

Abstract
Hepatocyte growth factor activator inhibitor type 2 (HAI‐2), encoded by the SPINT2 gene, is a membrane‐anchored protein that inhibits proteases involved in the activation of hepatocyte growth factor (HGF), a ligand of MET receptor. Epigenetic silencing of the SPINT2 gene has been reported in a human glioblastoma cell line (U87) and glioblastoma‐derived cancer stem cells. However, the incidence of SPINT2 methylation in tumor tissues obtained from glioma patients is unknown. In this study, we analyzed the methylation status of the SPINT2 gene of eight human glioblastoma cell lines and surgically resected glioma tissues of different grades (II, III, and IV) by bisulfite sequence analysis and methylation‐specific PCR. Most glioblastoma lines (7/8) showed methylation of the SPINT2 gene with a significantly reduced level of SPINT2 mRNA compared to cultured astrocytes and normal brain tissues. However, all glioblastoma lines expressed mRNA for HGF activator (HGFAC), a target protease of HAI‐2/SPINT2. Forced expression of SPINT2 reduced MET phosphorylation of U87 glioblastoma cells both in vitro and in intracranial xenografts in nude mice. Methylation‐specific PCR analysis of the resected glioma tissues indicated notable methylation of the SPINT2 gene in 33.3% (2/6), 71.4% (10/14), and 74.3% (26/35) of grade II, III, and IV gliomas, respectively. Analysis of RNA sequencing data in a public database indicated an increased HGFAC/SPINT2 expression ratio in high‐grade compared to low‐grade gliomas (P = .01). In summary, aberrant methylation of the SPINT2 gene is frequently observed in high‐grade gliomas and might confer MET signaling in the glioma cells.
Keywords: gene methylation, glioma, HAI‐2, HGF activator, SPINT2
1. INTRODUCTION
Gliomas are the most common primary neoplasm in the central nervous system (CNS) and are classified based on the cellular lineages involved: astrocytomas, ependymomas, and oligodendrogliomas. These gliomas are further separated into low‐grade (grades I and II) and high‐grade tumors (grades III and IV) based on cell morphology, mitotic activities, and molecular markers. Glioblastoma is the most malignant form (ie, grade IV), whereas pilocytic astrocytoma is a benign localized‐type glioma (grade I).1, 2 Glioblastoma is one of the most deadly forms of cancer in humans, with a median survival of 12‐15 months and a 5‐year survival rate of <5%.2 Despite ongoing research, including comprehensive genomic analyses, there has not been an improvement in the survival of patients suffering from glioblastoma.1 The aggressive nature of glioblastoma is reflected by its extensive invasive growth. Intratumoral heterogeneity confers malignant phenotypes on glioblastoma. This heterogeneity is caused by mixed gene mutations, including amplification, and by regional differences in gene expression.3
Hepatocyte growth factor activator inhibitor (HAI) is a type I transmembrane serine protease inhibitor. At present, two HAIs are known: HAI‐1 (encoded by the SPINT1 gene) and HAI‐2 (SPINT2). Both have two extracellular Kunitz‐type serine protease inhibitor domains, a transmembrane domain, and a C‐terminal short intracytoplasmic domain, and they are expressed in most epithelial tissues and the placenta.4 However, HAI‐2/serine peptidase inhibitor, Kunitz type 2 (SPINT2) is preferentially expressed in CNS tissues, and in murine CNS, HAI‐2/SPINT2 is highly expressed in the glomerular layer of the olfactory bulb, in the cerebral cortex, and in the striatum, whereas HAI‐1/SPINT1 is hardly detectable.5 Both HAIs were initially identified as endogenous cellular inhibitors of hepatocyte growth factor activator (HGFA).6, 7 However, it is now well known that HAIs also regulate other cellular proteases responsible for the activation of the proform of hepatocyte growth factor (HGF), such as matriptase and hepsin.4, 8 The extracellular activation of proHGF is critical for the biological activity of HGF through its specific receptor tyrosine kinase (MET); MET‐induced signals are involved in invasive growth and drug resistance of various types of cancers.4, 9 Therefore, HAI‐1/SPINT1 and HAI‐2/SPINT2 have generally been implicated as suppressors of cancer progression by inhibiting HGF‐MET signal transduction,4 although contradicting results have also been reported for HAI‐2/SPINT2.10, 11, 12
Previously, we reported that SPINT2 mRNA levels were significantly reduced along with the progression of gliomas and HAI‐2/SPINT2 protein suppressed Matrigel invasion of glioblastoma cell lines.13 Subsequently, it was reported that the hypermethylation of the SPINT2 promoter region underpins reduced HAI‐2/SPINT2 expression in the U87 human glioblastoma cell line.14 Furthermore, methylation of the SPINT2 gene was also reported in glioblastoma‐derived cancer stem cells.15 Medulloblastoma, another highly malignant CNS tumor, also shows hypermethylation and silencing of the SPINT2 gene.16, 17 However, the methylation status of the SPINT2 gene in human glioma tissues and its relationship to glioma progression have not yet been clearly elucidated. In this study, we aimed to analyze methylation of the 5′‐CpG island of the SPINT2 gene in a series of human glioblastoma cell lines and surgically resected glioma tissues of different histopathological grades (grades II‐IV). We also analyzed the effect of the forced expression of HAI‐2/SPINT2 on the growth and MET phosphorylation of glioblastoma cell lines.
2. MATERIALS AND METHODS
2.1. Cell culture and human tissue samples
The human glioblastoma cell lines U251, YKG1, A172, T98G, KS1, U87, YH13, and NYGM were maintained in DMEM containing 10% FBS. U251, YKG1, T98G, A172, and KS1 were obtained from the Riken Cell Bank (Tsukuba, Japan). NYGM was established in our laboratory.18 YH13 was from the Health Science Research Resource Bank (Osaka, Japan). U87 was from ATCC through Dainippon Sumitomo Pharma (Osaka, Japan). Immortalized human astrocyte cell line (T0281) was purchased from Applied Biological Materials (Vancouver, Canada) and maintained using Prigrow IV medium and PriCoat T25 flasks (Applied Biological Materials).
The experimental protocol used to obtain clinical samples was approved by the Human Ethics Review Committees of Miyazaki University (Miyazaki, Japan) (approval number 2014‐023). Tissue samples were obtained from surgically resected low‐grade gliomas, anaplastic gliomas, and glioblastomas with written informed consent from each patient. The histological diagnosis and grading of tumors were assessed according to the WHO classification. Normal genomic DNA from the whole brain of a fetus and an adult was purchased from BioChain Institute (Newark, CA, USA) and Epigentek Group (Farmingdale, NY, USA), respectively. Total RNA from a normal brain was obtained from Takara Bio (Shiga, Japan).
2.2. Reverse transcription‐PCR and immunoblotting
Reverse transcription‐PCR was carried out as described elsewhere using the following primers:12 SPINT2, forward 5′‐CGGGGCAATAAGAACAGCT‐3′ and reverse 5′‐AGCTGCTCCTTGTCATCATCTCC‐3′; and β‐actin (ACTB), forward 5′‐ ATTGCCGACAGGATGCAGA ‐3′ and reverse 5′‐ GAGTACTTGCGCTCAGGAGGA ‐3′. Primer sequences for HGFA (HGFAC), matriptase (ST14), hepsin (HPN), TMPRSS2, TMPRSS13, human airway trypsin‐like protease (HAT: TMPRSS11D), HGF, MET, HAI‐1 (SPINT1), and GAPDH are described in Table S1.
For immunoblotting, cultured cells (approximately 70% confluency) were washed 3 times with PBS and the cellular proteins were extracted with 1% (v/v) Triton X‐100 in PBS with protease inhibitor cocktail (Merck & Co., Kenilworth, NJ, USA). For immunoblotting, each extracted protein was separated by SDS‐PAGE under nonreducing conditions, transferred onto an Immobilon membrane (Millipore, Bedford, MA, USA), and processed for HAI‐2/SPINT2 detection using mouse mAb 2A6121 as described elsewhere.12 For the detection of MET and phosphorylated MET, cells were extracted in the presence of 100 mmol/L NaF and 1 mmol/L Na3VO4 and SDS‐PAGE was carried out under reducing conditions. Anti‐human MET mouse mAb was kindly provided by Dr. D. Naka, Yokohama Research Center, Mitsubishi Pharma (Yokohama, Japan), and antiphosphorylated (Tyr1234/1235) MET rabbit mAb was purchased from Cell Signaling Technology (Boston, MA, USA).
2.3. Bisulfite modification and methylation analysis
The promoter region of the SPINT2 gene was identified with the human genome browser (http://genome.ucsc.edu), and the CpG island around the putative promoter region was predicted by Methyl Primer Express Software (Applied Biosystems Japan, Tokyo, Japan). One microgram of genomic DNA was subjected to sodium bisulfite conversion using an EpiTect Bisulfite Kit (Qiagen, Tokyo, Japan), and 1/20 of the converted DNA was used for each PCR. For bisulfite sequence analysis, the SPINT2 promoter region was amplified with PCR using DNA with bisulfite conversion as a template, using the following primers: forward 5′‐TAAGTTTAAGGGAAGGGTGGTA‐3′ and reverse 5′‐TACCTAAATCTACTCCTCACTC‐3′. The PCR products were subcloned into plasmids using a TOPO TA Cloning Kit (Life Technologies, Tokyo, Japan). The plasmid DNA from isolated colonies of transformed Escherichia coli was extracted, and DNA from multiple independent clones was sequenced to determine SPINT2 methylation status. A Web‐based tool, QUMA (http://quma.cdb.riken.jp) was used for the visualization of bisulfite sequence data.19
For methylation‐specific PCR (MSP), published primers designed to amplify either the unmethylated or methylated SPINT2 promoter region DNA after bisulfite conversion were used.20 The primer sequences are as follows: unmethylated DNA, forward 5′‐GGTTGGGTGTTTTTATATTGAAGGTTT‐3′ and reverse 5′‐TCAACACCACCAACCATTAAAATCTCA‐3′; and methylated DNA, forward 5′‐CGGGCGTTTTTATATTGAAGGTTC‐3′ and reverse 5′‐ACGCCACCAACCGTTAAAATCTCG‐3′. The annealing temperatures for unmethylated and methylated DNAs were 58°C and 54°C, respectively, for 30 seconds. Hot start PCR with a total of 32 cycle numbers was applied for the amplification. The reaction products were separated by electrophoresis on 5%‐12% gradient PAGE and visualized by ethidium bromide. The mean signal intensity and the total pixel number of each band image were measured by Photoshop software (Adobe Systems, San Jose, CA, USA) to calculate the signal level. Treatment of cultured cells with 5‐azacytidine (Sigma‐Aldrich, St. Louis, MO, USA) was carried out in accordance with the method described.21
2.4. Forced expression of HAI‐2/SPINT2 and cell proliferation assay
Generation of the HAI‐2/SPINT2 expression plasmid was described previously.13 Briefly, the full‐length coding region for HAI‐2/SPINT2 was cloned into XbaI‐SalI sites of a pCI‐neo expression vector (Promega, Madison, WI, USA). An empty vector (mock) or SPINT2 expression plasmid was linearized and transfected into U87, U251, and T98G to establish clones stably expressing HAI‐2/SPINT2. To determine growth curves of the mock‐ or SPINT2‐transfected subline, triplicate 35‐mm dishes were seeded at 1 × 104 cells/3 mL growth medium and the number of viable cells was counted at the indicated time period.
2.5. Intracranial implantation of glioblastoma cells in nude mice
All animal procedures were undertaken in accordance with institutional guidelines, and the protocol was approved by the Animal Care Committee of the University of Miyazaki. Six‐week‐old male athymic nude mice (BALB/cAJc1‐nu; mean body weight, 20 g) were obtained from CLEA Japan (Tokyo, Japan). For intracranial transplantation, 1 × 105 U87 cells with or without forced expression of SPINT2 were suspended in 10 μL PBS and stereotactically transplanted into the forebrain of mice as described previously.20, 22 The mice were carefully observed every day for 98 days to calculate the Kaplan‐Meier survival curves. Brain specimens were prepared after euthanasia from each dying mouse and from mice 98 days after the transplantation. The brain tissues were fixed with 4% formaldehyde in PBS and sectioned coronally at the point of cellular implantation, followed by embedding in paraffin.
2.6. Immunohistochemistry
Formalin‐fixed paraffin‐embedded tissue sections (4 μm) were processed for immunohistochemistry. The staining was carried out on the Leica Bond‐Max III automated immunostainer (Leica Biosystems, Tokyo, Japan) according to the manufacturer's instructions. Heat treatment for antigen retrieval lasted for 30 minutes. The primary antibody used for immunohistochemistry was antiphosphorylated MET (Tyr1235) rabbit polyclonal antibody (0.5 μg/mL) reported previously.23 To evaluate the immunoreactivity of phosphorylated MET, we graded the intensity of immunoreactivity (3, easily recognizable the cell surface immunoreactivity with a 4× objective lens; 2, recognizable cellular immunoreactivity with a 4× objective lens; 1, recognizable cellular immunoreactivity with a 200× objective lens; and 0, no immunoreactivity) and positive ratio in a hot spot (2, positive in ≥50% of tumor cells with a 10× objective lens; 1, positive in ≥10% and <50%; and 0, positive in <10%), and total score intensity grade + positive ratio grade was designated as the phosphorylated MET score.
2.7. The Cancer Genome Atlas data collection and analysis
SPINT2 RNA sequencing (RNA‐Seq) expression data, available for gliomas, were retrieved from The Cancer Genome Atlas (TCGA) Research Network (http://cancergenome.nih.gov/). Data were extracted from TCGA through the cBioPortal for Cancer Genomics website (http://www.cbioportal.org/) on November 17, 2016. In total, we selected 320 grade II gliomas, 206 grade III gliomas (anaplastic astrocytomas/oligodendrogliomas), and 160 grade IV gliomas (glioblastomas).
2.8. Statistical analysis
Data were analyzed with R (The R Foundation for Statistical Computing, Vienna, Austria) using EZR software (Saitama Medical Center, Jichi Medical University, Saitama, Japan) that has a graphical user interface for R.24 Comparison between two unpaired groups was made with the Mann‐Whitney U test or two‐way repeated‐measures ANOVA. The χ2 test was used for assessment of the relationship between variables. Survival analysis was assessed by estimating the Kaplan‐Meier survival curves, which were compared by log‐rank test. Significance was set at P < .05.
3. RESULTS
3.1. Silencing of the SPINT2 gene in glioblastoma cell lines by hypermethylation of 5′ CpG island
Initially, we analyzed the expression of HAI‐2/SPINT2 in eight glioblastoma cell lines, an immortalized astrocyte cell line, and adult brain tissue. Consistent with a previous study,13 SPINT2 mRNA was hardly detected in any of the glioblastoma cell lines except for YH13 (Figure 1A). Consequently, a notable level of HAI‐2/SPINT2 protein was detectable only in extracts from YH13 cells (Figure 1A). In contrast, astrocytes and adult brain tissue expressed substantial levels of SPINT2 mRNA (Figure 1A). We also examined the expression of genes for presumed target proteases of HAI‐2/SPINT2, all of which are known to activate proHGF.4 Of note, low but distinct levels of HGFAC mRNA that encodes HGFA were consistently expressed by the glioblastoma cells (Figure 1B). The mRNAs for other proHGF‐activating proteases were hardly detectable. We then examined the methylation status of a 5′‐CpG island (Figure 1C) of the SPINT2 gene. The results of both MSP (Figure 1D) and bisulfite sequencing (Figure 1E) were consistent with the SPINT2 expression status observed by RT‐PCR analysis, suggesting that HAI‐2/SPINT2 is epigenetically downregulated in glioblastoma cell lines by hypermethylation of the SPINT2 gene. Indeed, treatment of the glioblastoma cells with 5‐azacytidine restored the expression of SPINT2 mRNA (Figure S1).
Figure 1.
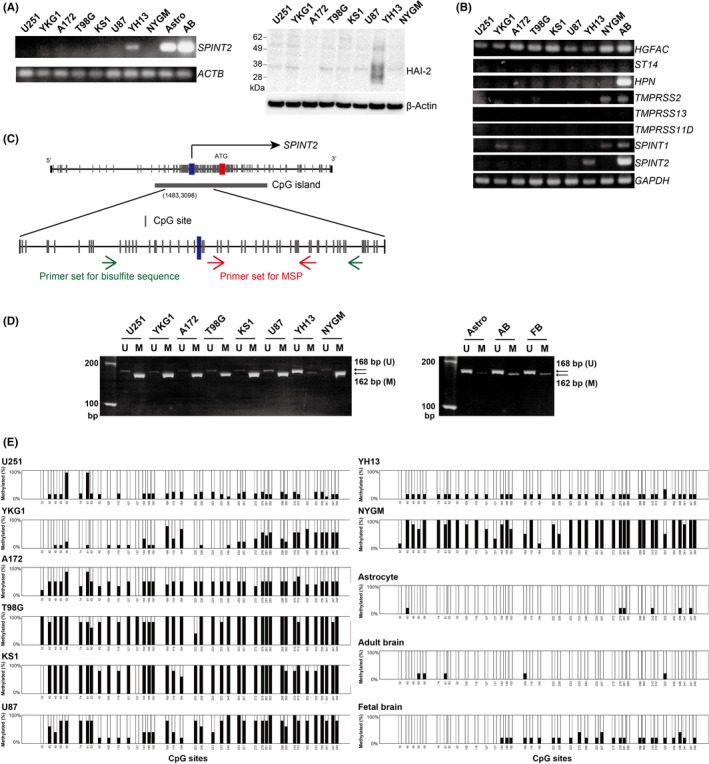
Expression and methylation status of SPINT2 in human glioblastoma cell lines. A, RT‐PCR analysis of SPINT2 mRNA (left panel) and immunoblot analysis of hepatocyte growth factor activator inhibitor type 2 (HAI‐2)/serine peptidase inhibitor, Kunitz type 2 (SPINT2) protein in glioblastoma cells. AB, adult brain; Astro, astrocytes. B, RT‐PCR analysis of mRNAs for HAI‐2‐target proteases: HGFA (HGFAC), matriptase (ST14), hepsin (HPN), TMPRSS2 (TMPRSS2), TMPRSS13 (TMPRSS13), and HAT (TMPRSS11D). RT‐PCR data for HAI‐1/SPINT1 (SPINT1) and HAI‐2/SPINT2 (SPINT2) are also shown. AB, adult brain. C, Presumed CpG island in the promoter region and exon 1 of the SPINT2 gene. Positions of the primers for methylation‐specific PCR (MSP; red arrows) and bisulfite sequence (green arrows) are also indicated. D, MSP analysis for the SPINT2 gene of glioblastoma cell lines, immortalized human astrocyte cell line (Astro), normal adult brain tissue (AB), and fetal brain tissue (FB). Positions of unmethylated (U; 168 bp) and methylated (M; 162 bp) bands are indicated by arrows. E, Results of bisulfite sequencing. Methylation status of 40 CpG dinucleotides around the transcription start site of the SPINT2 gene. Values are % of methylated clone of 10 (U251), 9 (YKG1), or 5 (others) clones analyzed
3.2. Forced expression of HAI‐2/SPINT2 suppressed proliferation of glioblastoma cells
Previously, we reported that transient overexpression of SPINT2 suppressed Matrigel invasion of U251 and YKG1 glioblastoma cell lines.13 Moreover, a recent study indicated that the U87 glioblastoma cell line stably overexpressing SPINT2 showed reduced cell growth and anchorage‐independent colony formation.15 In this study, we used U87, U251, and T98G cells to test the effect of forced SPINT2 expression on the growth in vitro. In all cell lines, stable transfection of the SPINT2 expression vector resulted in overexpression of HAI‐2/SPINT2 protein (Figure 2A) and significantly reduced the growth rate (Figure 2B). Then, we analyzed the effects of HAI‐2/SPINT2 on tumorigenicity of U87 cells in vivo by intracranial implantation. The tumorigenicity rates during the observation period (98 days) were 57.1% (8/14) and 46.7% (7/15) for mock‐transfected U87 and SPINT2 expression vector‐transfected U87 cells, respectively. The forced SPINT2 expression alleviated the mortality, as observed in the Kaplan‐Meier survival curves of the implanted mice, although the difference was not statistically significant as determined with a log‐rank test (Figure 2C).
Figure 2.
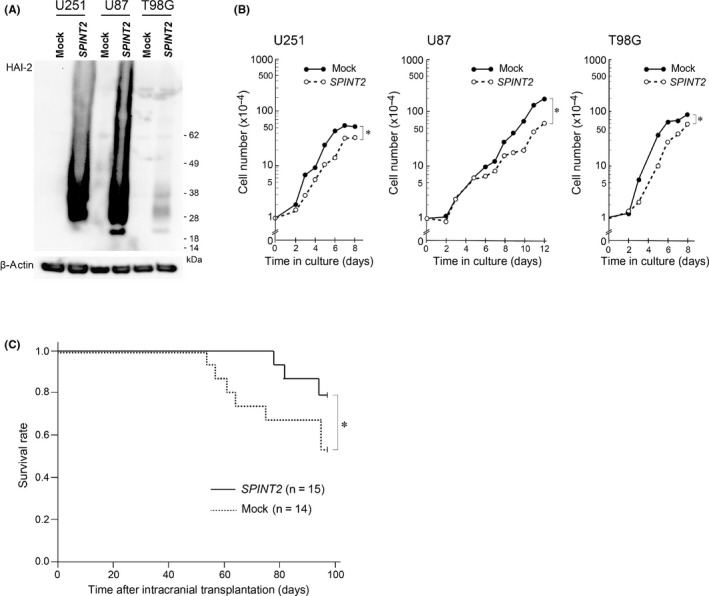
Effects of forced SPINT2 expression on proliferation of glioblastoma cells in vitro and in intracranial transplantation in nude mice. A, Immunoblot analysis for hepatocyte growth factor activator inhibitor type 2 (HAI‐2)/serine peptidase inhibitor, Kunitz type 2 (SPINT2) of cellular extracts from mock‐transfected (mock) or SPINT2‐transfected (SPINT2) glioblastoma cells (U251, U87, and T98G). B, Reduced cellular growth in vitro by forced expression of SPINT2 in glioblastoma cell lines. The growth curve is shown on a semilogarithmic graph. *P < .01 compared to mock (two‐way ANOVA). C, Kaplan‐Meier survival curves of mice after intracranial transplantation of U87 glioblastoma cell line transfected with (SPINT2) or without (mock) SPINT2 expression vector. *P = .135 (log‐rank test)
3.3. HAI‐2/SPINT2 reduced MET phosphorylation of U87 cells in vitro and in vivo
To study the role of HAI‐2/SPINT2 in HGF‐MET signaling in glioblastoma, we asked whether the forced expression of HAI‐2/SPINT2 altered MET activation in vitro and in tumors generated by intracranial implantation of U87 cells in vivo. This cell line reportedly has an HGF‐MET autocrine loop,25 and we confirmed concomitant expression of HGF and MET in this cell line (Figure 3A). The activation of MET was verified by an antibody that specifically recognized phosphorylated (Tyr1234/1235) MET. Immunoblot analysis showed that HAI‐2/SPINT2 reduced the phosphorylation of MET in cultured U87 cells in vitro (Figure 3B). For the analysis of in vivo tumors, we undertook immunohistochemical staining using antiphosphorylated MET antibody.23 The intracranial tumor tissues generated by mock‐transfected U87 (n = 7) and SPINT2 vector‐transfected U87 (n = 7) cells were processed for immunohistochemistry. As shown in Figure 3C,D, forced expression of HAI‐2/SPINT2 protein significantly reduced MET phosphorylation in vivo.
Figure 3.
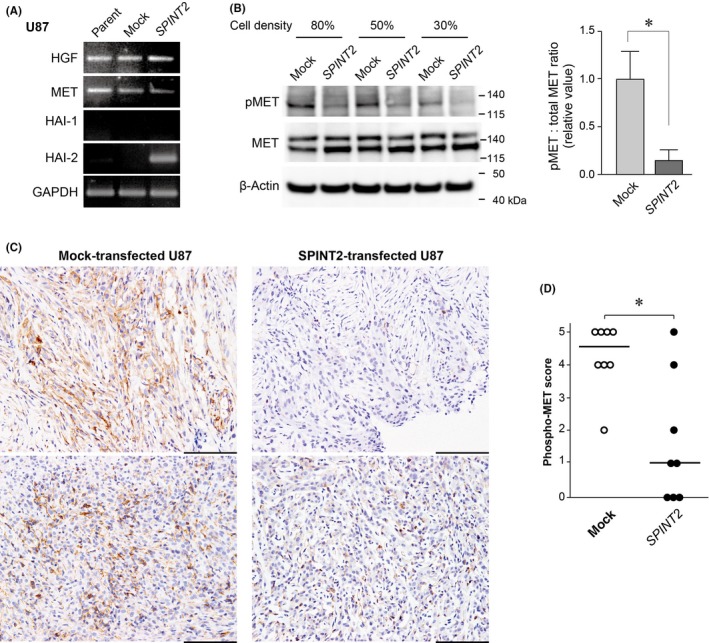
Decreased phosphorylation of MET in response to hepatocyte growth factor activator inhibitor type 2 (HAI‐2)/SPINT2 expression in U87 glioblastoma cells. A, Expression of hepatocyte growth factor (HGF), MET, HAI‐1, and HAI‐2 in the cultured U87 glioblastoma cell line and its mock‐transfected (mock) and SPINT2 expression vector‐transfected (SPINT2) sublines. B, Effect of HAI‐2/serine peptidase inhibitor, Kunitz type 2 (SPINT2) expression on MET phosphorylation in U87 cells. Immunoblot data of three independent experiments using different culture densities are shown in the left panel. pMET, phosphorylated (Tyr1234/1235) MET. The band signal of pMET relative to that of corresponding total MET at 80% culture density was calculated and indicated in the right panel. Values are mean ± SD of triplicate experiments. *P = .008 (Student's t test). C, Immunohistochemistry of phosphorylated (Tyr1235) MET of intracranial tumors of mock‐ or SPINT2‐transfected U87 cells. Representative images from two independent tumors of each subline are shown. Bar = 100 μm. D, Immunostaining score for phosphorylated MET. *P = .013 (Mann‐Whitney U test). Bar, median
3.4. SPINT2 is highly methylated in high‐grade glioma tissues
Whereas methylation of the SPINT2 gene has been reported in cultured glioblastoma cells, its incidence in human glioma tissues in vivo is unknown. Therefore, we analyzed the methylation status of SPINT2 in 56 cases of resected human glioma tissues, consisting of 6 cases of grade II, 14 cases of grade III, and 36 cases of grade IV tumors, by MSP. An easily visible methylation‐specific PCR band with a signal level at least more than 10% of the corresponding nonmethylated PCR product was judged as methylation‐positive in this analysis. Representative results of MSP are shown in Figure 4, and the whole data regarding the SPINT2 methylation status with some genetic characteristics (MGMT gene methylation status, TERT gene promoter mutation, and IDH1 gene mutation) are indicated in Table S2. Informative signals were obtained from all cases except for one glioblastoma patient (IV‐17; Table S2) who was excluded from the following analyses. Methylation of the SPINT2 gene was observed in 33.3% (2/6), 71.4% (10/14), and 74.3% (26/35) of grade II, grade III, and grade IV tumors, respectively (Table 1). Therefore, the SPINT2 gene is highly methylated in high‐grade gliomas (grades III and IV). Unmethylated specific PCR products were also amplified in most high‐grade gliomas that were methylated. This result was not unexpected as there was contamination of non‐neoplastic cells in the collected tissue samples.
Figure 4.
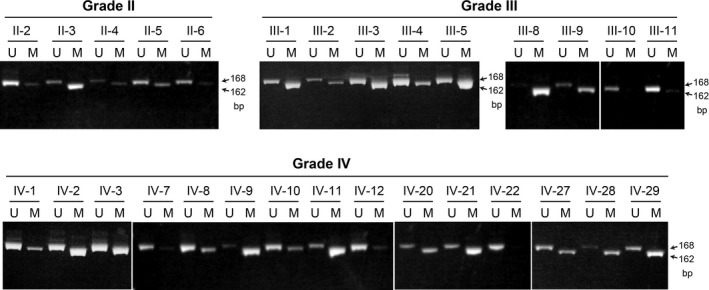
Results of methylation‐specific PCR analysis of SPINT2 in human glioma tissues. Representative data of grade II, III, and IV gliomas are shown. Positions of unmethylated (U; 168 bp) and methylated (M; 162 bp) bands are indicated by arrows
Table 1.
SPINT2 methylation status in glioma tissues
| Grade | Pathological diagnosis | SPINT2 methylation | |
|---|---|---|---|
| II | Diffuse astrocytoma | 2/5 | 2/6 (33.3%) |
| Oligodendroglioma | 0/1 | ||
| III | Anaplastic astrocytoma | 6/10 | 10/14 (71.4%) |
| Anaplastic oligodendroglioma | 4/4 | ||
| IV | Glioblastoma | 25/34 | 26/35 (74.3%) |
| Diffuse midline glioma | 1/1 | ||
3.5. Increased HGFAC/SPINT2 mRNA ratio in high‐grade gliomas
We determined the relationship between the methylation status and overall survival of patients after surgery. SPINT2 methylation status was not associated with the patients' survival in this study (Figure S2). A significant relationship was also not observed between SPINT2 methylation and MGMT methylation or TERT mutation status (χ2 test). However, most cases of MGMT‐methylated high‐grade gliomas showed SPINT2 methylation (14/16) (Table 2). It is reasonable to speculate that the biological role of protease inhibitors might depend on the level of its target protease. Among target proteases of HAI‐2/SPINT2, only HGFA was consistently expressed in cultured glioblastoma cell lines (Figure 1B). Therefore, we asked whether the expression ratio of HGFAC to SPINT2 (HGFAC:SPINT2) was increased along with glioma progression. For this purpose, we undertook a gene expression analysis using RNA‐Seq data in TCGA public database. The HGFAC RNA‐Seq tended to increase along with glioma progression, whereas SPINT2 RNA‐Seq decreased in high‐grade gliomas compared to low‐grade gliomas (Figure 5A). As a result, HGFAC:SPINT2 was increased in high‐grade gliomas (n = 366) compared to low‐grade gliomas (n = 320) at a statistically significant level (P = .039) (Figure 5B). In addition, although a higher HGFAC:SPINT2 ratio did not show significant impact on the overall survival of patients suffering from high‐grade glioma (P = .105, log‐rank test) (Figure 5C, left panel), there was a relationship between higher HGFAC expression and shorter overall survival (P = .010) (Figure 5C, right panel). In contrast, the SPINT2 RNA‐Seq level was not associated with overall survival of high‐grade glioma patients (P = .773, log‐rank test).
Table 2.
Correlation between SPINT2 methylation status and MGMT methylation status in high‐grade gliomas (grades III and IV)
| MGMT unmethylated | MGMT methylated | |
|---|---|---|
| SPINT2 unmethylated | 11 (8) | 2 (1) |
| SPINT2 methylated | 22 (15) | 14 (11) |
Numbers in parentheses represent the number of grade IV glioma cases.
P = .121 for grade III + IV gliomas; P = .089 for grade IV gliomas (χ2 test).
Figure 5.
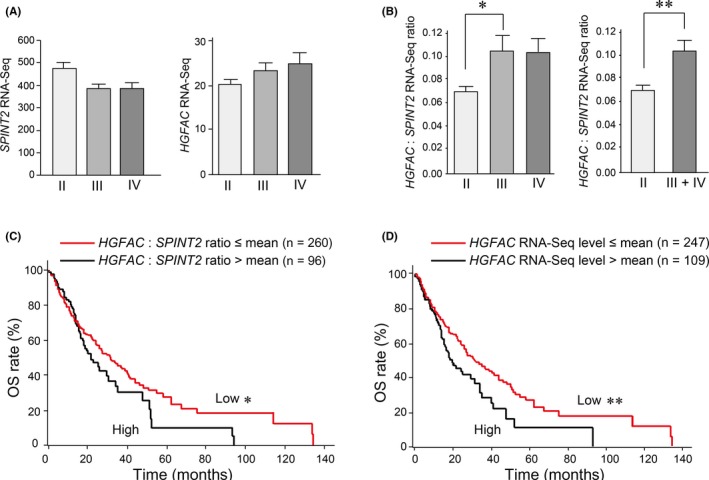
HGFAC:SPINT2 expression ratio and its relationship to the survival of high‐grade glioma patients. A, Expression of SPINT2 and HGFAC in grade II (n = 320), grade III (n = 206), and grade IV (n = 160) gliomas. The expression levels (RNA sequencing data) of each gene were retrieved from The Cancer Genome Atlas. Differences between groups were not statistically significant in either graph. B, HGFAC/SPINT2 expression ratio in low‐grade (II) and high‐grade (III + IV) gliomas. *P = .027; **P = .039 (Mann‐Whitney U test). Error bar indicates SEM. C, Kaplan‐Meier analysis of the HGFAC:SPINT2 expression ratio (left panel) or HGFAC RNA sequencing data (right panel) and outcomes from 356 high‐grade glioma cases. *P = .105; **P = .010 (log‐rank test). High expression of HGFAC (ie, >mean) was associated with decreased overall survival (OS)
4. DISCUSSION
In this study, we analyzed the methylation status of the 5′‐CpG island of the SPINT2 gene in a series of human glioblastoma cell lines and surgically resected glioma tissues. In accordance with previous reports using U87 cells,14, 15 most glioblastoma cell lines showed methylation of the SPINT2 gene, and that reversion of SPINT2 expression in glioblastoma cell lines (U87, U251, and T98G) suppressed the growth rate in vitro. Moreover, forced expression of HAI‐2/SPINT2 reduced MET phosphorylation of U87 cells both in vitro and in vivo. The U87 cell line is known to express proHGF and MET concomitantly to establish an autocrine HGF‐MET signaling loop.25 Therefore, HAI‐2/SPINT2 might suppress HGF‐MET signaling by inhibiting the proHGF‐activating protease. Several serine proteases are known to activate proHGF in the pericellular microenvironment: HGFA (HGFAC), matriptase (ST14), hepsin (HPN), TMPRSS2, TMPRSS13, and HAT (TMPRSS11D).4 Among these proteases, only HGFAC mRNA was consistently detected in most glioblastoma cell lines in vitro with the sensitive RT‐PCR method used in this study. Whereas HGFA is mainly produced by the liver and circulates in plasma as its zymogen form, extrahepatic expression of HGFAC mRNA has also been reported and HGFAC mRNA was detected in 4 of 5 high‐grade glioma tissues.26, 27 Therefore, we assume that both circulating and tumor cell‐derived HGFA might be responsible for the activation of proHGF in human glioblastoma tissues. In fact, a previous report revealed that HGFA enhances MET phosphorylation of glioblastoma cells in vivo.21 Our current analysis of TCGA database also supports this hypothesis, showing an increased HGFAC:SPINT2 RNA‐Seq ratio in high‐grade gliomas compared to low‐grade ones. Interestingly, a recent study suggested that epigenetic silencing of SPINT2 occurred in glioblastoma stem cells.15 As MET signaling is required for the maintenance of cancer stem cells,9, 28, 29 insufficiency of HAI‐2/SPINT2 might contribute to the maintenance of cancer stem cells in glioblastoma through dysregulated proteolytic activation of proHGF. Further studies to explore the proHGF‐activating mechanism in glioblastoma stem cells will be required.
Epigenetic silencing of the SPINT2 gene by hypermethylation has been reported in several malignancies, including hepatocellular carcinoma, renal cell carcinoma, medulloblastoma, melanoma, esophageal carcinoma, and gastric carcinoma.16, 21, 30, 31, 32, 33, 34 Patients suffering from esophageal cancer with SPINT2 hypermethylation had shorter survival.33 In this study, we also undertook MSP analyses of a series of resected human glioma tissues. The analyses confirmed the high incidence of SPINT2 methylation in high‐grade gliomas (grades III and IV). However, we did not observe a correlation between the methylation status and patients' overall survival in the current analysis. This might be due to the limited number of cases analyzed, and we could not undertake detailed stage‐controlled or genetic signature‐controlled subgroup analysis of the glioma cases in this study. Further analysis of the clinical relevance of the SPINT2 methylation status will require a larger cohort of glioma cases in a future study. As SPINT2 silencing might confer resistance to therapies through activation of HGF/MET signaling in glioma cells,9 analysis of the impact of SPINT2 methylation status on resistance to radiation or drugs such as temozolomide will also be required. Alternatively, the expression level of the target proteases of HAI‐2/SPINT2, such as HGFA, in glioma cells with SPINT2 methylation could be an important determinant in the patient's prognosis. In fact, our analysis of TCGA database suggested that higher HGFAC expression level predicted shorter survival of patients with high‐grade glioma.
In summary, the SPINT2 gene is highly methylated in high‐grade glioma. Forced overexpression of HAI‐2/SPINT2 protein in glioblastoma cells suppressed the growth in vitro and downregulated MET phosphorylation both in vitro and in vivo. Further studies of the roles of HAI‐2/SPINT2 protein in gliomas and identification of the protease regulated by HAI‐2/SPINT2 in glioma cells could supply novel molecular targets for glioma treatment.
CONFLICT OF INTEREST
The authors have no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
The authors would like to thank Dr. T. Shimomura for his invaluable discussions and Ms. J. Kurogi and K. Kanemaru for their skillful technical assistance. The study was funded by the Japan Society for the Promotion of Science KAKENHI (Grant No. 16H05175 to H.K. and 17K08764 to T.F.) from the Japanese Government and a Grant for Clinical Research from Miyazaki University Hospital (to T.F.).
Fukushima T, Kawaguchi M, Yamamoto K, et al. Aberrant methylation and silencing of the SPINT2 gene in high‐grade gliomas. Cancer Sci. 2018;109:2970–2979. 10.1111/cas.13732
REFERENCES
- 1. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803‐820. [DOI] [PubMed] [Google Scholar]
- 2. Cloughesy TF, Cavenee WK, Mischel PS. Glioblastoma: from molecular pathology to targeted treatment. Annu Rev Pathol. 2014;9:1‐25. [DOI] [PubMed] [Google Scholar]
- 3. Patel MA, Kim JE, Ruzevick J, Li G, Lim M. The future of glioblastoma therapy: synergism of standard of care and immunotherapy. Cancers (Basel). 2014;6:1953‐1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kataoka H, Kawaguchi M, Fukushima T, Shimomura T. Hepatocyte growth factor activator inhibitors (HAI‐1 and HAI‐2): emerging key players in epithelial integrity and cancer. Pathol Int. 2018;68:145‐158. [DOI] [PubMed] [Google Scholar]
- 5. Szabo R, Hobson JP, List K, Molinolo A, Lin CY, Bugge TH. Potent inhibition and global co‐localization implicate the transmembrane Kunitz‐type serine protease inhibitor hepatocyte growth factor activator inhibitor‐2 in the regulation of epithelial matriptase activity. J Biol Chem. 2008;283:29495‐29504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shimomura T, Denda K, Kitamura A, et al. Hepatocyte growth factor activator inhibitor, a novel Kunitz‐type serine protease inhibitor. J Biol Chem. 1997;272:6370‐6376. [DOI] [PubMed] [Google Scholar]
- 7. Kawaguchi T, Qin L, Shimomura T, et al. Purification and cloning of hepatocyte growth factor activator inhibitor type 2, a Kunitz‐type serine protease inhibitor. J Biol Chem. 1997;272:27558‐27564. [DOI] [PubMed] [Google Scholar]
- 8. Kawaguchi M, Kataoka H. Mechanisms of hepatocyte growth factor activation in cancer tissues. Cancers (Basel). 2014;6:1890‐1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Matsumoto K, Umitsu M, De Silva DM, Roy A, Bottaro DP. Hepatocyte growth factor/MET in cancer progression and biomarker discovery. Cancer Sci. 2017;108:296‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Muller‐Pillasch F, Wallrapp C, Bartels K, et al. Cloning of a new Kunitz‐type protease inhibitor with a putative transmembrane domain overexpressed in pancreatic cancer. Biochim Biophys Acta. 1998;1395:88‐95. [DOI] [PubMed] [Google Scholar]
- 11. Generali D, Fox SB, Berruti A, et al. Regulation of hepatocyte growth factor activator inhibitor 2 by hypoxia in breast cancer. Clin Cancer Res. 2007;13:550‐558. [DOI] [PubMed] [Google Scholar]
- 12. Yamamoto K, Kawaguchi M, Shimomura T, et al. Hepatocyte growth factor activator inhibitor type‐2 (HAI‐2)/SPINT2 contributes to invasive growth of oral squamous cell carcinoma cells. Oncotarget. 2018;9:11691‐11706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hamasuna R, Kataoka H, Meng JY, et al. Reduced expression of hepatocyte growth factor activator inhibitor type‐2/placental bikunin (HAI‐2/PB) in human glioblastomas: implication for anti‐invasive role of HAI‐2/PB in glioblastoma cells. Int J Cancer. 2001;93:339‐345. [DOI] [PubMed] [Google Scholar]
- 14. Schuster JM, Longo M, Nelson PS. Differential expression of bikunin (HAI‐2/PB), a proposed mediator of glioma invasion, by demethylation treatment. J Neurooncol. 2003;64:219‐225. [DOI] [PubMed] [Google Scholar]
- 15. Lee EJ, Rath P, Liu J, et al. Identification of gobal DNA methylation signatures in glioblastoma‐derived cancer stem cells. J Genet Genomics. 2015;42:355‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kongkham PN, Northcott PA, Ra YS, et al. An epigenetic genome‐wide screen identifies SPINT2 as a novel tumor suppressor gene in pediatric medulloblastoma. Cancer Res. 2008;68:9945‐9953. [DOI] [PubMed] [Google Scholar]
- 17. Onvani S, Terakawa Y, Smith C, Northcott P, Taylor M, Rutka J. Molecular genetic analysis of the hepatocyte growth factor/MET signaling pathway in pediatric medulloblastoma. Genes Chromosomes Cancer. 2012;51:675‐688. [DOI] [PubMed] [Google Scholar]
- 18. Nuki Y, Uchinokura S, Miyata S, et al. Establishment and characterization of a new human glioblastoma cell line, NYGM. Hum Cell. 2004;17:145‐150. [DOI] [PubMed] [Google Scholar]
- 19. Kumaki Y, Oda M, Okano M. A web‐based tool, QUMA (X123) was used for visualization of bisulfite sequence data. Nucleic Acids Res. 2008;36:W170‐W175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Uchinokura S, Miyata S, Fukushima T, et al. Role of hepatocyte growth factor activator (HGF activator) in invasive growth of human glioblastoma cells in vivo. Int J Cancer. 2006;118:583‐592. [DOI] [PubMed] [Google Scholar]
- 21. Moris MR, Gentle D, Abdulrahman M, et al. Tumor suppressor activity and epigenetic inactivation of hepatocyte growth factor activator inhibitor type 2/SPINT2 in papillary and clear cell renal cell carcinoma. Cancer Res. 2005;65:4598‐4606. [DOI] [PubMed] [Google Scholar]
- 22. Fukushima T, Kawaguchi M, Yorita K, et al. Antitumor effect of dehydroxymethylepoxyquinomicin, a small molecule inhibitor of nuclear factor‐kappaB, on glioblastoma. Neuro Oncol. 2012;14:19‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Inoue T, Kataoka H, Goto K, et al. Activation of c‐Met (hepatocyte growth factor receptor) in human gastric cancer tissue. Cancer Sci. 2004;95:803‐808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kanda Y. Investigation of the freely available easy‐to‐use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013;48:452‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xie Q, Bradley R, Kang L, et al. Hepatocyte growth factor (HGF) autocrine activation predicts sensitivity to MET inhibition in glioblastoma. Proc Natl Acad Sci USA. 2012;109:570‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kataoka H, Kawaguchi M. Hepatocyte growth factor activator (HGFA): pathophysiological functions in vivo. FEBS J. 2010;277:2230‐2237. [DOI] [PubMed] [Google Scholar]
- 27. Moriyama T, Kataoka H, Tsubouchi H, Koono M. Concomitant expression of hepatocyte growth factor (HGF), HGF activator and c‐met genes in human glioma cells in vitro. FEBS Lett. 1995;372:78‐82. [DOI] [PubMed] [Google Scholar]
- 28. Vermeulen L, De Sousa EMF, van der Heijden M, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12:468‐476. [DOI] [PubMed] [Google Scholar]
- 29. Boccaccio C, Comoglio PM. The MET oncogene in glioblastoma stem cells: implications as a diagnostic marker and a therapeutic target. Cancer Res. 2013;73:3193‐3199. [DOI] [PubMed] [Google Scholar]
- 30. Fukai K, Yokosuka O, Chiba T, et al. Hepatocyte growth factor activator inhibitor 2/placental bikunin (HAI‐2/PB) gene is frequently hypermethylated in human hepatocellular carcinoma. Cancer Res. 2003;63:8674‐8679. [PubMed] [Google Scholar]
- 31. Tung EK, Wong CM, Yau TO, Lee JM, Ching YP, Ng IO. HAI‐2 is epigenetically downregulated in human hepatocellular carcinoma, and its Kunitz domain type 1 is critical for anti‐invasive functions. Int J Cancer. 2009;124:1811‐1819. [DOI] [PubMed] [Google Scholar]
- 32. Hwang S, Kim HE, Min M, et al. Epigenetic silencing of SPINT2 promotes cancer cell motility via HGF‐MET pathway activation in melanoma. J Invest Dermatol. 2015;135:2283‐2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yue D, Fan Q, Chen X, et al. Epigenetic inactivation of SPINT2 is associated with tumor suppressive function in esophageal squamous cell carcinoma. Exp Cell Res. 2014;322:149‐158. [DOI] [PubMed] [Google Scholar]
- 34. Qu Y, Dang S, Hou P. Gene methylation in gastric cancer. Clin Chim Acta. 2013;424:53‐65. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials