

Abstract
Recent years have seen a number of regulatory approvals for immune oncology or immunotherapies based on their ability to enhance antitumor immune responses. Nevertheless, the majority of patients remain refractory to these treatments; hence, new therapies that augment current immunotherapies are required. Innate immune receptors that recognize nucleic acids are potent activators of subsequent T‐cell responses and, as a result, can evoke potent antitumor immune responses. Herein, we present a novel compound N‐{3‐[(1,4′‐bipiperidin)‐1′‐yl]propyl}‐6‐[4‐(4‐methylpiperazin‐1‐yl)phenyl]picolinamide (SINCRO; STING‐mediated interferon‐inducing and cytotoxic reagent, original) as an anticancer drug that activates the cytosolic DNA‐sensing STING (stimulator of interferon genes) signaling pathway leading to the induction of type I interferon (IFN) genes. Indeed, IFN‐β gene induction by SINCRO is abolished in STING‐deficient cells. In addition to its IFN‐inducing activity, SINCRO shows STING‐independent cytotoxic activity against cancer cells. SINCRO does not evoke DNA double‐strand break or caspase‐3 cleavage. Thus, SINCRO induces cell death in a method different from conventional apoptosis‐inducing pathways. Finally, we provide evidence that giving SINCRO significantly attenuates in vivo tumor growth by both type I IFN‐dependent and independent mechanisms. Thus, SINCRO is an attractive anticancer compound with dual function in that it evokes type I IFN response to promote antitumor immunity as well as inducing tumor cell death. SINCRO may provide a new platform for the development of drugs for effective cancer therapy.
Keywords: anticancer drug, cancer immunity, cytotoxicity, STING, type I interferon
Abbreviations
- BMDC
bone marrow‐derived dendritic cells
- CDN
cyclic dinucleotide
- CMA
10‐carboxymethyl‐9‐acridanone
- DMXAA
5,6‐dimethylxanthenone‐4‐acetic acid
- DSB
double‐strand break
- GM‐CSF
granulocyte macrophage colony‐stimulating factor
- IFN
interferon
- LMP
lysosomal membrane permeabilization
- MAVS/IPS‐1
mitochondrial antiviral signaling protein/IFN‐β promoter stimulator‐1
- MEF
mouse embryonic fibroblasts
- MyD88
myeloid differentiation primary response gene 88
- NAC
N‐acetyl‐l‐cysteine
- PAMP
pathogen‐associated molecular pattern
- PI
propidium iodide
- PRR
pattern recognition receptor
- qRT‐PCR
quantitative polymerase chain reaction with reverse transcription
- RANTES
regulated on activation, normal T cell expressed and secreted
- ROS
reactive oxygen species
- SINCRO
N‐{3‐[(1,4′‐bipiperidin)‐1′‐yl]propyl}‐6‐[4‐(4‐methylpiperazin‐1‐yl)phenyl]picolinamide, STING‐mediated interferon‐inducing and cytotoxic reagent, original
- STING
stimulator of interferon genes
- TLR
Toll‐like receptor
- TNF‐α
tumor necrosis factor alpha
- TRIF
TIR‐domain‐containing adapter‐inducing IFN‐β
1. INTRODUCTION
The development of effective strategies to activate the immune system for the treatment of cancer has become a major focus of research in cancer therapeutics. Indeed, remarkable progress has been made in cancer immunotherapies as typically exemplified by cancer‐targeting monoclonal antibody treatment, immune checkpoint blockade, cancer vaccine, and chimeric antigen receptor T‐cell therapy.1, 2 However, these therapies are not always effective, leaving a number of patients in need of alternatives. In this context, several major steps may be needed for the effective control of cancer by the immune system, and these include activation of innate immune responses necessary for activation of innate antitumor responses and priming of anticancer T‐cell responses.3, 4
The innate immune system constitutes the first line of defense against invading pathogens. Recognition by one of several classes of PRR of PAMP results in immune activation.5 Among PRR, STING is a particular focus of attention in antitumor immune responses.6 STING is a PRR for CDN and also functions as an adaptor protein for multiple cytosolic receptors of double‐stranded DNA.7 The hallmark of STING pathway activation is the induction of type I IFN production,8, 9, 10 and there is good evidence that type I IFN exert antitumor activities.11 Therefore, the search for drugs that activate the STING‐type I IFN axis has been an attractive area for cancer immunotherapy.7
To date, two synthetic small compounds, DMXAA and CMA, have been developed as STING agonists inhibiting tumor development.12, 13 Both compounds induce type I IFN production in antigen‐presenting cells and giving these compounds to mice suppresses tumor growth.14, 15 However, DMXAA and CMA function only in mouse cells, not in human cells.12, 13 In addition to these small compounds, ML RR‐S2, an analog of CDN, has been synthesized as another STING agonist.14, 16 Although ML RR‐S2 is effective both in mouse and in human cells and suppresses tumor growth in mice, type I IFN induction is restricted to antigen‐presenting cells and not to cancer cells.14 As type I IFN often exert antitumor activities by directly acting on cancer cells,17 it would be interesting to search drugs that induce type I IFN in cancer cells.
In the present study, we found that a synthetic small compound, SINCRO, activates the STING pathway in mouse and human cells, inducing IFN‐β gene expression. In addition, we also show that SINCRO treatment of cancer cells triggers cell death. Interestingly, SINCRO‐triggered cytotoxicity is independent of the STING pathway and conventional apoptosis‐inducing pathway, suggesting the dual function of SINCRO. We discuss the potential significance of the discovery and characterization of SINCRO for future drug development for cancer therapy.
2. MATERIALS AND METHODS
2.1. Reagents and cells
SINCRO was kindly provided by Kowa Company, Ltd., Aichi, Japan. Mouse melanoma B16F1 cells, mouse colon carcinoma SL4 cells, human breast cancer HBC4 cells, human cervical carcinoma HeLa cells, mouse fibroblast NIH/3T3 cells, and HEK293T cells were maintained as described previously.18, 19 Mouse T‐lymphoma EL4 cells were obtained from RIKEN BioResource Research Center (Ibaraki, Japan) and maintained in DMEM (Nacalai Tesque, Kyoto, Japan) supplemented with 10% FBS (Hyclone, Logan, UT, USA). GM‐CSF‐induced BMDC, peritoneal macrophages, and MEF were prepared as described previously.18, 20
2.2. Mice
C57BL/6 mice were purchased from CLEA Japan (Tokyo, Japan). Tmem173 −/− mice (STING KO mice), Myd88 −/− mice (MyD88 KO mice), and Trif −/− mice (TRIF KO mice) described previously were maintained as C57BL/6 background.21, 22 Mavs −/− mice (MAVS KO mice) were kindly provided by S. Akira (Osaka University). Ifnar1 −/− mice (IFNAR1 KO mice) were described previously.17 All animal experiments were approved and carried out according to the guidelines of the animal research committee of the University of Tokyo.
2.3. Quantification of mRNA expression
EL4 cells (5 × 105 cells), BMDC (5 × 105 cells), peritoneal macrophages (5 × 105 cells), or other cancer cells (8 × 104 cells) were treated with SINCRO (2.5, 5, or 10 μg/mL) or DMSO for 0, 4, 6, 8, 9, or 12 hours. In some experiments, the cells were stimulated with poly dA:dT (B‐DNA) (10 μg/mL) for 9 hours as described previously.21 Total RNA was extracted from the cells using RNAiso Plus (TaKaRa, Shiga, Japan) and reverse‐transcribed into cDNA with PrimeScript RT Reagent Kit with gDNA Eraser (Perfect Real Time) (TaKaRa) according to the manufacturer's instructions. qRT‐PCR was carried out as described previously.23 Expression levels of mouse and human mRNA were normalized to those of Gapdh and GAPDH, respectively. Primer sequences are as follows: Ifnb forward 5′‐ACGCCTGGATGGTGGTCCGA‐3′; Ifnb reverse 5′‐TGCCTGCAACCACCACTCATTCT‐3′; Tnf forward 5′‐TCATACCAGGAGAAAGTCAACCTC‐3′; Tnf reverse 5′‐GTATATGGGCTCATACCAGGGTTT‐3′; Rantes forward 5′‐ACGTCAAGGAGTATTTCTACAC‐3′; Rantes reverse 5′‐GATGTATTCTTGAACCCACT‐3′; Sting forward 5′‐AATAACTGCCGCCTCATTGT‐3′; Sting reverse 5′‐TCCTCCTTTTCTTCCTGACG‐3′; Gapdh forward 5′‐CTCATGACCACAGTCCATGC‐3′; Gapdh reverse 5′‐CACATTGGGGGTAGGAACAC‐3′; IFNB forward 5′‐AGCACTGGCTGGAATGAGAC‐3′; IFNB reverse 5′‐CTATGGTCCAGGCACAGTGA‐3′; STING forward 5′‐ GAGCAGGCCAAACTCTTCTG‐3′; STING reverse 5′‐ TGCCCACAGTAACCTCTTCC‐3′; GAPDH forward 5′‐CCTCCAAGGAGTAAGACCCC‐3′; GAPDH reverse 5′‐TGTGAGGAGGGGAGATTCAG‐3′.
2.4. Enzyme‐linked immunosorbent assay
B16F1 cells (8 × 104 cells) were cultured in the presence of SINCRO (2.5, 5, or 10 μg/mL), DMSO, or incubated with B‐DNA (10 μg/mL) as described above for 24 hours. IFN‐β concentration in the culture supernatant was measured by VeriKine Mouse IFN Beta ELISA Kit (PBL Assay Science, Piscataway, NJ, USA).
2.5. Optical characterization of SINCRO
Absorbance spectrum of SINCRO (10, 25, 50, 100 or 200 μmol/L) or DMSO was measured using an ND‐1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA). Fluorescence emission spectrum of SINCRO (2 μmol/L) or DMSO with an excitation wavelength of 325 nm was obtained using a fluorescence spectrophotometer F‐7000 (HITACHI, Tokyo, Japan).
2.6. Immunoprecipitation assay
cDNA encoding mouse STING tagged with human influenza hemagglutinin molecule corresponding to amino acids 98‐106 (HA‐STING) was cloned into pCXNII vector24 and expressed in HEK293T cells. Whole cell lysate was extracted using RIPA lysis buffer20 and was subjected to immunoprecipitation with anti‐HA antibody (12CA5; Roche, Basel, Switzerland) and Dynabeads Protein G (Life Technologies, Carlsbad, CA, USA). Then, HA‐STING‐bound beads were incubated with SINCRO (100 μg/mL) in PBS for 2 hours at 4°C and boiled in 15 μL PBS. Absorbance of 325 nm light was measured using an ND‐1000 spectrophotometer.
2.7. Confocal microscopy analysis
B16F1 cells (1.5 × 106 cells) on glass‐bottom 35 mm dish (AGC TECNO GLASS, Shizuoka, Japan) were stimulated with SINCRO (10 μg/mL) for 3 hours and incubated with LysoTracker Deep Red (Molecular Probes, Eugene, OR, USA) according to the manufacturer's instructions. Confocal fluorescence images were obtained using a BZ‐X700 fluorescence microscope (KEYENCE, Osaka, Japan). For SINCRO visualization, 520 nm fluorescence emission by 325 nm excitation laser was detected.
2.8. Cell viability analysis
EL4 cells (5 × 104 cells), BMDC (7 × 104 cells), or other cells (1 × 104 cells) were incubated with SINCRO (2.5, 5, or 10 μg/mL) or DMSO for 40 hours and subsequently cultured in the presence of MTT; Dojindo, Kumamoto, Japan) (0.5 mg/mL) for 4 hours. After cells were lysed with DMSO, absorbance at 595 nm was measured. EC50 of SINCRO for cell killing was calculated using Image J (National Institutes of Health). For the inhibition of caspase activity, B16F1 cells were treated with Caspase Inhibitor Z‐VAD‐FMK (Promega, Madison, WI, USA) (20 or 40 μmol/L) or DMSO for 1 hour before SINCRO treatment. Inhibition of oxidative stress in B16F1 cells was carried out by treatment to the cells with NAC (Nacalai Tesque; 1 or 3 mmol/L) at the same time as SINCRO treatment.
2.9. Flow cytometry analysis
B16F1 cells (8 × 104 cells) were treated with SINCRO (10 μg/mL) for 0, 12, 24, or 36 hours and were stained with Annexin V and PI using an Annexin V‐FITC Apoptosis Detection Kit (Biovision, Milpitas, CA, USA). Proportion of Annexin V+ PI+ dead cells was analyzed using BD LSRII Fortessa (BD Biosciences, San Jose, CA, USA).
2.10. Immunoblot analysis
B16F1 cells (2 × 106 cells) were treated with SINCRO (10 μg/mL) or cisplatin (50 μmol/L) for 0, 6, or 12 hours. Whole cell lysates were prepared and immunoblot analysis was carried out as described previously.20 Antibodies for γH2AX (20E3), H2AX (D17A3), and cleaved caspase‐3 were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti‐LC3 antibody (8E10) and anti‐p62 polyclonal antibody were obtained from MBL (Aichi, Japan). Each protein level was quantified by analyzing its band intensity using Image J (National Institutes of Health).
2.11. In vivo tumor growth
B16F1 cells (1 × 106 cells) were inoculated s.c. into C57BL/6 or IFNAR1 KO mice. From day 9, SINCRO (10 μg) or DMSO in PBS was injected into the tumor every 2 days. Tumor volume was calculated as ab2/2 (where a represents longer axis of tumor and b represents shorter axis of tumor).
2.12. Statistical analysis
Data were analyzed by two‐tailed, unpaired Student's t test. P‐value <0.05 was considered significant.
3. RESULTS
3.1. SINCRO induces IFN‐β expression in cancer cells and immune cells
SINCRO is a small molecule compound (504.71 molecular weight) discovered for its potency in activating innate immune receptors (Figure 1A). SINCRO treatment of B16F1 mouse melanoma cells resulted in the induction of IFN‐β mRNA, a hallmark of innate immune activation, as well as TNF‐α and RANTES mRNA (Figures 1B and S1). In addition, SINCRO‐induced IFN‐β mRNA and protein expression in B16F1 cells in a dose‐dependent way albeit the induction levels were much lower compared to the same cells stimulated with B‐form DNA (B‐DNA), a well‐characterized STING agonist25 (Figure 1C,D). Of note, SINCRO‐induced IFN‐β mRNA was observed in human cancer cell lines, although interestingly not in all examined cells (Figure 1E). Perhaps expectedly, SINCRO also induced IFN‐β mRNA in immune cells such as GM‐CSF‐induced BMDC and peritoneal macrophages (Figure 1F). Collectively, these observations indicate that SINCRO has IFN‐β‐inducing activity in both malignant and normal cells.
Figure 1.
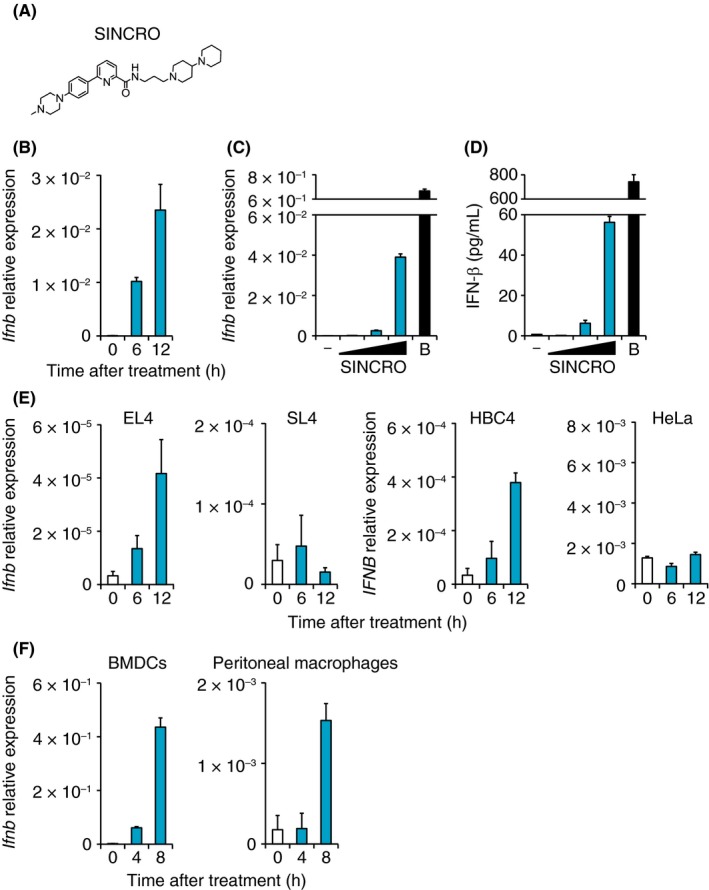
N‐{3‐[(1,4′‐bipiperidin)‐1′‐yl]propyl}‐6‐[4‐(4‐methylpiperazin‐1‐yl)phenyl]picolinamide, STING‐mediated interferon‐inducing and cytotoxic reagent, original (SINCRO) induces interferon‐β (IFN‐β) expression in cancer cells and immune cells. A, Structural formula of SINCRO is shown. B, B16F1 cells (8 × 104 cells) were treated with SINCRO (10 μg/mL) for the indicated time. IFN‐β mRNA expression was quantified by qRT‐PCR analysis. C,D, B16F1 cells (8 × 104 cells) were cultured in the presence of SINCRO (2.5, 5 or 10 μg/mL), DMSO, or B‐DNA (10 μg/mL; B). C, IFN‐β mRNA expression was determined by qRT‐PCR analysis after incubation for 9 h. D, IFN‐β protein levels in the culture supernatant were measured by ELISA after 24 h treatment. E, EL4 cells (5 × 105 cells), SL4 cells (8 × 104 cells), HBC4 cells (8 × 104 cells), or HeLa cells (8 × 104 cells) were treated with SINCRO (10 μg/mL) for the indicated period. IFN‐β mRNA levels were quantified by qRT‐PCR analysis. F, Bone marrow‐derived dendritic cells (BMDC; 5 × 105 cells) or peritoneal macrophages (5 × 105 cells) were treated with SINCRO (10 μg/mL). IFN‐β mRNA levels at the indicated time points were quantified by qRT‐PCR analysis. Data are shown as mean ± SEM
We next examined the mechanism(s) for how SINCRO regulates IFN‐β expression. As dead cell‐derived and exosomal nucleic acids have the potential to induce type I IFN,10, 26 we asked whether SINCRO induced the release of nucleic acids. To test this, we first stimulated B16F1 cells with SINCRO for 6 hours, removed the compound by washing, and then collected the medium from the cells incubated for another 6 hours. When the resulting conditioned medium was mixed with B16F1 cells, IFN‐β mRNA was not significantly induced in the B16F1 cells (Figure S2A) whereas, in contrast, the mRNA was induced in the B16F1 cells used to prepare the conditioned media (Figure S2B). Thus, we surmise that IFN‐β was not triggered by SINCRO‐mediated release of nucleic acids.
We next examined subcellular localization of SINCRO using confocal microscopy. First, the absorption and fluorescence properties of SINCRO were determined (Figure S2C,D). In B16F1 cells treated with SINCRO, fluorescence associated with SINCRO was mainly detected within the endosome/lysosome of the cells based on colocalization with a LysoTracker cell dye (Molecular Probes) (Figure S2E). Thus, these observations indicate that SINCRO is internalized into cells through the endocytosis pathway.
3.2. Involvement of the STING pathway for SINCRO‐mediated IFN‐β gene induction
To further look for how SINCRO regulates IFN‐β, we considered the activation of innate immune receptors. TLR regulate expression of IFN‐β by signaling through MyD88 or TRIF adaptor molecules. In addition, cytosolic receptors such as MAVS/IPS‐1 and STING also induce expression of IFN‐β.27, 28 Bone marrow‐derived dendritic cells generated from mice deficient in either the common TLR adaptors or the cytosolic receptors were examined for IFN‐β expression by treatment with SINCRO. As shown in Figure 2, IFN‐β expression was abrogated in the BMDC lacking STING. In contrast, IFN‐β mRNA was observed to be normal in TRIF‐ or MAVS‐deficient BMDC, while marginally decreased in MyD88‐deficient cells as compared to BMDC derived from WT mice (Figure 2). Thus, these results indicate that SINCRO induces IFN‐β expression mainly through the STING pathway.
Figure 2.
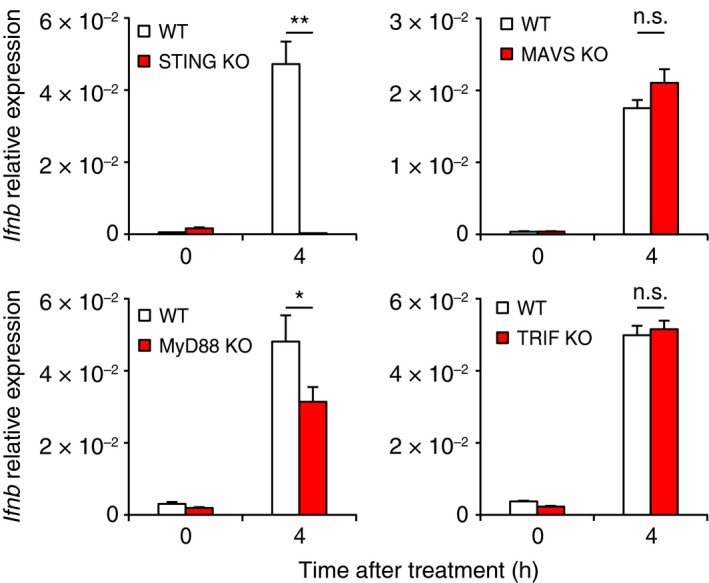
N‐{3‐[(1,4′‐bipiperidin)‐1′‐yl]propyl}‐6‐[4‐(4‐methylpiperazin‐1‐yl)phenyl]picolinamide, STING‐mediated interferon‐inducing and cytotoxic reagent, original (SINCRO) induces STING‐dependent interferon (IFN)‐β expression. Bone marrow‐derived dendritic cells (BMDC; 5 × 105 cells) prepared from WT, Sting‐deficient (STING KO), Mavs‐deficient (MAVS KO), MyD88‐deficient (MyD88 KO), or Trif‐deficient (TRIF KO) mice were stimulated with SINCRO (10 μg/mL) for the indicated times. IFN‐β mRNA expression was quantified by qRT‐PCR analysis. Data are shown as mean ± SEM. *P < 0.05. **P < 0.01. n.s., not significant. MAVS, mitochondrial antiviral signaling protein; MyD88, myeloid differentiation primary response gene 88; STING, stimulator of interferon genes; TRIF, TIR‐domain‐containing adapter‐inducing IFN‐β; WT, wild‐type
DMXAA and CMA are compounds that directly bind to STING to induce antitumor activity.12, 13 We next asked whether SINCRO also interacts with STING. Mouse HA‐STING was prepared from the whole cell lysate of HEK293T cells expressing HA‐STING and incubated with SINCRO in vitro. Here, light absorption consistent with SINCRO (325 nm) was observed to be increased when HA‐STING was incubated with SINCRO (Figure S2F, left). Expectedly, the increased absorption was observed only when SINCRO was present in the in vitro assay (Figure S2F, right). As such, although further verification will be required, SINCRO may directly interact with STING for the activation of its downstream signaling pathway(s) (see Discussion).
3.3. SINCRO induces cell death in cancer cells
In addition to immune cell activation, we also considered cytotoxic effects of SINCRO on cancer cells. SINCRO‐mediated cytotoxicity against B16F1 cells was analyzed by a MTT cell viability assay and Annexin V/PI staining. As shown in Figure 3A,B, SINCRO treatment decreased cell viability and increased Annexin V+ PI+ dead cell staining. Similar results were obtained across several mice and human cancer cell lines (Figure 3C). The cytocidal effect of SINCRO was observed to be most pronounced in the EL4 mouse lymphoma cells of those examined. In addition, SINCRO induced cell death in nontransformed cells such as BMDC, MEF, and NIH/3T3 cells; however, these cells are less sensitive to SINCRO compared to the above cancer cells (Figure S3A). Although the cancer cells respond to SINCRO at low concentrations (ie, 2.5 or 5 μg/mL) (Figure 3), the nontransformed cells remain unresponsive (Figure S3A). Indeed, EC50 of SINCRO for cell killing was almost 2‐fold lower in B16F1 cells compared to MEF (Figure S3B).
Figure 3.
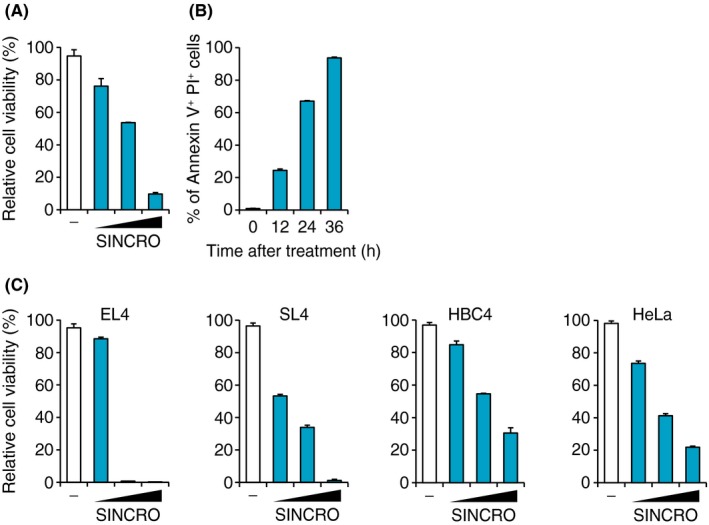
N‐{3‐[(1,4′‐bipiperidin)‐1′‐yl]propyl}‐6‐[4‐(4‐methylpiperazin‐1‐yl)phenyl]picolinamide, STING‐mediated interferon‐inducing and cytotoxic reagent, original (SINCRO) induces cell death in cancer cells. A, B16F1 cells (1 × 104 cells) were cultured in the presence of SINCRO (2.5, 5 or 10 μg/mL) or DMSO for 40 h. Cell viability was evaluated by MTT assay. B, B16F1 cells (8 × 104 cells) were stimulated with SINCRO (10 μg/mL) for the indicated time. Proportion of Annexin V+ PI + dead cells among whole cells was analyzed by flow cytometry. C, EL4 cells (5 × 104 cells), SL4 cells (1 × 104 cells), HBC4 cells (1 × 104 cells), or HeLa cells (1 × 104 cells) were treated with SINCRO (2.5, 5 or 10 μg/mL) or DMSO and cell viabilities were evaluated by MTT assay 40 h later. Data are shown as mean ± SEM
3.4. SINCRO induces cell death in a STING‐ and caspase‐independent pathway
To determine the mechanism of SINCRO‐mediated cell death, we first examined the involvement of the STING pathway using BMDC collected from STING‐deficient mice. Figure 4A shows that the viability of STING‐deficient BMDC decreased in the presence of SINCRO as observed in WT cells, suggesting that SINCRO triggers cell death distinctly from the STING pathway. In this context, it is interesting that SINCRO‐mediated cell death was observed in SL4 cells and HeLa cells, although IFN‐β mRNA induction by SINCRO was not observed in these cells (Figures 3C and 1E). Sensitivity to cell death by SL4 cells and HeLa cells was not due to their lack of functional STING as these cells expressed STING mRNA even at higher levels compared to B16F1 and HBC4 cells, respectively (Figure S4A). Moreover, SL4 and HeLa cells responded to B‐DNA, a STING agonist, for the induction of IFN‐β mRNA25 (Figure S4B). Thus, SINCRO exerts its cytocidal activity independently of the STING‐IFN pathway.
Figure 4.
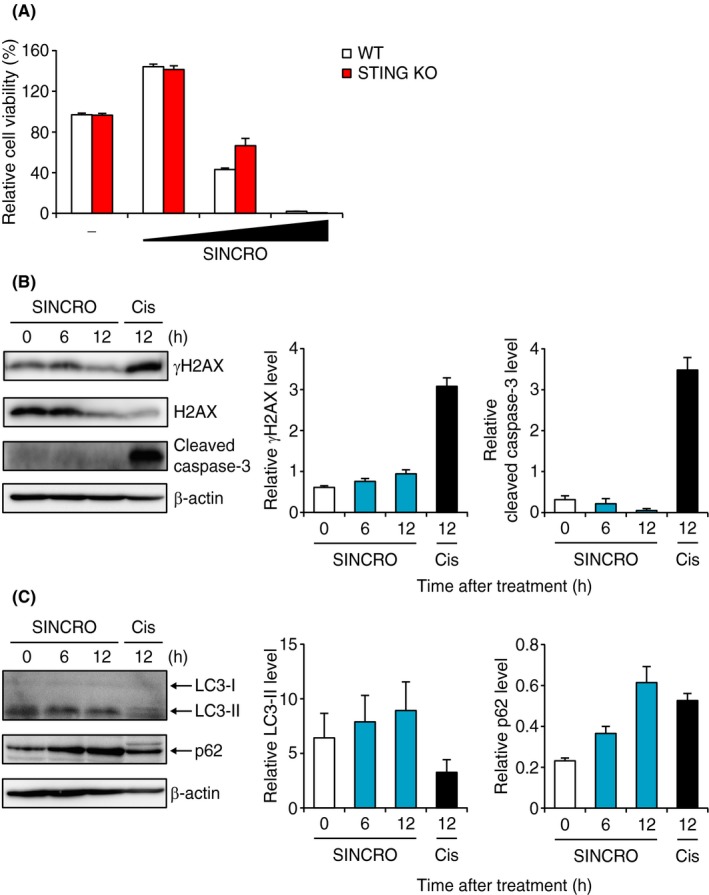
N‐{3‐[(1,4′‐bipiperidin)‐1′‐yl]propyl}‐6‐[4‐(4‐methylpiperazin‐1‐yl)phenyl]picolinamide, STING‐mediated interferon‐inducing and cytotoxic reagent, original (SINCRO) triggers STING‐independent cell death without caspase‐3 activation. A, WT or Sting‐deficient (STING KO) mice‐derived bone marrow‐derived dendritic cells (BMDC) (7 × 104 cells) were incubated with SINCRO (2.5, 5 or 10 μg/mL) or DMSO for 40 h. Cell viability was evaluated by MTT assay. B, B16F1 cells (2 × 106 cells) were stimulated with SINCRO (10 μg/mL) or cisplatin (50 μmol/L; Cis) for the indicated times. Protein expression of γH2AX, H2AX, cleaved caspase‐3 and β‐actin was measured by immunoblot analysis. B, images for each protein are shown (left panel). Relative band intensities of γH2AX normalized with H2AX (middle panel) and cleaved caspase‐3 normalized with β‐actin (right panel) are shown. C, B16F1 cells (2 × 106 cells) were treated with SINCRO (10 μg/mL) for the indicated period. Protein expression of LC3, p62, and β‐actin was detected by immunoblot analysis. Left panel shows band images for each protein. Relative band intensities of LC3‐II normalized with LC3‐I (middle panel) and p62 level normalized with β‐actin (right panel) are shown. Data are shown as mean ± SEM
Many classical anticancer agents, such as cisplatin, induce apoptosis of cancer cells by causing DNA DSB and by activating effector caspases.29 We therefore next examined whether SINCRO induces DSB and caspase activation. Although cisplatin treatment of B16F1 cells increased the protein level of γH2AX, a well‐known indicator for DSB, and cleaved caspase‐3, a marker of apoptosis, SINCRO treatment did not affect levels of either (Figure 4B). In addition, a pan‐caspase inhibitor z‐VAD‐fmk treatment did not suppress SINCRO‐triggered cell death (Figure S4C). These results indicate that SINCRO induces cell death through a mechanism independent of caspase‐mediated apoptotic pathways, including DSB‐induced apoptosis.
Notably, we found that SINCRO treatment of B16F1 cells increased p62 protein expression level, although not significantly affecting the level of an autophagosome marker LC3‐I/II (Figure 4C). p62 is known to be a substrate of autophagic protein degradation and to be upregulated in response to oxidative stress.30 In addition, ROS produced during oxidative stress induce caspase‐independent cell death.31, 32, 33, 34 Indeed, SINCRO‐induced B16F1 cell death was suppressed, albeit not completely, by treatment with NAC, a ROS scavenger (Figure S4D). Thus, SINCRO induces cancer cell death through oxidative stress and other yet unknown mechanism(s), wherein STING and caspases are not involved.
3.5. Suppression of in vivo tumor growth by SINCRO
The antitumor properties of SINCRO in vitro prompted us to examine whether SINCRO possesses antitumor activity in vivo. To test this, B16F1 cells were s.c. inoculated into C57BL/6 mice and then dosed with SINCRO intratumorally. As shown in Figure 5A, tumor volume became approximately 4‐fold smaller upon SINCRO treatment, indicating that SINCRO exerts antitumor activity in vivo. Interestingly, the decrease in tumor volume by SINCRO treatment was less when B16F1 cells were inoculated into mice deficient in one of the essential receptor components, IFNAR1, for type I IFN signaling (Figure 5B). These results suggest that both the type I IFN pathway and other pathway(s) in the recipient mice contribute to SINCRO‐mediated suppression of tumor growth.
Figure 5.
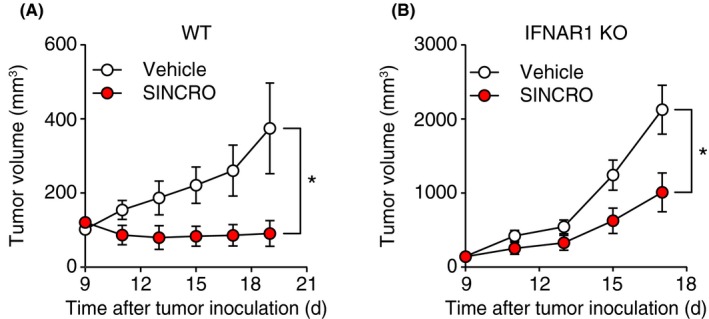
N‐{3‐[(1,4′‐bipiperidin)‐1′‐yl]propyl}‐6‐[4‐(4‐methylpiperazin‐1‐yl)phenyl]picolinamide, STING‐mediated interferon‐inducing and cytotoxic reagent, original (SINCRO) suppresses tumor growth in vivo. A, WT mice were s.c. inoculated with B16F1 cells (1 × 106 cells) and then treated with SINCRO (10 μg) or vehicle intratumorally every 2 days from day 9. Tumor volume was measured every 2 d. B, Ifnar1‐deficient (IFNAR1 KO) mice were inoculated with B16F1 cells and treated with SINCRO or vehicle as (A). Data are shown as mean ± SEM. *P < 0.05
4. DISCUSSION
With recent advances in T‐cell–mediated cancer immunotherapies, much attention has been focused on ways to harness immune responses against cancers.1, 2 In the context of the regulation of antitumor immunity by innate immune receptors, STING has been shown to play critical roles in the activation of antitumor T‐cell responses.8, 9, 10 Based on these findings, agonists for STING, such as DMXAA, CMA and ML RR‐S2, have been developed and found to be effective in several tumor models.14, 15, 16 A limitation to advancement of some of these molecules for treatment of human malignancy is the lack of cross‐reactivity to humans.12, 13 In the present study, we report that SINCRO is a novel chemical compound that activates the STING pathway, inducing expression of IFN‐β and other cytokines (Figures 1B‐F and S1). Thus, it is likely that SINCRO‐STING engagement activates both IRF and nuclear factor kappa B (NF‐κB) pathways.7 Unlike the above‐mentioned agonists, our results suggest that SINCRO can function in both mouse and human cells, although not all of the cells were responsive (Figure 1E,F).
We presented evidence that SINCRO activates the STING pathway for IFN‐β induction activity (Figure 2) and that SINCRO may directly bind to STING for its activation (Figure S2F). However, as some cell lines show differential responses between SINCRO‐ and B‐DNA‐mediated induction of IFN‐β mRNA (Figures 1E, S4A) suggests factors other than STING, presumably inactivated in these cells, are also required for SINCRO to activate this pathway.25 These observations raise the possibility that SINCRO interacts with other components upstream of the STING signaling pathway, such as cGAS (cyclic GMP‐AMP synthase), DDX41 (DEAD box protein 41), and IFI16 (IFN‐γ‐inducible protein 16) or as yet unknown molecules that lead to STING activation.28 SINCRO may therefore act on one or more of these molecules to activate the pathway and induce IFN‐β mRNA expression. Thus, further work will be required to clarify the exact mechanism for how SINCRO activates the STING pathway. It also remains to be examined whether STING activation by SINCRO promotes the expansion of tumor antigen‐specific T cells as do the other above‐mentioned STING agonists.
We also found that SINCRO exerts cytocidal activity on cancer cells. Perhaps expectedly, SINCRO showed a similar activity on untransformed cells such as BMDC particularly at high concentrations (Figure S3A). The detailed mechanism for how SINCRO induces cell death remains unclear. STING deficiency in BMDC did not significantly affect SINCRO‐induced cell death (Figure 4A). Moreover, cell death was induced by SINCRO in SL4 cells and HeLa cells (Figure 3C), whereas IFN‐β induction by SINCRO was not observed (Figure 1E). These results indicate that SINCRO does not target the STING pathway to induce cell death. Moreover, as indicated in Figures 4B and S4C, caspases were not substantially involved in SINCRO‐triggered cell death.
Instead, our results showed that the expression level of an autophagy substrate p62 was elevated by SINCRO treatment despite no downregulation of autophagy (Figure 4C). In this context, it has been reported that p62 expression levels are increased during oxidative stress,30 and oxidative stress is induced by LMP, the release of lysosomal components into the cytosol.35, 36 As SINCRO accumulates in the endosome/lysosome compartment (Figure S2E), the aberrantly or prolonged SINCRO accumulation may cause LMP to increase oxidative stress in the cells. Thus, SINCRO stimulation possibly leads to the production of ROS to inactivate caspases and induce caspase‐independent cell death as reported previously.31, 32, 33, 34 Consistent with this notion, treatment with ROS scavenger NAC increased B16F1 cell viability upon SINCRO stimulation (Figure S4D). As NAC treatment does not completely suppress SINCRO‐induced cell death, another mechanism(s) is clearly involved in the cytocidal activity of SINCRO (Figure S4D). Further study is required to elaborate on the detailed mechanism of SINCRO‐induced cell death.
Finally, in our subcutaneous tumor growth model, SINCRO treatment actually suppressed tumor development (Figure 5A). We also suggested the effectiveness of IFN‐β‐inducing activity and cytocidal property of SINCRO on in vivo tumor suppression (Figure 5B). It has been known that immune checkpoint blockade therapy, which is notable for providing durable responses through reactivation of antitumor immune responses,37 shows more potent antitumor activity when combined with a STING agonist or cytotoxic drug treatment.16, 38, 39 In this context, activation of the IFN‐β pathway and induction of cancer cell death may enhance the therapeutic efficacy of immune checkpoint modulators.
In summary, herein, we have identified a novel synthetic small compound SINCRO that mediates antitumor activity by at least two mechanisms. One is through activation of STING‐mediated innate immune responses, whereas the other is through direct cytocidal activity on cancer cells. Despite the unique functions of SINCRO, it may have potential to cause adverse side‐effects due to its cytocidal activity in untransformed cells (Figure S3A). Thus, our study may offer a new avenue of research to modify SINCRO structure so as to further potentiate its agonistic potential for STING, while enhancing cytocidal activity that is more selective to cancer cells.
DISCLOSURE
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
We specially thank S. Akira for MAVS KO mice, T. Tamura and T. Ban for IFNAR1 KO mice, and T. Tatsuma and K. Ishii for helpful comments and advice. We also thank M. Sugahara, Y. Kuroiwa and Y. Ikeuchi for technical assistance and Kowa Company Ltd, Japan for kindly providing us with SINCRO compound. This work is supported in part by Grant‐In‐Aid for Scientific Research (S) JP15H05787 from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), AMED (Japan Agency for Medical Research and Development) under grant numbers JP17ek0109107, JP18ek0109319 and JP20gm6110008, the Uehara Memorial Foundation, the Japan Rheumatism Foundation and the Princess Takamatsu Cancer Research Fund. The Department of Molecular Immunology is supported by BONAC Corporation and Kyowa Hakko Kirin Co., Ltd.
Kimura Y, Negishi H, Matsuda A, et al. Novel chemical compound SINCRO with dual function in STING‐type I interferon and tumor cell death pathways. Cancer Sci. 2018;109:2687–2696. 10.1111/cas.13726
Kimura and Negishi equally contributed to this work.
REFERENCES
- 1. Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol. 2016;13(6):394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smyth MJ, Ngiow SF, Ribas A, Teng MW. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol. 2016;13(3):143‐158. [DOI] [PubMed] [Google Scholar]
- 3. Chen DS, Mellman I. Elements of cancer immunity and the cancer‐immune set point. Nature. 2017;541(7637):321‐330. [DOI] [PubMed] [Google Scholar]
- 4. Ghiringhelli F, Apetoh L, Housseau F, Kroemer G, Zitvogel L. Links between innate and cognate tumor immunity. Curr Opin Immunol. 2007;19(2):224‐231. [DOI] [PubMed] [Google Scholar]
- 5. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783‐801. [DOI] [PubMed] [Google Scholar]
- 6. Corrales L, McWhirter SM, Dubensky TW Jr, Gajewski TF. The host STING pathway at the interface of cancer and immunity. J Clin Invest. 2016;126(7):2404‐2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15(12):760‐770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Woo SR, Fuertes MB, Corrales L, et al. STING‐dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41(5):830‐842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deng L, Liang H, Xu M, et al. STING‐dependent cytosolic DNA sensing promotes radiation‐induced type I interferon‐dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41(5):843‐852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Klarquist J, Hennies CM, Lehn MA, Reboulet RA, Feau S, Janssen EM. STING‐mediated DNA sensing promotes antitumor and autoimmune responses to dying cells. J Immunol. 2014;193(12):6124‐6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15(7):405‐414. [DOI] [PubMed] [Google Scholar]
- 12. Conlon J, Burdette DL, Sharma S, et al. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6‐dimethylxanthenone‐4‐acetic acid. J Immunol. 2013;190(10):5216‐5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cavlar T, Deimling T, Ablasser A, Hopfner KP, Hornung V. Species‐specific detection of the antiviral small‐molecule compound CMA by STING. EMBO J. 2013;32(10):1440‐1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Corrales L, Glickman LH, McWhirter SM, et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 2015;11(7):1018‐1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Storch E, Kirchner H, Schirrmacher V. Prolongation of survival of mice bearing the Eb and ESb lymphoma by treatment with interferon inducers alone or in combination with Corynebacterium parvum. Cancer Immunol Immunother. 1986;23(3):179‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fu J, Kanne DB, Leong M, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD‐1 blockade. Sci Transl Med 2015;7(283):283ra52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takaoka A, Hayakawa S, Yanai H, et al. Integration of interferon‐α/β signalling to p53 responses in tumour suppression and antiviral defence. Nature. 2003;424(6948):516‐523. [DOI] [PubMed] [Google Scholar]
- 18. Chiba S, Ikushima H, Ueki H, et al. Recognition of tumor cells by Dectin‐1 orchestrates innate immune cells for anti‐tumor responses. Elife. 2014;3:e04177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yanai H, Ban T, Wang Z, et al. HMGB proteins function as universal sentinels for nucleic‐acid‐mediated innate immune responses. Nature. 2009;462(7269):99‐103. [DOI] [PubMed] [Google Scholar]
- 20. Hangai S, Ao T, Kimura Y, et al. PGE2 induced in and released by dying cells functions as an inhibitory DAMP. Proc Natl Acad Sci USA. 2016;113(14):3844‐3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Negishi H, Matsuki K, Endo N, et al. Beneficial innate signaling interference for antibacterial responses by a Toll‐like receptor‐mediated enhancement of the MKP‐IRF3 axis. Proc Natl Acad Sci USA. 2013;110(49):19884‐19889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ouyang X, Negishi H, Takeda R, Fujita Y, Taniguchi T, Honda K. Cooperation between MyD88 and TRIF pathways in TLR synergy via IRF5 activation. Biochem Biophys Res Commun. 2007;354(4):1045‐1051. [DOI] [PubMed] [Google Scholar]
- 23. Kimura Y, Inoue A, Hangai S, et al. The innate immune receptor Dectin‐2 mediates the phagocytosis of cancer cells by Kupffer cells for the suppression of liver metastasis. Proc Natl Acad Sci USA. 2016;113(49):14097‐14102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Negishi H, Yanai H, Nakajima A, et al. Cross‐interference of RLR and TLR signaling pathways modulates antibacterial T cell responses. Nat Immunol. 2012;13(7):659‐666. [DOI] [PubMed] [Google Scholar]
- 25. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455(7213):674‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kitai Y, Kawasaki T, Sueyoshi T, et al. DNA‐containing exosomes derived from cancer cells treated with topotecan activate a STING‐dependent pathway and reinforce antitumor immunity. J Immunol. 2017;198(4):1649‐1659. [DOI] [PubMed] [Google Scholar]
- 27. Kawai T, Akira S. The role of pattern‐recognition receptors in innate immunity: update on Toll‐like receptors. Nat Immunol. 2010;11(5):373‐384. [DOI] [PubMed] [Google Scholar]
- 28. Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol. 2014;32:461‐488. [DOI] [PubMed] [Google Scholar]
- 29. Cheung‐Ong K, Giaever G, Nislow C. DNA‐damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol. 2013;20(5):648‐659. [DOI] [PubMed] [Google Scholar]
- 30. White E. Deconvoluting the context‐dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12(6):401‐410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Seiler A, Schneider M, Forster H, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15‐lipoxygenase dependent‐ and AIF‐mediated cell death. Cell Metab. 2008;8(3):237‐248. [DOI] [PubMed] [Google Scholar]
- 32. Shindo R, Kakehashi H, Okumura K, Kumagai Y, Nakano H. Critical contribution of oxidative stress to TNFα‐induced necroptosis downstream of RIPK1 activation. Biochem Biophys Res Commun. 2013;436(2):212‐216. [DOI] [PubMed] [Google Scholar]
- 33. Hanus J, Zhang H, Wang Z, Liu Q, Zhou Q, Wang S. Induction of necrotic cell death by oxidative stress in retinal pigment epithelial cells. Cell Death Dis. 2013;4:e965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Carmody RJ, Cotter TG. Oxidative stress induces caspase‐independent retinal apoptosis in vitro. Cell Death Differ. 2000;7(3):282‐291. [DOI] [PubMed] [Google Scholar]
- 35. Stern ST, Adiseshaiah PP, Crist RM. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Part Fibre Toxicol. 2012;9:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27(50):6434‐6451. [DOI] [PubMed] [Google Scholar]
- 37. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pfirschke C, Engblom C, Rickelt S, et al. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity. 2016;44(2):343‐354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Twyman‐Saint Victor C, Rech AJ, Maity A, et al. Radiation and dual checkpoint blockade activate non‐redundant immune mechanisms in cancer. Nature. 2015;520(7547):373‐377. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials