042707.50032.19. [DOI] [PubMed] [Google Scholar]
- Otero M., Lago R., Gomez R., Lago F., Dieguez C., Gomez-Reino J.J., Gualillo O. Changes in plasma levels of fat-derived hormones adiponectin, leptin, resistin and visfatin in patients with rheumatoid arthritis. Ann Rheum Dis. 2006;65:1198–1201. doi: 10.1136/ard.2005.046540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park P.H., McMullen M.R., Huang H., Thakur V., Nagy L.E. Short-term treatment of RAW264.7 macrophages with adiponectin increases tumor necrosis factor-alpha (TNF-alpha). expression via ERK1/2 activation and Egr-1 expression: role of TNF-alpha in adiponectin-stimulated interleukin-10 production. J Biol Chem. 2007;282:21695–21703. doi: 10.1074/jbc.M701419200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattar A.A., Sattar R. Globular adiponectin activates Akt in cultured myocytes. Biochemi Biophys Res Commun. 2012;424:753–757. doi: 10.1016/j.bbrc.2012.07.027. [DOI] [PubMed] [Google Scholar]
- Schlegel A. Adiponectin and risk of coronary heart disease. JAMA. 2004;292:40. doi: 10.1001/jama.292.1.40-a. author reply 40. [DOI] [PubMed] [Google Scholar]
- Senolt L., Pavelka K., Housa D., Haluzik M. Increased adiponectin is negatively linked to the local inflammatory process in patients with rheumatoid arthritis. Cytokine. 2006;35:247–252. doi: 10.1016/j.cyto.2006.09.002. [DOI] [PubMed] [Google Scholar]
- Takeda K., Akira S. TLR signaling pathways. Seminars in immunology. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Tanianskii D.A., Denisenko A.D. [Molecular forms of adiponectin: comparative evaluation of their correlations with parameters of carbohydrate and lipid metabolism]. Biomeditsinskaia khimiia. 2012;58:457–466. doi: 10.18097/pbmc20125804457. [DOI] [PubMed] [Google Scholar]
- Thomas J.A., Allen J.L., Tsen M., Dubnicoff T., Danao J., Liao X.C., Cao Z., Wasserman S.A. Impaired cytokine signaling in mice lacking the IL-1 receptor-associated kinase. J Immunol. 1999;163:978–984. [PubMed] [Google Scholar]
- Tilg H., Moschen A.R. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6:772–783. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- Tsatsanis C., Zacharioudaki V., Androulidaki A., Dermitzaki E., Charalampopoulos I., Minas V., Gravanis A., Margioris A.N. Adiponectin induces TNF-alpha and IL-6 in macrophages and promotes tolerance to itself and other pro-inflammatory stimuli. Biochem Biophys Res Commun. 2005;335:1254–1263. doi: 10.1016/j.bbrc.2005.07.197. [DOI] [PubMed] [Google Scholar]
- Tsatsanis C., Zacharioudaki V., Androulidaki A., Dermitzaki E., Charalampopoulos I., Minas V., Gravanis A., Margioris A.N. Peripheral factors in the metabolic syndrome: the pivotal role of adiponectin. Ann N Y Acad Sci. 2006;1083:185–195. doi: 10.1196/annals.1367.013. [DOI] [PubMed] [Google Scholar]
- Turner J.J., Smolinska M.J., Sacre S.M., Foxwell B.M. Induction of TLR tolerance in human macrophages by adiponectin: does LPS play a role? Scand J Immunol. 2009;69:329–336. doi: 10.1111/j.1365-3083.2008.02224.x. [DOI] [PubMed] [Google Scholar]
- Waki H., Yamauchi T., Kamon J., Kita S., Ito Y., Hada Y., Uchida S., Tsuchida A., Takekawa S., Kadowaki T. Generation of globular fragment of adiponectin by leukocyte elastase secreted by monocytic cell line THP-1. Endocrinology. 2005;146:790–796. doi: 10.1210/en.2004-1096. [DOI] [PubMed] [Google Scholar]
- Wolf A.M., Wolf D., Rumpold H., Enrich B., Tilg H. Adiponectin induces the anti-inflammatory cytokines IL-10 and IL-1RA in human leukocytes. Biochem Biophys Res Commun. 2004;323:630–635. doi: 10.1016/j.bbrc.2004.08.145. [DOI] [PubMed] [Google Scholar]
- Yamaguchi N., Argueta J.G., Masuhiro Y., Kagishita M., Nonaka K., Saito T., Hanazawa S., Yamashita Y. Adiponectin inhibits Toll-like receptor family-induced signaling. FEBS Lett. 2005;579:6821–6826. doi: 10.1016/j.febslet.2005.11.019. [DOI] [PubMed] [Google Scholar]
- Yamauchi T., Kamon J., Waki H., Terauchi Y., Kubota N., Hara K., Mori Y., Ide T., Murakami K., Tsuboyama-Kasaoka N., et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7:941–946. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- Yamin T.T., Miller D.K. The interleukin-1 receptor-associated kinase is degraded by proteasomes following its phosphorylation. J Biol Chem. 1997;272:21540–21547. doi: 10.1074/jbc.272.34.21540. [DOI] [PubMed] [Google Scholar]
- Yoon H.I., Li Y., Man S.F.P., Tashkin D., Wise R.A., Connett J.E., Anthonisen N.A., Churg A., Wright J.L., Sin D.D. The complex relationship of serum adiponectin to COPD outcomes COPD and adiponectin. Chest. 2012;142:893–899. doi: 10.1378/chest.11-2173. [DOI] [PubMed] [Google Scholar]
- Zacharioudaki V., Androulidaki A., Arranz A., Vrentzos G., Margioris A.N., Tsatsanis C. Adiponectin promotes endotoxin tolerance in macrophages by inducing IRAK-M expression. J Immunol. 2009;182:6444–6451. doi: 10.4049/jimmunol.0803694. [DOI] [PubMed] [Google Scholar]
- Zhao L., Lee J.Y., Hwang D.H. The phosphatidylinositol 3-kinase/Akt pathway negatively regulates Nod2-mediated NF-kappaB pathway. Biochem Pharmacol. 2008;75:1515–1525. doi: 10.1016/j.bcp.2007.12.014. [DOI] [PubMed] [Google Scholar]
- Zhou H., Yu M., Fukuda K., Im J., Yao P., Cui W., Bulek K., Zepp J., Wan Y., Kim T.W., et al. IRAK-M mediates Toll-like receptor/IL-1R-induced NFκB activation and cytokine production. EMBO J. 2013;32:583–596. doi: 10.1038/emboj.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]