

Abstract
Tetraploidy, a condition in which a cell has four homologous sets of chromosomes, is often seen as a natural physiological condition but is also frequently seen in pathophysiological conditions such as cancer. Tetraploidy facilitates chromosomal instability (CIN), which is an elevated level of chromosomal loss and gain that can cause production of a wide variety of aneuploid cells that carry structural and numerical aberrations of chromosomes. The resultant genomic heterogeneity supposedly expedites karyotypic evolution that confers oncogenic potential in spite of the reduced cellular fitness caused by aneuploidy. Recent studies suggest that tetraploidy might also be associated with aging; mice with mutations in an intermediate filament protein have revealed that these tetraploidy‐prone mice exhibit tissue disorders associated with aging. Cellular senescence and its accompanying senescence‐associated secretory phenotype have now emerged as critical factors that link tetraploidy and tetraploidy‐induced CIN with cancer, and possibly with aging. Here, we review recent findings about how tetraploidy is related to cancer and possibly to aging, and discuss underlying mechanisms of the relationship, as well as how we can exploit the properties of cells exhibiting tetraploidy‐induced CIN to control these pathological conditions.
Keywords: aging, aneuploidy, cancer, chromosomal instability, senescence, senescence‐associated secretory phenotype, tetraploidy
1. INTRODUCTION
Ploidy is the number of complete sets of chromosomes in the cell. Eukaryotes generally have two sets, as pairs of homologous chromosomes, and this situation is referred to as diploidy. However, there are occasions in which cells have more than two sets of chromosomes and this is called polyploidy. Both diploid and polyploid cells can also have multiples of one complete (haploid) set of chromosomes, collectively called euploidy. In contrast, aneuploidy is a deviation from a multiple of the haploid chromosome number in which some of the chromosomes are missing or present in excess. Aneuploidy is generally considered a pathological condition, which is seen in most cancer cells and also suggested to be implicated in aging.1 Polyploidy, however, is not necessarily a pathological condition and can sometimes be seen in physiological conditions. Polyploidy is commonly seen in plants, and some specific mammalian cell types become polyploid during terminal differentiation, for example, megakaryocytes in the bone marrow and trophoblasts in the placenta.2, 3
Tetraploidy is a type of polyploidy in which a single cell has four sets of chromosomes. Tetraploidy is formed from diploid cells through mechanisms such as cell fusion, endoreduplication, mitotic slippage, or cytokinetic failure, the latter two being the main routes (Figure 1).2, 3 Mitotic slippage is a phenomenon in which mitotic cells enter the next cell cycle without undergoing chromosome segregation and cell division (Figure 2A). The timing of chromosome segregation is regulated by the spindle assembly checkpoint (SAC), which halts chromosome segregation until all the chromosomes establish biorientation, that is the attachment of kinetochores on replicated chromosomes (sister chromatids) to microtubules from opposite spindle poles (Figure 2A).4 Mitotic slippage occurs when the SAC‐dependent mitotic arrest is sustained due to problems in biorientation establishment. It also happens when the SAC is defective. Cytokinetic failure occurs when the cleavage furrow formation or resolution is disturbed, resulting in binucleated cells (Figure 2B). It is known that the chromosomes left at the spindle center when chromosome segregation occurs due to defective kinetochore‐microtubule attachments, known as lagging chromosomes, can disturb cleavage furrow formation, and cause cytokinetic failure (Figure 2B).5
Figure 1.
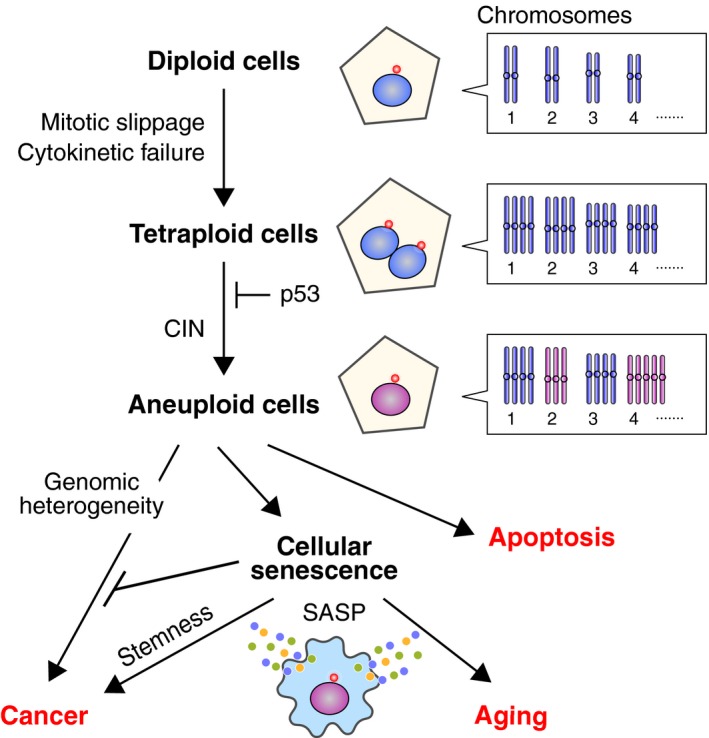
Plausible routes from tetraploidy to cancer and aging. Tetraploid cells are formed from diploid cells mainly through mitotic slippage and cytokinetic failure. Proliferation of tetraploid cells is suppressed by p53, but cells that have overcome this barrier show chromosomal instability (CIN) and develop into aneuploid cells. Genomic heterogeneity caused by CIN accelerates karyotypic evolution that confers tumorigenic potential. However, aneuploidy is generally detrimental to cellular fitness, leading to apoptosis or cellular senescence. Cellular senescence is primarily an anticancer mechanism, but senescent cells increase in number with age, and this might contribute to tissue disorders associated with aging by compromising functionality and reducing the regenerative potential. Secretion of various pro‐inflammatory proteins from senescent cells, referred to as the senescence‐associated secretory phenotype (SASP), also promotes both tissue disorders and tumorigenesis. It was recently shown that senescent cells show stem cell‐like gene expression patterns and exert aggressive growth potential following release from senescence.95
Figure 2.
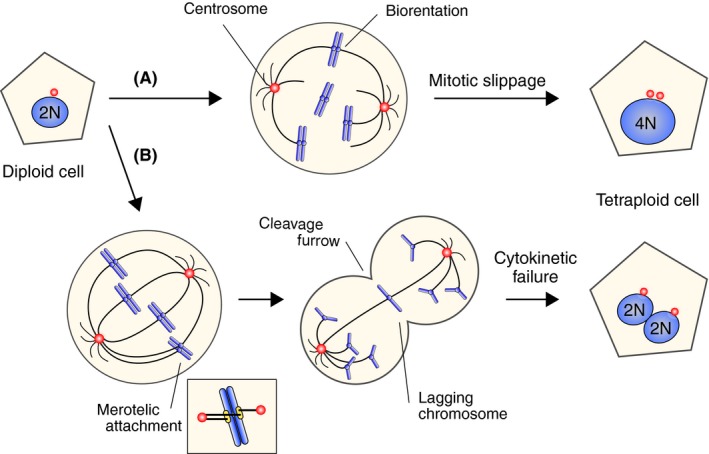
Formation of tetraploid cells. A, Mitotic slippage is a phenomenon in which mitotic cells enter into the next cell cycle without chromosome segregation and cell division, which can be caused by prolonged mitotic arrest due to defective kinetochore‐microtubule attachments. B, Cytokinetic failure occurs when cleavage furrow formation or resolution is disturbed, which can be caused by lagging chromosomes
Tetraploidy is a balanced state in terms of gene dosage, in contrast to aneuploidy, which is accompanied by unbalanced expression of genes on the affected chromosomes.6 Tetraploidy is commonly seen in a subset of hepatocytes and cardiomyocytes in physiological conditions.7, 8 Nevertheless, it is an uncommon and unfavorable event in other somatic cells; tetraploid fetuses inevitably result in spontaneous miscarriages.2, 9 It has been shown that tetraploidization facilitates oncogenic transformation and cancer progression.3 Furthermore, our unique mouse model has also suggested an unprecedented link between tetraploidy and aging.10, 11 In this review, we will introduce the emerging relationship of tetraploidy with cancer and its possible association with aging, and discuss its role in the pathogenesis of these states as well as potential therapeutic strategies for their control by exploiting the properties of tetraploid cells.
2. TETRAPLOIDY AND CANCER
It has been reported that approximately 26% of solid tumors show tetraploid or near‐tetraploid karyotypes.12 Based on bioinformatic analyses, whole‐genome doubling is estimated to occur in a significant proportion of representative solid tumors (range, 11%‐64%; overall average, ~37%).13 This event is highly associated with copy number aberration13, 14 and poor prognosis.15, 16 Under in vitro cultivation, tetraploid cells often develop into aneuploid cells.17, 18, 19, 20 Chromosomal instability (CIN), a condition in which chromosome missegregation occurs at high rates, underlies the appearance of aneuploid cells, and recent data suggest that CIN resulting from tetraploidy, but not tetraploidy itself, plays a major role in oncogenesis (Figure 1).
One important cause of CIN in tetraploid cells is the increase in the number of centrosomes.21, 22 The centrosome is the main microtubule organizing center in animal cells and comprises the poles of the bipolar spindle in mitotic cells (Figures 2A and 3A). In cases of mitotic slippage and cytokinetic failure, the resultant tetraploid cells contain twice as many centrosomes as diploid cells, thus undergoing mitosis with four centrosomes. To avoid formation of multipolar spindles and subsequent deleterious multipolar divisions,23 cells cluster the multiple centrosomes into two spindle poles, to allow bipolar division on the pseudobipolar spindle (Figure 3).18, 24, 25, 26 However, the centrosome clustering process increases the formation of erroneous kinetochore‐microtubule attachments, such as syntelic attachment in which sister kinetochores attach to microtubules from the same spindle pole, and merotelic attachment where a single kinetochore attaches to microtubules from both spindle poles (Figures 2B and 3B).4 Such erroneous attachments, if they are not corrected before anaphase onset, result in chromosome missegregation, leading to CIN. In addition to promoting CIN, a recent report suggested that centrosome amplification also promotes cellular invasion through increased centrosomal microtubule nucleation resulting in degradation of the ECM.27
Figure 3.
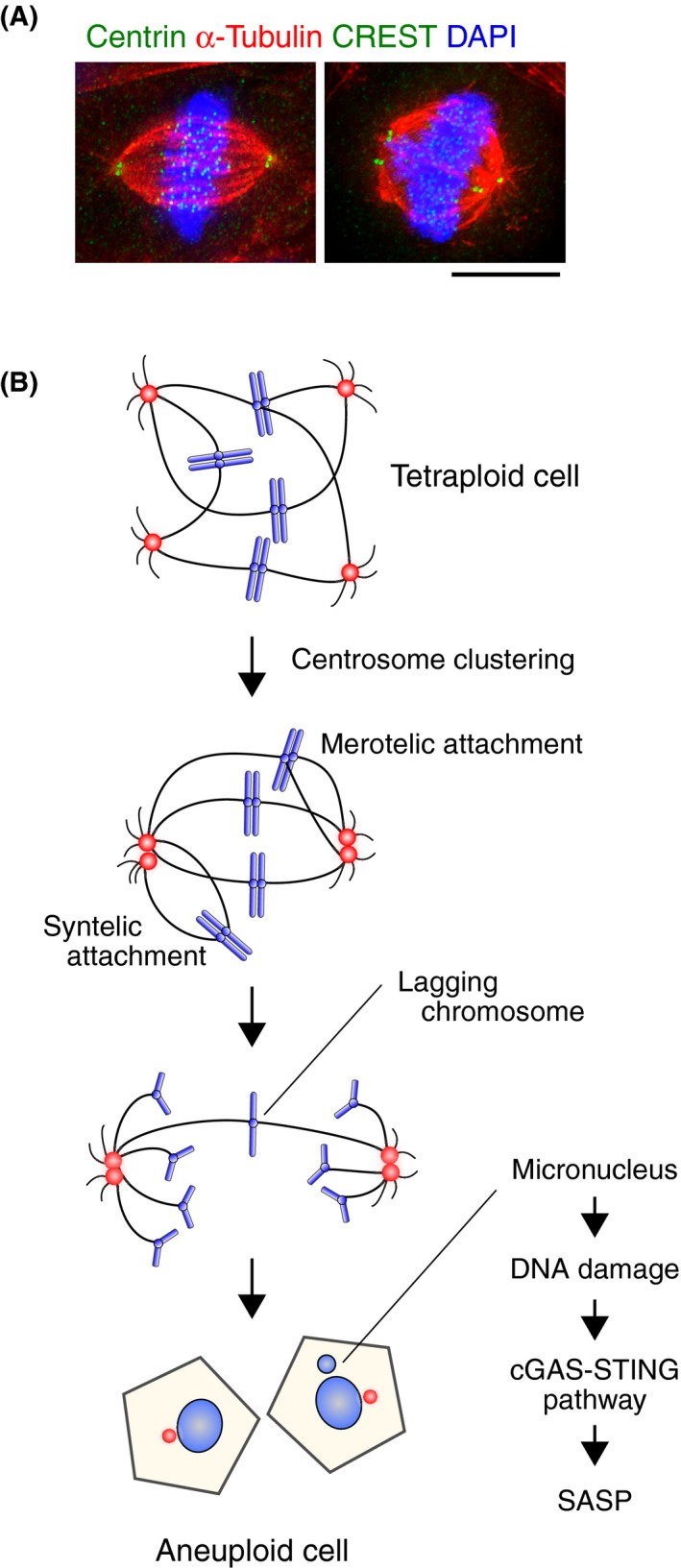
Tetraploidy‐induced aneuploidization. A, Left panel: bipolar spindle of a diploid cell (a primary fibroblast from mouse neonatal skin). Right panel: pseudobipolar spindle of a tetraploid cell containing an excessive number of chromosomes (a primary fibroblast from neonatal skin of a vimentin phosphodeficient mouse (VIMSA / SA)).11 Scale bar = 5 μm. B, Tetraploid cells containing excessive numbers of chromosomes avoid deleterious multipolar mitosis by clustering centrosomes and forming a pseudobipolar spindle. However, centromere clustering facilitates the formation of erroneous kinetochore‐microtubule attachments such as syntelic or merotelic attachments, resulting in the production of aneuploid cells. Chromosomes forming merotelic attachments can result in the formation of micronuclei through lagging chromosomes. Chromosomes in micronuclei are often damaged, which triggers activation of the cyclic GMP‐AMP synthase‐stimulator of interferon genes (cGAS‐STING) pathway that leads to senescence‐associated secretory phenotype (SASP) induction
Another reason for the prevalence of CIN in tetraploid cells is their tolerance to aneuploidy compared to diploid cells.3, 16 Aneuploidy is generally disruptive to cellular fitness due to severe imbalances in gene expression.6 The detrimental effect of gene expression imbalance is more severe in diploid cells in that the loss or gain of one chromosome in diploid cells results in 50% decrease or increase in copy number (from 2 to 1 or 3) of genes on the chromosome, whereas the change is 25% in tetraploid cells (from 4 to 3 or 5). Therefore, tetraploid cells are more resistant to aneuploidy than diploid cells by buffering the detrimental effects of chromosomal loss and gain. In a recent stochastic model recapitulating karyotypic evolution in cancer cells, the near‐triploid state commonly seen in cancer cells was shown to be achieved from both diploid and tetraploid precursors, but tetraploid cells reached the state more efficiently than diploid cells because the survival cost to achieve the state is smaller in tetraploid cells compared to diploid cells.28
In general, tetraploidization leads to p53 stabilization, resulting in cell cycle arrest,29, 30, 31, 32, 33, 34 cellular senescence,35 or apoptosis,36 thus suppressing the proliferation of tetraploid cells (Figure 1). Consequently, aneuploidization after tetraploidization is observed mainly in p53‐inactivated cells.17, 18 Under some culture conditions, p53‐positive tetraploid cells can divide and then develop into aneuploid ones,20, 37, 38 partly because enhancement of growth factor signaling can bypass cell cycle arrest.37 Tetraploid cells containing an excessive number of centrosomes are gradually lost from cell populations,18, 38 but tetraploid cells can exhibit CIN even after multiple passages when the centrosome number is supposedly normal.16 This suggests that CIN might be driven by the presence of defects other than excessive centrosome numbers. Tetraploidization enhances drug resistance not only in cancerous cells,20, 39 but also in non‐transformed cells.20 Also, tetraploidization confers the ability of non‐transformed cells to form tumors in grafted mice.17, 19, 40 These above effects are probably mediated by tetraploidy‐induced aneuploidization,17, 19, 20, 39 suggesting that tetraploidy in oncogenic cells results in karyotypic complexity in the cell population.41, 42
In summary, tetraploidy is likely a transient intermediate in the development of CIN. Chromosomal instability is related to drug resistance, metastasis, and poor prognosis, which may be caused by intratumor heterogeneity (Figure 1);43, 44 CIN leads to continuous chromosome missegregation and formation of a variety of aneuploid cells. This increases the chance of gaining additional oncogenic potential, even though aneuploid cells often suffer from various stresses that reduce their cellular fitness.45 Chromosome missegregation promotes not only changes in chromosome number, but also structural changes, such as deletion, amplification, and translocation. One mechanism is through the formation of micronuclei, which are compartmentalized chromosome masses isolated from the main nucleus (Figure 3B). Common origins for micronuclei are lagging chromosomes resulting from merotelic attachment. Chromosomes in micronuclei are subjected to DNA damage due to a defective nuclear membrane of the micronuclei, and fragmented DNAs are lost or erroneously ligated in the next cell cycle (Figure 3B). This is considered to be one of the mechanisms generating chromothripsis, which is characterized by extensive genomic rearrangements confined to one or few chromosomal regions.46 Chromosomal instability also promotes DNA damage and genomic instability by several mechanisms, such as cleavage furrow ingression in the presence of lagging chromosomes and telomere deprotection during prolonged mitotic arrest.47, 48
The idea that genomic heterogeneity caused by CIN facilitates oncogenesis and cancer progression fits well with the cancer clonal evolution model first proposed by Peter Nowell,49 a theory similar to Darwinian natural selection.50 Sequencing of whole cancer genomes has revealed that chromosomal regions enriched in oncogenes tend to be amplified, whereas those enriched in tumor suppressor genes tend to be lost.51 The aforementioned stochastic model recapitulated karyotypic evolution of cancer cells under conditions in which the chromosome missegregation rate was optimal for balancing genomic heterogeneity and clonal survival.28
3. POTENTIAL ASSOCIATION OF TETRAPLOIDY WITH AGING
In addition to the relationship between tetraploidy and cancer, an unexpected link between tetraploidy and aging has been recently proposed in a mouse model.10, 11 The tetraploidy‐prone mice had not existed before 2013,10 partly because whole‐genome doubling leads to spontaneous miscarriages.2, 9 To overcome this difficulty, we used an alternative strategy in which unscheduled tetraploidy was induced in a tissue‐specific manner.10 Before explaining our strategy, we will briefly introduce intermediate filaments (IFs) and their constituent proteins (IF proteins).
Together with microtubules and actin filaments, IFs form the cytoskeletal network in the cytoplasm of virtually all vertebrate cells.52, 53 Unlike the other two major cytoskeletal network proteins, IF proteins are expressed in a cell‐type and tissue‐specific manner.52, 53 Intermediate filament reorganization is regulated largely by IF protein phosphorylation.54, 56, 57 A series of studies using cultured cells have shown that compromising IF protein phosphorylation during mitosis induces cytokinetic failure by the retention of an IF bridge‐like structure (referred to as an IF bridge) connecting the two daughter cells, resulting in binucleation/tetraploidization (Figure 4A,B).56, 57, 58, 59
Figure 4.
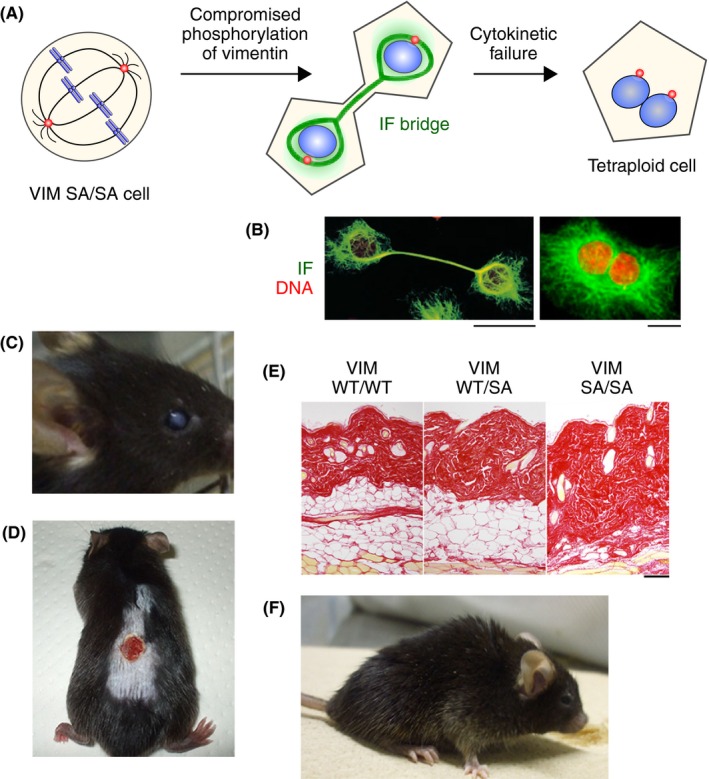
Tissue‐specific phenotypes associated with aging in vimentin phosphodeficient mice (VIMSA / SA). A, Schematic diagram depicting tetraploidization by a phosphodefective vimentin mutant. B, Cultured cells expressing phosphodefective intermediate filament (IF) mutants. Left, cells with an IF bridge.57 Right, binucleated cell.59 C, Lens cataract in a VIMSA / SA mouse.10, 56 D, Impaired wound healing in a VIMSA / SA mouse. 11 E, Subcutaneous fat loss in a VIMSA / SA mouse (14 months).11 F, Lordokyphosis in a VIMSA / SA mouse. Scale bar = 10 μm (B) or 200 μm (E)
As the IF protein vimentin is expressed in all mesenchymal cells, with the eye lens being the tissue containing by far the highest levels,52, 53, 60 we produced genetically modified mice in which vimentin mutated at mitosis‐specific phosphorylation sites is expressed instead of WT vimentin.10 In these phosphodeficient mice, unscheduled binucleation (tetraploidization) is induced in cells in which vimentin is highly expressed, such as lens epithelial cells10 and subcutaneous fibroblastic/adipose cells.11 The level of p53 is elevated in these tetraploid cells,11 suggesting that the p53 pathway likely functions in these mice. However, in the phosphodeficient mice, the tetraploid cells continue to divide and develop aneuploidy,10, 11 a similar phenomenon observed in cancer (see section 2). Such aneuploid cells accumulate DNA damage and, in turn, become senescent.10, 11 Our mice, in which cytokinetic failure induces unscheduled tetraploidy, show several tissue‐specific phenotypes such as lens cataracts,10 impaired wound healing, subcutaneous fat loss,11 and lordokyphosis (Figure 4C‐F; M. Inagaki, unpublished observation). These phenotypes were also observed in progeroid mice.61, 62
Cellular senescence is characterized by irreversible cell cycle arrest induced by various cellular stresses, such as oncogenic activation or DNA damage.63 Aneuploidy promotes cellular senescence supposedly through DNA damage (Figure 1).64, 65 Cellular senescence works primarily as an anticancer mechanism by preventing the proliferation of damaged, precancerous cells and promoting their removal by the immune system (Figure 1).66 However, senescent cells accumulate with age due to their increased production and reduced clearance and might contribute to tissue disorders by compromising functionality and reducing the regenerative potential (Figure 1). In addition, it has been revealed that secretion of various pro‐inflammatory proteins by senescent cells, referred to as the senescence‐associated secretory phenotype (SASP), plays a crucial role in tissue disorders (Figure 1).67
In support of our observations in the tetraploidy‐prone mice, the population of tetraploid cells is elevated at several organs during normal aging.68, 69 For example, some hepatocytes increase their ploidy, becoming tetraploid and octoploid, during the aging process.70, 71 In the liver, some polyploid cells are likely to develop into aneuploid ones.12 The phenotypes of our mice resemble those of aneuploidy‐prone mice such as BubR1‐hypomorphic61 or Bub3/Rae1‐haploinsufficient72 mice. Therefore, tetraploidization contributes to progeroid phenotype probably through aneuploidization. It should be noted, however, that it has not been evaluated in detail whether tetraploid cells increase with age in other organs in general.
The link between tetraploidy and cellular senescence is also suggested in zebrafish, an attractive animal model for various human diseases including cancer73 and age‐related disorders.74 It has been reported that tetraploid cardiomyocytes increase in prevalence with age and that cardiomyocyte regeneration is lost with age in mammals.75, 76 A recent report verified the relationship between tetraploidy and cardiomyocyte regeneration in zebrafish by inhibiting epithelial cell transforming sequence 2 oncogene (ECT2), a major player in cytokinesis regulated by Rho GTPases77 and in DNA damage responses.78 Knockdown of ECT2 induced senescence markers in a spontaneously immortalized non‐transformed mammary epithelial cell line with a KRAS mutation.79 In adult zebrafish, transient inhibition of Ect2 increased tetraploidy in the heart and inhibited regeneration after apical ventricular resection.80 These findings suggest that increased tetraploidy of cardiomyocytes and resultant decreased regeneration is a phenotype related to cellular senescence. Another example is condensin, altered expression of which has been linked to cancer and cellular senescence. Condensin is a highly conserved complex composed of two subunits of the structural maintenance of chromosome (SMC) family of proteins plus three non‐SMC subunits. The expression of condensin is often increased in human cancer cells, potentially conferring resistance to DNA replication stress and DNA damage.81 During aging, the expression of Alu and SINE retrotransposon RNAs is increased in human stem cells, which prevents recruitment of condensin to the chromatin structure and causes persistent DNA damage checkpoint activation and senescence.82 In zebrafish, mutation of the non‐SMC subunit of condensin increased tetraploidy and the rate of apoptosis in retinal progenitor cells, but not in postmitotic retinal cells at 3 days postfertilization.83 The results are consistent with the functional role of condensin in the regulation of chromosomal organization of mitotic cells.81 The results are also consistent with previous reports showing that tetraploidization can induce apoptosis.36 These results suggest that apoptosis in the retina of developing zebrafish is one of the phenotypes caused by unscheduled tetraploidy that may be related to cellular senescence.
4. TETRAPLOIDY‐INDUCED TISSUE DISORDERS AND CANCER
Tetraploidy is found in various human tissues with varying frequencies that can be greater than those of chromosome missegregation.84 Therefore, tetraploidy through whole‐genome duplication is supposed to be an efficient and important route to aneuploidization by promoting the CIN that contributes to the pathogenesis of cancer and tissue disorders associated with aging. Cellular senescence is a key phenomenon that links cancer and aging. Although cellular senescence is an anticancer mechanism, increased senescent cells in aged tissues can provide a milieu for tumor progression by secreting SASP factors,85 which trigger inflammation that can promote tumorigenesis (Figure 1).86
Recent studies have revealed that activation of the cyclic GMP‐AMP synthase‐stimulator of interferon genes (cGAS‐STING) cytosolic DNA‐sensing pathway is a mechanism for SASP induction in cells showing CIN. The cGAS‐STING pathway is involved in the innate immune response, which is triggered by dsDNA, derived from pathogens, in the cytoplasm.87 In cells showing CIN, an origin of fragmented DNA that can activate the cGAS‐STING pathway is the DNA in micronuclei that is exposed to the cytoplasm due to nuclear membrane collapse (Figure 3B),88, 89 which occurs in more than half of the micronuclei.90 Cytoplasmic chromatin fragments released from nuclei through the disintegrated nuclear membrane after DNA damage in senescent cells can also activate the pathway.91 The cGAS‐STING pathway promotes the SASP program and triggers inflammation in senescence and cancer. Another report has shown that CIN also facilitates cancer metastasis through SASP induction by fragmented DNA in micronuclei.92 These findings suggest an important role for CIN in cancer and tissue disorders associated with aging through SASP induction.
Many chemotherapeutic drugs act by inducing severe DNA damage in tumor cells and thus triggering apoptosis or cellular senescence. Considering that cancer cells showing CIN suffer from reduced cellular fitness, promoting CIN over a tolerable level for cancer cells is now proposed as a promising anticancer strategy to selectively eradicate cancer cells.93 For tetraploid cancer cells carrying extra centrosomes, perturbing centrosome clustering can cause multipolar spindle formation followed by catastrophic mitosis.94 However, promoting CIN is a double‐edged sword and can accelerate production of a malignant phenotype by increasing genomic heterogeneity and inducing SASP. Intriguingly, it was recently reported that senescent cells show a stem cell‐like gene expression pattern, which exerts aggressive growth potential following release from senescence (Figure 1).95 In this regard, purging of senescent cells, termed senolysis, or inhibition of SASP induction, have now emerged as novel strategies for cancer treatment.85 These approaches also show promise for suppressing aging‐related disorders66 but, again, we have to bear in mind that senescence and SASP also have beneficial functions, such as wound healing and tissue regeneration. Furthermore, inflammation caused by SASP is implicated in the efficacy of anticancer therapy itself.96 Another caveat is that the response to tetraploidy and tetraploidy‐induced aneuploidy is highly context‐dependent. Whole‐genome doubling (tetraploidization) widely occurred during evolution.97, 98, 99 In hepatocytes and neurons, it has been speculated that tetraploidy and tetraploidy‐induced aneuploidy provide beneficial phenotypic diversity.100, 101 At least in yeast, polyploidization (tetraploidization) can induce beneficial mutations leading to environmental adaptation.102 A recent report suggested that tetraploidy in hepatocytes is rather tumor‐suppressive,103 indicating a complex relationship between tetraploidy and cancer.
5. CONCLUSION
There has been a long‐standing debate as to whether CIN is a cause of cancer. From the current viewpoint, we can understand that previous conflicting results on the effect of CIN on tumorigenesis represented two sides of a coin: reduced cellular fitness of aneuploid cells and genomic heterogeneity, both resulting from CIN. Induction of SASP has now been recognized as an additional critical role of CIN in tumorigenesis. Whether CIN promotes or suppresses tumorigenesis could be determined by cellular context. As tetraploidy both promotes CIN and ameliorates decline in cellular fitness, it is suggested to be a faster path to tumorigenesis. The relationship of tetraploidy with aging is less well understood, as is the link between aneuploidy and aging. For further understanding of these, a comprehensive characterization of tetraploidy in physiological aging and elucidation of its underlying cause is necessary. At the same time, studies using animal models will facilitate the unraveling of the relationship of tetraploidy with aging and tissue disorders associated with aging. In particular, analysis of different IF protein mutant mice would be informative, especially because differential expression patterns of respective IF proteins provide an opportunity to dissect the effects of tetraploidy on disorders associated with aging in different tissues.56 The reason why tetraploidy is both beneficial and detrimental for cellular fitness, depending on cellular context, is also an important unanswered question. For the treatment of cancer, tetraploidy and CIN are promising targets for selective therapy, and clarifying the complicated relationship between CIN, cancer, and aging is crucial for developing optimal anticancer strategies as well as for mitigating aging‐related disorders.
CONFLICT OF INTEREST
K.T., H.G., Y.N., A.M., and M.I. report receiving research grants from Takeda Science Foundation. A.M. or M.I. also report receiving research grants from JCR Pharmaceuticals Co. Ltd. and the Naito Foundation, respectively. K.K. has no conflict of interest to declare.
ACKNOWLEDGMENTS
This work was supported in part by Grants‐in‐Aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Japan Society for the Promotion of Science KAKENHI Grant Nos. 15H04368, 16H01296, 16K14604, 18H02434, and 18H04896 [K.T.]; 15K08324 and 18K06927 [H.G.]; 16K08547 [Y. N.]; 17K08269 [K.K.]; 16H06461 [A.M.]; and 15H02398 [M.I]), by Takeda Science Foundation (K.T., H.G., Y.N., A.M., and M.I.), and by the Naito Foundation (M.I.). We deeply apologize to all colleagues whose work we were unable to cite due to space limitations.
Tanaka K, Goto H, Nishimura Y, Kasahara K, Mizoguchi A, Inagaki M. Tetraploidy in cancer and its possible link to aging. Cancer Sci. 2018;109:2632–2640. 10.1111/cas.13717
Contributor Information
Kozo Tanaka, Email: kozo.tanaka.d2@tohoku.ac.jp.
Masaki Inagaki, Email: minagaki@doc.medic.mie-u.ac.jp.
REFERENCES
- 1. Naylor RM, van Deursen JM. Aneuploidy in cancer and aging. Annu Rev Genet. 2016;50:45‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Storchova Z, Kuffer C. The consequences of tetraploidy and aneuploidy. J Cell Sci. 2008;121:3859‐3866. [DOI] [PubMed] [Google Scholar]
- 3. Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev. 2007;17:157‐162. [DOI] [PubMed] [Google Scholar]
- 4. Tanaka K. Regulatory mechanisms of kinetochore‐microtubule interaction in mitosis. Cell Mol Life Sci. 2013;70:559‐579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shi Q, King RW. Chromosome nondisjunction yields tetraploid rather than aneuploid cells in human cell lines. Nature. 2005;437:1038‐1042. [DOI] [PubMed] [Google Scholar]
- 6. Santaguida S, Amon A. Short‐ and long‐term effects of chromosome mis‐segregation and aneuploidy. Nat Rev Mol Cell Biol. 2015;16:473‐485. [DOI] [PubMed] [Google Scholar]
- 7. Gentric G, Desdouets C. Polyploidization in liver tissue. Am J Pathol. 2014;184:322‐331. [DOI] [PubMed] [Google Scholar]
- 8. Mollova M, Bersell K, Walsh S, et al. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc Natl Acad Sci U S A. 2013;110:1446‐1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dewhurst S, Swanton C. Tetraploidy and CIN: a dangerous combination. Cell Cycle. 2015;14:3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Matsuyama M, Tanaka H, Inoko A, et al. Defect of mitotic vimentin phosphorylation causes microophthalmia and cataract via aneuploidy and senescence in lens epithelial cells. J Biol Chem. 2013;288:35626‐35635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tanaka H, Goto H, Inoko A, et al. Cytokinetic failure‐induced tetraploidy develops into aneuploidy, triggering skin aging in phosphovimentin‐deficient mice. J Biol Chem. 2015;290:12984‐12998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zasadil LM, Britigan EM, Weaver BA. 2n or not 2n: aneuploidy, polyploidy and chromosomal instability in primary and tumor cells. Semin Cell Dev Biol. 2013;24:370‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zack TI, Schumacher SE, Carter SL, et al. Pan‐cancer patterns of somatic copy number alteration. Nat Genet. 2013;45:1134‐1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lundberg G, Jin Y, Sehic D, Ora I, Versteeg R, Gisselsson D. Intratumour diversity of chromosome copy numbers in neuroblastoma mediated by on‐going chromosome loss from a polyploid state. PLoS One. 2013;8:e59268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Carter SL, Cibulskis K, Helman E, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dewhurst SM, McGranahan N, Burrell RA, et al. Tolerance of whole‐genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 2014;4:175‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53‐null cells. Nature. 2005;437:1043‐1047. [DOI] [PubMed] [Google Scholar]
- 18. Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lv L, Zhang T, Yi Q, et al. Tetraploid cells from cytokinesis failure induce aneuploidy and spontaneous transformation of mouse ovarian surface epithelial cells. Cell Cycle. 2012;11:2864‐2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kuznetsova AY, Seget K, Moeller GK, et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle. 2015;14:2810‐2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pihan GA, Purohit A, Wallace J, et al. Centrosome defects and genetic instability in malignant tumors. Cancer Res. 1998;58:3974‐3985. [PubMed] [Google Scholar]
- 22. Chan JY. A clinical overview of centrosome amplification in human cancers. Int J Biol Sci. 2011;7:1122‐1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pihan GA. Centrosome dysfunction contributes to chromosome instability, chromoanagenesis, and genome reprograming in cancer. Front Oncol. 2013;3:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Quintyne NJ, Reing JE, Hoffelder DR, Gollin SM, Saunders WS. Spindle multipolarity is prevented by centrosomal clustering. Science. 2005;307:127‐129. [DOI] [PubMed] [Google Scholar]
- 25. Kwon M, Godinho SA, Chandhok NS, et al. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008;22:2189‐2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Silkworth WT, Nardi IK, Scholl LM, Cimini D. Multipolar spindle pole coalescence is a major source of kinetochore mis‐attachment and chromosome mis‐segregation in cancer cells. PLoS One. 2009;4:e6564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Godinho SA, Picone R, Burute M, et al. Oncogene‐like induction of cellular invasion from centrosome amplification. Nature. 2014;510:167‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Laughney AM, Elizalde S, Genovese G, Bakhoum SF. Dynamics of tumor heterogeneity derived from clonal karyotypic evolution. Cell Rep. 2015;12:809‐820. [DOI] [PubMed] [Google Scholar]
- 29. Lanni JS, Jacks T. Characterization of the p53‐dependent postmitotic checkpoint following spindle disruption. Mol Cell Biol. 1998;18:1055‐1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Andreassen PR, Lohez OD, Lacroix FB, Margolis RL. Tetraploid state induces p53‐dependent arrest of nontransformed mammalian cells in G1. Mol Biol Cell. 2001;12:1315‐1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Margolis RL, Lohez OD, Andreassen PR. G1 tetraploidy checkpoint and the suppression of tumorigenesis. J Cell Biochem. 2003;88:673‐683. [DOI] [PubMed] [Google Scholar]
- 32. Margolis RL. Tetraploidy and tumor development. Cancer Cell. 2005;8:353‐354. [DOI] [PubMed] [Google Scholar]
- 33. Ganem NJ, Pellman D. Limiting the proliferation of polyploid cells. Cell. 2007;131:437‐440. [DOI] [PubMed] [Google Scholar]
- 34. Kuffer C, Kuznetsova AY, Storchova Z. Abnormal mitosis triggers p53‐dependent cell cycle arrest in human tetraploid cells. Chromosoma. 2013;122:305‐318. [DOI] [PubMed] [Google Scholar]
- 35. De Santis Puzzonia M, Gonzalez L, Ascenzi S, Cundari E, Degrassi F. Tetraploid cells produced by absence of substrate adhesion during cytokinesis are limited in their proliferation and enter senescence after DNA replication. Cell Cycle. 2016;15:274‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Castedo M, Coquelle A, Vivet S, et al. Apoptosis regulation in tetraploid cancer cells. EMBO J. 2006;25:2584‐2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ganem NJ, Cornils H, Chiu SY, et al. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell. 2014;158:833‐848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Potapova TA, Seidel CW, Box AC, Rancati G, Li R. Transcriptome analysis of tetraploid cells identifies cyclin D2 as a facilitator of adaptation to genome doubling in the presence of p53. Mol Biol Cell. 2016;27:3065‐3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee AJ, Endesfelder D, Rowan AJ, et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011;71:1858‐1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Davoli T, de Lange T. Telomere‐driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell. 2012;21:765‐776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Davoli T, de Lange T. The causes and consequences of polyploidy in normal development and cancer. Annu Rev Cell Dev Biol. 2011;27:585‐610. [DOI] [PubMed] [Google Scholar]
- 42. Coward J, Harding A. Size does matter: why polyploid tumor cells are critical drug targets in the war on cancer. Front Oncol. 2014;4:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McGranahan N, Burrell RA, Endesfelder D, Novelli MR, Swanton C. Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Rep. 2012;13:528‐538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168:613‐628. [DOI] [PubMed] [Google Scholar]
- 45. Zhu J, Tsai HJ, Gordon MR, Li R. Cellular stress associated with aneuploidy. Dev Cell. 2018;44:420‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stephens PJ, Greenman CD, Fu B, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Janssen A, van der Burg M, Szuhai K, Kops GJ, Medema RH. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science. 2011;333:1895‐1898. [DOI] [PubMed] [Google Scholar]
- 48. Hayashi MT, Cesare AJ, Fitzpatrick JA, Lazzerini‐Denchi E, Karlseder J. A telomere‐dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat Struct Mol Biol. 2012;19:387‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23‐28. [DOI] [PubMed] [Google Scholar]
- 50. Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Davoli T, Xu AW, Mengwasser KE, et al. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell. 2013;155:948‐962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fuchs E, Weber K. Intermediate filaments: structure, dynamics, function, and disease. Annu Rev Biochem. 1994;63:345‐382. [DOI] [PubMed] [Google Scholar]
- 53. Eriksson JE, Dechat T, Grin B, et al. Introducing intermediate filaments: from discovery to disease. J Clin Invest. 2009;119:1763‐1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Inagaki M, Nishi Y, Nishizawa K, Matsuyama M, Sato C. Site‐specific phosphorylation induces disassembly of vimentin filaments in vitro. Nature. 1987;328:649‐652. [DOI] [PubMed] [Google Scholar]
- 55. Snider NT, Omary MB. Post‐translational modifications of intermediate filament proteins: mechanisms and functions. Nat Rev Mol Cell Biol. 2014;15:163‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Goto H, Inagaki M. New insights into roles of intermediate filament phosphorylation and progeria pathogenesis. IUBMB Life. 2014;66:195‐200. [DOI] [PubMed] [Google Scholar]
- 57. Yasui Y, Goto H, Matsui S, et al. Protein kinases required for segregation of vimentin filaments in mitotic process. Oncogene. 2001;20:2868‐2876. [DOI] [PubMed] [Google Scholar]
- 58. Yasui Y, Amano M, Nagata K, et al. Roles of Rho‐associated kinase in cytokinesis; mutations in Rho‐associated kinase phosphorylation sites impair cytokinetic segregation of glial filaments. J Cell Biol. 1998;143:1249‐1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yamaguchi T, Goto H, Yokoyama T, et al. Phosphorylation by Cdk1 induces Plk1‐mediated vimentin phosphorylation during mitosis. J Cell Biol. 2005;171:431‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Song S, Landsbury A, Dahm R, Liu Y, Zhang Q, Quinlan RA. Functions of the intermediate filament cytoskeleton in the eye lens. J Clin Invest. 2009;119:1837‐1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Baker DJ, Jeganathan KB, Cameron JD, et al. BubR1 insufficiency causes early onset of aging‐associated phenotypes and infertility in mice. Nat Genet. 2004;36:744‐749. [DOI] [PubMed] [Google Scholar]
- 62. Kubben N, Misteli T. Shared molecular and cellular mechanisms of premature ageing and ageing‐associated diseases. Nat Rev Mol Cell Biol. 2017;18:595‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lopez‐Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194‐1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bartkova J, Rezaei N, Liontos M, et al. Oncogene‐induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633‐637. [DOI] [PubMed] [Google Scholar]
- 65. Di Micco R, Fumagalli M, Cicalese A, et al. Oncogene‐induced senescence is a DNA damage response triggered by DNA hyper‐replication. Nature. 2006;444:638‐642. [DOI] [PubMed] [Google Scholar]
- 66. Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age‐related disease: from mechanisms to therapy. Nat Med. 2015;21:1424‐1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547‐556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yang D, McCrann DJ, Nguyen H, et al. Increased polyploidy in aortic vascular smooth muscle cells during aging is marked by cellular senescence. Aging Cell. 2007;6:257‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hanselmann RG, Oberringer M. Polyploidization: a Janus‐faced mechanism. Med Hypotheses. 2001;56:58‐64. [DOI] [PubMed] [Google Scholar]
- 70. Gentric G, Desdouets C, Celton‐Morizur S. Hepatocytes polyploidization and cell cycle control in liver physiopathology. Int J Hepatol. 2012;2012:282430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wang MJ, Chen F, Lau JTY, Hu YP. Hepatocyte polyploidization and its association with pathophysiological processes. Cell Death Dis. 2017;8:e2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Baker DJ, Jeganathan KB, Malureanu L, Perez‐Terzic C, Terzic A, van Deursen JM. Early aging‐associated phenotypes in Bub3/Rae1 haploinsufficient mice. J Cell Biol. 2006;172:529‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ratner N, Miller SJ. A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer. 2015;15:290‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Carneiro MC, de Castro IP, Ferreira MG. Telomeres in aging and disease: lessons from zebrafish. Dis Model Mech. 2016;9:737‐748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kajstura J, Gurusamy N, Ogorek B, et al. Myocyte turnover in the aging human heart. Circ Res. 2010;107:1374‐1386. [DOI] [PubMed] [Google Scholar]
- 76. Richardson GD, Laval S, Owens WA. Cardiomyocyte regeneration in the mdx mouse model of nonischemic cardiomyopathy. Stem Cells Dev. 2015;24:1672‐1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cook DR, Rossman KL, Der CJ. Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene. 2014;33:4021‐4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. He D, Xiang J, Li B, Liu H. The dynamic behavior of Ect2 in response to DNA damage. Sci Rep. 2016;6:24504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Cairney CJ, Godwin LS, Bilsland AE, et al. A ‘synthetic‐sickness’ screen for senescence re‐engagement targets in mutant cancer backgrounds. PLoS Genet. 2017;13:e1006942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gonzalez‐Rosa JM, Sharpe M, Field D, et al. Myocardial polyploidization creates a barrier to heart regeneration in zebrafish. Dev Cell. 2018;44:433‐436 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kagami Y, Yoshida K. The functional role for condensin in the regulation of chromosomal organization during the cell cycle. Cell Mol Life Sci. 2016;73:4591‐4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Ohsawa R, Seol JH, Tyler JK. At the intersection of non‐coding transcription, DNA repair, chromatin structure, and cellular senescence. Front Genet. 2013;4:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Seipold S, Priller FC, Goldsmith P, Harris WA, Baier H, Abdelilah‐Seyfried S. Non‐SMC condensin I complex proteins control chromosome segregation and survival of proliferating cells in the zebrafish neural retina. BMC Dev Biol. 2009;9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Biesterfeld S, Gerres K, Fischer‐Wein G, Bocking A. Polyploidy in non‐neoplastic tissues. J Clin Pathol. 1994;47:38‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Watanabe S, Kawamoto S, Ohtani N, Hara E. Impact of senescence‐associated secretory phenotype and its potential as a therapeutic target for senescence‐associated diseases. Cancer Sci. 2017;108:563‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Trinchieri G. Cancer and inflammation: an old intuition with rapidly evolving new concepts. Annu Rev Immunol. 2012;30:677‐706. [DOI] [PubMed] [Google Scholar]
- 87. Margolis SR, Wilson SC, Vance RE. Evolutionary origins of cGAS‐STING signaling. Trends Immunol. 2017;38:733‐743. [DOI] [PubMed] [Google Scholar]
- 88. Mackenzie KJ, Carroll P, Martin CA, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548:461‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548:466‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell. 2013;154:47‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Dou Z, Ghosh K, Vizioli MG, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550:402‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bakhoum SF, Ngo B, Laughney AM, et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. 2018;553:467‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Tanaka K, Hirota T. Chromosomal instability: a common feature and a therapeutic target of cancer. Biochim Biophys Acta. 2016;1866:64‐75. [DOI] [PubMed] [Google Scholar]
- 94. Watts CA, Richards FM, Bender A, et al. Design, synthesis, and biological evaluation of an allosteric inhibitor of HSET that targets cancer cells with supernumerary centrosomes. Chem Biol. 2013;20:1399‐1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Milanovic M, Fan DNY, Belenki D, et al. Senescence‐associated reprogramming promotes cancer stemness. Nature. 2018;553:96‐100. [DOI] [PubMed] [Google Scholar]
- 96. Haschka M, Karbon G, Fava LL, Villunger A. Perturbing mitosis for anti‐cancer therapy: is cell death the only answer? EMBO Rep 2018;19:e45440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Otto SP, Whitton J. Polyploid incidence and evolution. Annu Rev Genet. 2000;34:401‐437. [DOI] [PubMed] [Google Scholar]
- 98. Semon M, Wolfe KH. Consequences of genome duplication. Curr Opin Genet Dev. 2007;17:505‐512. [DOI] [PubMed] [Google Scholar]
- 99. Hufton AL, Panopoulou G. Polyploidy and genome restructuring: a variety of outcomes. Curr Opin Genet Dev. 2009;19:600‐606. [DOI] [PubMed] [Google Scholar]
- 100. Duncan AW, Hanlon Newell AE, Bi W, et al. Aneuploidy as a mechanism for stress‐induced liver adaptation. J Clin Invest. 2012;122:3307‐3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Muotri AR, Gage FH. Generation of neuronal variability and complexity. Nature. 2006;441:1087‐1093. [DOI] [PubMed] [Google Scholar]
- 102. Selmecki AM, Maruvka YE, Richmond PA, et al. Polyploidy can drive rapid adaptation in yeast. Nature. 2015;519:349‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zhang S, Zhou K, Luo X, et al. The polyploid state plays a tumor‐suppressive role in the liver. Dev Cell. 2018;44:447‐459 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]