

Abstract
Reprogramming technology has enabled the fate conversion of terminally differentiated somatic cells into pluripotent stem cells or into another differentiated state. A dynamic reorganization of epigenetic regulation takes place during cellular reprogramming. Given that reprogramming does not require changes in the underlying genome, the technology can be used to actively modify epigenetic regulation. Although reprogramming has been investigated mostly at the cellular level in vitro, studies have reported that somatic cells are reprogrammable in multicellular organisms in vivo. In vivo reprogramming provides a potential strategy for regenerative medicine. Notably, recent studies using in vivo reprogramming technology to alter epigenetic regulation at organismal levels have revealed unappreciated epigenetic mechanisms in various biological phenomena, including cancer development, tissue regeneration, aging, and rejuvenation in mammals. Moreover, in vivo reprogramming technology can be applied to abrogate epigenetic aberrations associated with aging and cancer, which raises the possibility that the technology could provide a potential strategy to control the fate of detrimental cells such as senescent cells and cancer cells in vivo. Here, we review recent progress and future perspectives of in vivo reprogramming.
Keywords: aging, cancer, cellular senescence, epigenetics, in vivo reprogramming, reprogramming technology
1. INTRODUCTION
Mammalian organisms consist of various types of differentiated cells, all of which originate from a single fertilized egg. After fertilization, a single zygote divides and differentiates into a range of cell types. For a long time, it had been believed that once a cell differentiates into a final cell state, it permanently loses the potential to diversify its functions and stably maintains its identity. Similarly, it is generally accepted that age‐related changes including cellular senescence are irreversible processes. The classic concept of unidirectional differentiation was presented by Waddington and is known as the epigenetic landscape model. Consistent with the stable maintenance of cellular identity, cell fate conversion, such as dedifferentiation and transdifferentiation, are restricted to certain conditions, such as tissue regeneration and cancer development. However, we now know that this dogma is not always true and that cell fate can be altered by artificial reprogramming technology.
In past decades, advances in reprogramming technology have provided various strategies to alter nuclear information and somatic cell identity. The nucleus of a somatic cell can be converted into a totipotent state when it is transferred into an enucleated oocyte.1, 2 Similarly, when a somatic cell is fused with an embryonic stem cell, a subset of the embryonic program is initiated in its nucleus.3, 4 Finally, the landmark study by Takahashi and Yamanaka revealed that the induction of a certain set of transcription factors (Oct3/4, Sox2, Klf4 and c‐Myc; OSKM) highly expressed in embryonic stem cells can confer the gene expression profiles and the epigenetic landscape of pluripotent stem cells on differentiated cells, resulting in induced pluripotent stem cells (iPSCs).5, 6 In addition, starting with the first report, which showed that the ectopic expression of MyoD, a myoblast determination gene, is sufficient to reprogram mouse fibroblasts into myoblasts in vitro, 7 a number of experiments have shown that the forced expression of lineage‐specific transcription factors successfully induces direct reprogramming, which converts one differentiated cell type into another without dedifferentiating to the pluripotent state.8, 9 Based on these studies, the Waddington model of cell differentiation has been revised, and it is now widely accepted that cell fate can be converted by artificial manipulations. Although most cellular reprogramming strategies have been developed in vitro, in vivo reprogramming strategies, which describe the fate conversion of differentiated somatic cells to another cell type in a mammalian body, have been developing rapidly in recent years. In vivo reprogramming technology can serve as a potential novel strategy for regenerative medicine. Moreover, recent studies suggest that the technology could be a powerful tool for unveiling biological phenomena related to epigenetic regulation in tissue regeneration, aging, rejuvenation, and cancer development in multicellular organisms.
2. EPIGENETIC REGULATION ENSURES STABLE MAINTENANCE OF CELL FATE
Epigenetics is defined as the meitotically and mitotically inherited regulation of gene expressions that are not accompanied by alteration of the DNA sequence.10 The stable maintenance of somatic cell identity is governed by various epigenetic modifications, such as DNA methylation, histone modifications, and chromatin structure.11 During development, epigenetic regulation plays a central role in the establishment and maintenance of stable gene expression signatures. Once cells differentiate and lineage commitment occurs, DNA methylation and histone modification profiles are faithfully maintained as the cells divide. It is known that multiple enzymatic systems, which include DNA methyltransferases, histone acetylases, deacetylases, methylases, and demethylases, as well as protein complexes implicated in chromatin remodeling, maintain epigenetic regulation.11 Overall, cell fate conversion requires a dynamic reorganization of the epigenetic regulation that overcomes this maintenance machinery.
Because somatic cell fate is stably maintained, it is assumed that epigenetic regulation in somatic cells is stably maintained as well. Given that epigenetic regulation interconnects environmental stimuli with the genome, epigenetic alterations could occur when cells in tissues are exposed to continuous and/or strong external signals. Epigenetic alterations include alterations in DNA methylation patterns, post‐translational modification of histones and chromatin remodeling. Interestingly, previous studies have reported that epigenetic alterations occur in various pathophysiological conditions, such as aging and cancer.
3. HISTONE ALTERATIONS DURING AGING
Aging can be defined as the progressive decline in the ability of a cell or organism to resist stress, damage, and disease.12 This deterioration is considered to be the primary risk factor for major human pathologies, including cancer, diabetes, cardiovascular disorders, and neurodegenerative diseases. Aging research has experienced an unprecedented advance over recent years.13 A number of molecular and cellular hallmarks of aging have been proposed, including genomic instability, telomere attrition, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication, most of which are shared among different organisms. Importantly, recent studies suggest that these age‐related phenotypes are often linked with epigenetic regulation. During aging, in addition to the gradual accumulation of genetic mutations, a variety of epigenetic alterations have been observed in multiple organisms. The general loss of histones, which is accompanied by local and global chromatin remodeling, altered histone modifications, and concomitant transcriptional changes, has been reported in various aging models from yeast to humans.14 Although accumulating evidence suggests that epigenetic changes are an important aspect of the progression of aging, it is still unclear exactly how these changes arise and to what extent they affect lifespan and age‐related phenotypes.
The overexpression of histones ameliorates many age‐related changes and extends the lifespan of yeast;15 however, whether histone expression has a similar contribution to aging in human tissues is unclear. Methylation at certain amino acids of a histone tail is associated with transcriptional activity. For example, transcription start sites of transcriptionally active genes are marked with H3K4 methylation, whereas H3K9 and H2K27 methylation are enriched at transcriptionally repressed loci. Histone methylations have been linked to lifespan regulation in multiple organisms, but with different effects.14, 16 Knockdown of H3K4me3 methyltransferase subunits including SET‐2 extends lifespan, whereas knockdown of the demethylase shortens the lifespan of Caenorhabditis elegans.16 Similarly, knockdown of lsd‐1, an H3K4 mono‐ and di‐demethylase, improves longevity.17 The upregulation of utx‐1 removes H3K27me3, and the concomitant decrease in H3K27me3 takes place with aging in C. elegans.18 Conversely, the depletion of utx‐1 increases H3K27me3 levels and extends lifespan.19 Together, these results support a model that claims a gain of activating histone marks and loss of repressive histone marks, which are both representative epigenetic alterations during aging, play a role in lifespan (Figure 1).
Figure 1.
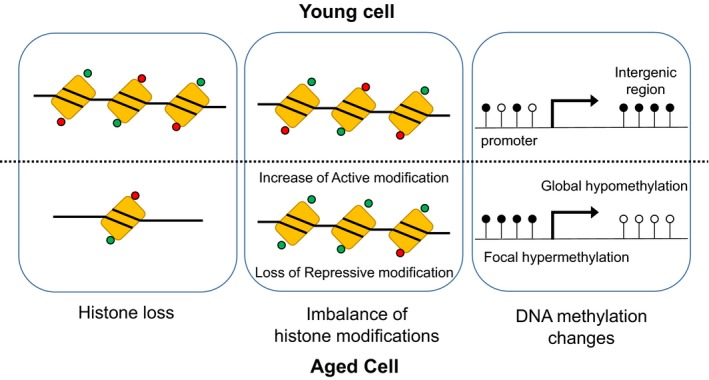
Epigenetic alterations during aging. A general loss of histones along with altered histone modifications and alterations in DNA methylation patterns are detectable in aged cells. Red circle, repressive modification; green circle, active modification; black circle, methylated DNA; white circle, unmethylated DNA
4. HISTONE MODIFICATIONS IN SENESCENT CELLS
Two hallmarks of aging are an increase in the number of senescent cells and decline in tissue regeneration ability due to the loss of stem cell proliferation.13 Cellular senescence can be defined as a stable arrest of the cell cycle coupled to stereotyped phenotypic changes.20, 21 This phenomenon was originally described by Hayflick in human fibroblasts that are serially passaged in culture.22 Today, we know that such replicative senescence is caused by telomere attrition,23 but there are other aging‐associated stimuli that trigger senescence. Indeed, oxidative stress, genotoxic stress, cytokines, and chromatin perturbation can induce senescence.24 Cell cycle arrest is also apparent in oncogene‐induced senescence, where cells stop proliferation by unrestricted activation of an oncogene, underscoring the tumor‐suppressive role of senescence.25
Consistent with the functional involvement of altered histone modifications in worm lifespan, similar alterations in histone modifications are detectable in human cultured cells from aged individuals, which include reduced H3K9me3.26 In contrast, tissues in aged rat harbor increased H4K20me3.27 In addition, promoters of active genes are exceptionally enriched in H4K16 acetylation in human senescent cells.28 It has been shown that histone chaperone HIRA, which deposits variant histone H3.3 as well as histone H4 into chromatin, is required for the retention of H4K16 acetylation.28 Importantly, genetic ablation of HIRA leads to enhanced skin tumor development in a mouse model expressing the oncogene BRAF, 28 which raised the possibility that HIRA‐mediated H4K16 acetylation is involved in the induction of oncogene‐induced senescence. Similarly, the NAD‐dependent deacetylase Sirtuins, which is implicated in aging in multiple model systems, increases the lifespan of yeast, possibly through reduced H4K16 acetylation at subtelomeric loci.29
Senescence is also characterized by a reduction of EZH2, a polycomb‐repressive protein, with a concomitant decrease of H3K27me3.14 A genome‐wide study of human senescent cells revealed that senescent cells harbor the generation of large‐scale domains of H3K27me3 over lamin‐associated domains (LADs) and large losses of H3K27me3 outside LADs.14, 30 Site‐specifically, the loss of H3K27me3 takes place around the INK4A‐ARF locus, which plays a critical role on the induction of senescence.31 Altered H3K27me3 is also linked with the senescence‐associated secretory phenotype (SASP), which has noncell autonomous functions in senescent cells.32, 33, 34 Senescence‐associated secretory phenotype can explain the diverse functions of senescent cells in multicellular organs in vivo, including enhanced tumorigenesis,35 tissue repair,36 immune surveillance,37, 38 and embryonic development39, 40 (Figure 2). Notably, the increased expression of SASP genes in senescent cells often correlates with decreased H3K27me3 deposition.30 Additionally, the inhibition of the H3K4 methyltransferase MLL1 inhibits SASP,41 suggesting that SASP is governed by altered histone modifications. The impact of H3K27me3 on senescence is further highlighted by the fact that the overexpression of BMI1, which causes increased EZH2 and increased H3K27me3 deposition at the INK4A‐ARF locus, ameliorates senescence‐related phenotypes.31 Taken together, altered histone deposition and modifications that are associated with transcriptional changes have a profound impact on organismal lifespan and senescence‐associated phenotypes in diverse organisms.
Figure 2.
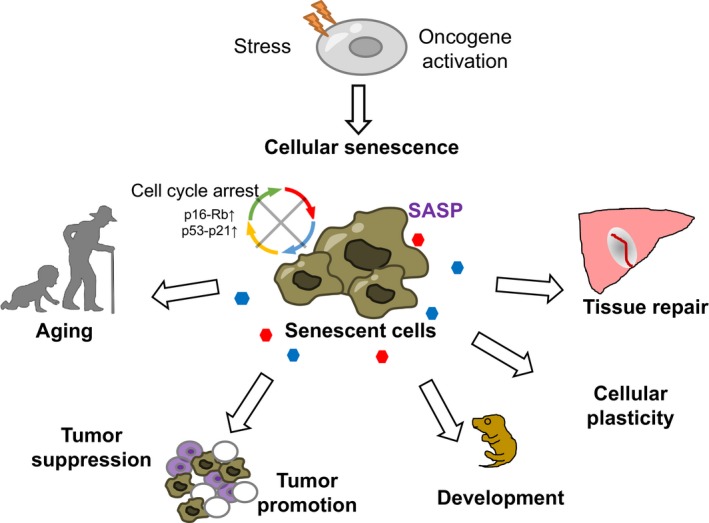
Diverse roles of cellular senescence in pathophysiological conditions. Cellular senescence is a state of a stable cell cycle arrest regulated by the p53‐p21 and p16‐Rb pathway, and can be induced by a range of cellular stresses. Senescent cells have functions not only in aging but also in various pathophysiological conditions, such as normal development, tissue repair, and cancer prevention, as well as cancer promotion through both cell autonomous and noncell autonomous mechanisms. Senescent cells exert diverse effects on the neighboring cells and the tissue microenvironment although the senescence‐associated secretory phenotype (SASP)
5. DNA METHYLATION IN SENESCENT CELLS
An alteration of DNA methylation patterns occurs during aging and senescence in mammals. These senescence‐associated DNA methylation changes are significantly enriched in genomic regions with repressive histone marks and at target sites of Polycomb group proteins.42, 43 As cells undergo aging, DNA methylation levels are decreased gradually during premature and replicative senescence.44 Although DNA hypomethylation is common with aging, some regions actually become hypermethylated.45 The loci that display age‐dependent DNA hypermethylation include tissue‐specific genes, genes involved in differentiation and development, genes encoding transcription factors, and transcription factor binding sites.45 Indeed, centenarian DNA has lower DNA methylation content globally, but higher DNA methylation at CpG island promoters.46 Consistent with the fact that constitutive heterochromatin structures are disorganized in senescent cells, global DNA hypomethylation in senescent cells is preferentially detectable at repetitive regions of the genome,26, 47 which are heavily methylated in normal cells but are hypomethylated in many cancers. Consistently, transposons are derepressed in both senescent cells and cancer cells.48 Notably, replicative senescence and aging are both accompanied by highly reproducible epigenetic changes, particularly alterations in the DNA methylation pattern of developmental genes.44
6. EPIGENETIC ALTERATIONS DURING CANCER INITIATION AND PROMOTION
It is widely accepted that an accumulation of genetic mutations causes the development of cancer.49 Indeed, genome‐wide sequencing analyses have unveiled a number of oncogenic mutations in various types of cancer. Studies using forward/reverse genetics have revealed the functional significance of oncogenic mutations in vivo. Rodents harboring these mutations develop cancer as seen in human patients. However, in addition to genetic aberrations, many studies have revealed that cancer cells harbor epigenetic modifications that differ from those in normal cells.11 Various types of cancers show epigenetic alterations including aberrant DNA methylation and altered histone modifications. Epigenetic changes in human cancer were first described as a reduction of global DNA methylation levels.50 A majority of cancers harbor changes in DNA methylation, which include genome‐wide DNA hypomethylation as well as site‐specific de novo DNA methylation.11 Mechanistically, such hypomethylation has been connected with genomic instability.51 In contrast, DNA hypermethylation has been often linked to the silencing of neighboring genes that are functionally related to tumor suppression in many cancers.52 Taken together, the current consensus holds that epigenetic alterations promote cancer development.
7. IN VIVO STUDIES THAT INVESTIGATED THE ROLE OF EPIGENETIC REGULATION IN CANCER DEVELOPMENT
The functional involvement of epigenetic regulation in both cancer initiation and progression in vivo has been highlighted by previous studies. Particularly noteworthy are detailed studies using Apc‐mutant mice that have revealed the consequence of modified DNA methylation in colon tumor development.53, 54, 55, 56 These studies suggest that forced reduction in DNA methylation causes chromosomal instability, resulting in the loss of Apc heterozygosity, and eventually promotes neoplastic transformation of colonic mucosa; however, at the same time, it suppresses the progression of early microadenomas into macroscopic tumors. In addition, the overexpression of de novo DNA methyltransferase Dnmt3b accelerates the progression of colonic microadenoma to a macroscopic tumor, whereas deletion of Dnmt3b suppresses this progression. In contrast, the forced reduction in DNA methylation induces liver cancers in the same model. Together, these in vivo studies suggest that DNA methylation plays a critical role in cancer development but has different effects depending on the cellular context and the stage of tumorigenesis.
8. ENVIRONMENTAL FACTORS CAUSE EPIGENETIC ALTERATIONS IN CANCER
The available data suggest that cancer progresses through multistep processes involving both genetic mutations and epigenetic abnormalities. However, it is still unclear how epigenetic alterations occur during cancer development. Recent genome‐wide sequencing studies have elucidated that epigenetic modifier genes are often mutated in various types of cancers23, 57, 58, 59, 60 (Table S1, modified from Feinberg et al.59 and Ito et al.60). Additionally, given that epigenetic modifications are tightly coupled with the gene expression profile, altered transcriptional signatures by genetic mutations could cause secondary epigenetic alterations. Collectively, it is possible that most epigenetic alterations in cancer cells could be a consequence of genetic abnormality. Therefore, it remains unclear how epigenetic abnormalities independent of genetic aberrations have a primary and causal role on the initiation and progression of cancer. Given that epigenetic regulation can be modified with chemical compounds, the identification of bona fide primary epigenetic abnormalities is of particular importance because epigenetic abnormalities provide a promising target for efficient cancer treatment.
It should be noted that environmental factors affect epigenetic modifications. Chronic infections, inflammation such as Helicobacter pylori infection, and hypoxia are often associated with an increased risk of cancer development. Of interest, previous studies have reported such environmental factors induce dynamic changes in epigenetic modifications, especially in DNA methylation patterns,61 suggesting that epigenetic alterations by these environmental factors exert cancer‐promoting effects through mechanisms independent of genetic abnormalities. Intriguingly, some environmental factors that cause an increased risk of cancer development, such as chronic inflammation and reactive oxygen species, are also related to the induction of cellular senescence, which raises the possibility that epigenetic alterations in senescent cells might promote cancer development.
9. SENESCENCE AS A ROADBLOCK FOR BOTH REPROGRAMMING AND CANCER
Like the neoplastic transformation seen during cancer development, somatic cells acquire unlimited proliferation capacity when reprogrammed to iPSCs. Previous studies have shown that key tumor‐suppressors, such as members of the p53, p21, and p16INK4a/retinoblastoma networks, decrease the efficiency of iPSC generation by activating cell‐intrinsic programs. Notably, such tumor‐suppressors are also involved in the induction of cellular senescence, suggesting that senescence is a barrier to iPSC derivation. Consistently, senescence‐related mechanisms are activated during the reprogramming process, which might explain the considerably low efficiency of iPSC derivation. Indeed, the expression of OSKM triggers senescence by upregulating p53, p16INK4a, and p21CIP1 (reprogramming‐induced senescence).62, 63 Furthermore, several groups have shown that knocking down p53 in human or mouse cells can significantly increase the efficiency of reprogramming. Similarly, low levels or the outright absence of p16INK4a or p21CIP1 expression leads to more efficient and faster reprogramming in both human and mouse cells.64, 65 In contrast, fibroblasts from older mice, which express higher levels of Ink4b/Arf/Ink4a locus products, are less efficiently reprogrammable to iPSCs,66 further linking aging/senescence with decreased reprogramming efficiency.
Notably, the induction of senescence is similarly observed in the early stage of cancer development where the increased expressions of tumor suppressor genes are detectable at preneoplastic lesions. It is therefore suggested that the proliferation arrest that is associated with senescence acts as a safeguard for malignant transformation, providing a close link between senescence escape and neoplastic growth. Collectively, senescence could be a critical roadblock for both cell reprogramming and cancer development, which underscores the similarity between these two processes.
10. SHARED EPIGENETIC ALTERATIONS BETWEEN SENESCENT CELLS AND CANCER CELLS
Although the molecular mechanisms underlying escape from senescence during cancer development are often explained by genetic aberrations such as inactivating mutations at the TP53 and CDKN2A genes, it is still possible that epigenetic regulation plays an important role, especially in cancers that lack genetic mutations in senescence‐associated genes. In contrast to the tumor‐inhibitory effects of senescence, the risk of cancer development increases with aging where senescent cells are accumulating. Considering the noncell autonomous effects of senescent cells through SASP, it is possible that the accumulated senescent cells could promote cancer development, thus explaining, at least in part, the increased incidence of cancer seen with aging.
It is also interesting to note that previous studies have reported partial but highly significant overlaps between altered DNA methylation patterns in cancer cells and senescent cells,67 which raises another possibility that the DNA methylome of senescent cells might promote malignant transformation when these cells escape the proliferative barrier. Indeed, the genome‐wide decrease in DNA methylation levels and site‐specific DNA hypermethylation detectable in senescent cells are mechanistically linked with genetic instability and the repression of tumor suppressor genes in cancer cells, respectively. Frequent overlaps between large‐scale domains of H3K4me‐enriched regions at LADs in senescent cells and cancer cells further support the hypothesis that premalignant senescent chromatin changes contribute to cancer development and thus are sustained in cancer cells.
Accordingly, epigenetic regulation in senescent cells might function as either an inhibitor or a promoter of cancer. Yet, how senescence‐related epigenetic regulation modulates cancer development remains largely unclear. Therefore, precise understanding of the epigenetic landscape in both senescent cells and cancer cells could eventually uncover the role of senescence in cancer development.
11. INTERVENTION FOR EPIGENETIC REGULATION IN SENESCENT CELLS AND CANCER CELLS WITH REPROGRAMMING TECHNOLOGY
By necessity, a dynamic reorganization of epigenetic regulation occurs during reprogramming, whereas changes in the underlying genomic information are not required. Therefore, reprogramming technology could be a useful strategy for actively manipulating epigenetic regulation. Of particular interest, previous studies have shown that age‐associated DNA methylation changes can be reverted by somatic cell reprogramming into iPSCs in vitro.43 Notably, reprogramming by OSKM resets cellular damage‐associated, stress‐associated, and senescence‐associated epigenetic marks in vitro.68, 69, 70 Consistently, the reprogramming of cells from elderly humans into pluripotent cells erases many of the hallmarks of aging and restores a youthful gene expression profile that is maintained during differentiation. This effect suggests that, in addition to telomere elongation, age/senescence‐associated epigenetic parameters can be reprogrammed with iPSC technology.71 These observations fit the general assumption that iPSCs are fully rejuvenated (Figure 3). It is noteworthy that some age‐associated phenotypes are sustained in neurons induced from old cells through direct reprogramming, suggesting that passage through the pluripotent state might be necessary for complete abrogation of the aging clock.72
Figure 3.
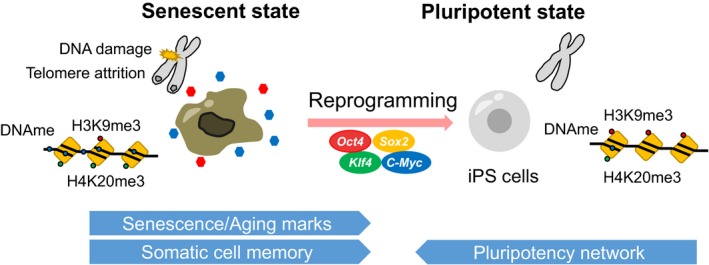
Erasure of age‐related epigenetic marks during cellular reprogramming in vitro. Reprogramming factors induce a loss of somatic cell identity and acquisition of self‐renewing capacity, leading to the derivation of induced pluripotent stem cells (iPSCs). During reprogramming, cellular marks of senescence and aging, which include epigenetic alterations, can be erased in vitro
With regard to altered epigenetic modifications in cancer, the altered DNA methylation in colon tumor cells is reprogrammable after the induction of pluripotency,73 suggesting that cancer‐associated epigenetic aberrations can be also erased with reprogramming technology. Moreover, partial reprogramming‐induced kidney cancer cells lose their tumorigenic potential after reprogramming into iPSCs and subsequent redifferentiation into kidney cells,74 indicating that the fate of particular types of cancer is controllable by epigenetic reorganization. The fact that cellular reprogramming is achievable in vivo further indicates that epigenetic regulation can be altered in multicellular organisms in vivo. Given that epigenetic alterations play a role in both senescence and cancer, reprogramming technology could provide a unique strategy for controlling the fate of detrimental cells in vivo, thus implying therapeutic implications. Such intervention on epigenetic regulation in detrimental cells should further contribute to better understanding of aging and cancer at the organismal level. In the following sections, we describe previous studies that revealed how in vivo reprogramming is applicable to abrogating detrimental phenotypes in mammals and other studies that took advantage of in vivo reprogramming systems to unveil epigenetic mechanisms in various pathophysiological phenomena.
12. ANTI‐AGING STRATEGY WITH IN VIVO REPROGRAMMING
Previous studies have described in vivo approaches to induce organismal rejuvenation and extend lifespans. Heterochronic parabiosis, a technique that links the circulatory systems of an old and young animal, can rejuvenate several hallmarks of aging in the older animal.75 The elimination of p16‐positive senescent cells is shown to extend lifespans in both progeria and normal mouse.76, 77
Considering that in vitro cell reprogramming abrogates senescence‐associated epigenetic alterations, researchers have investigated whether OSKM‐induced reprogramming has beneficial effects on aging phenotypes at organismal levels.12 Interestingly, cyclic short‐term induction (2 days) of OSKM in vivo dampens aging‐associated phenotypes and age‐associated histological changes in multiple organs. It also extends the lifespan of Hutchinson‐Gilford progeria mice, which harbor representative epigenetic alterations during aging, such as decreased levels of H3K9me3 and H3K27me3 as well as increased levels of H4K20me3. Additionally, the cyclic short‐term induction of OSKM in naturally aged mice promotes the expansion of beta cells after pancreatic injury and enhances glucose tolerance, providing additional evidence that in vivo reprogramming could be an alternative strategy for regenerative medicine. Similarly, the regenerative capacity of skeletal muscle after injury is enhanced in OSKM‐induced mice. However, it still remains to be fully understood how in vivo reprogramming affects the regeneration capacity, organismal aging, and lifespan (Figure 4). It would be therefore interesting to clarify the cell autonomous and non‐cell autonomous mechanisms that lead to cellular rejuvenation and increased longevity after OSKM expression in vivo.
Figure 4.
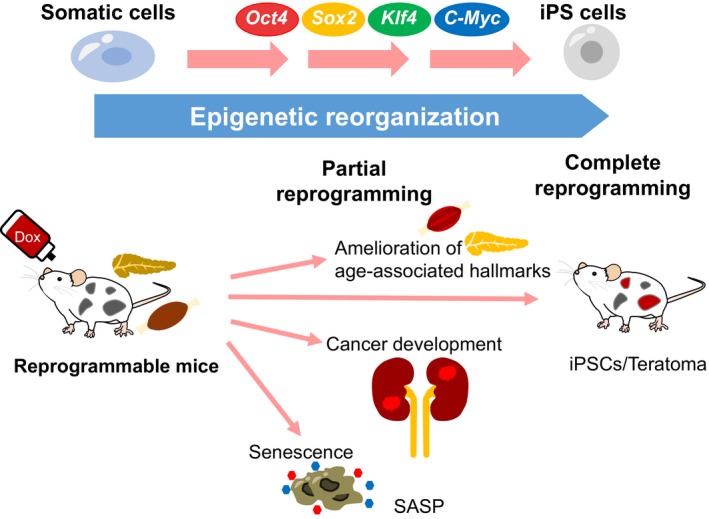
Uncovering the impact of epigenetic regulation on various organismal phenomena with in vivo reprogramming. During reprogramming, dynamic reorganization of epigenetic regulation takes place in vitro and in vivo. Short‐term induction of OSKM in vivo ameliorates aging‐associated phenotypes and extends lifespan. In contrast, premature termination of in vivo reprogramming can cause the development of cancer. The expression of OSKM in vivo simultaneously induces cellular senescence, indicating the complexity of in vivo reprogramming. iPSC, induced pluripotent stem cell; SASP, senescence‐associated secretory phenotype
13. UNCOVERING A UNIQUE CANCER DRIVER WITH IN VIVO REPROGRAMMING TECHNOLOGY
The reprogramming process toward the pluripotent state shares many properties with cancer development, including the acquisition of self‐renewing potential and the loss of original somatic cell identity. In addition, the acquisition of stem cell properties is often observed during carcinogenesis in many organs. Considering the number of shared aspects of the reprogramming process and cancer development, reprogramming technology in vivo could provide a unique experimental platform to elucidate the impact of reprogramming‐associated epigenetic regulation on cancer development. Indeed, previous studies have uncovered a link between reprogramming and cancer development using in vivo reprogrammable mice. Consistent with the fact that somatic cells acquire both self‐renewing activity and pluripotency during reprogramming, the long‐term expression of OSKM in vivo resulted in teratomas that contain iPSCs in various organs, indicating successful reprogramming in vivo.74, 78
In contrast, a shorter transient induction (3‐5 days) of the reprogramming factors often causes reversible dysplastic lesions in the pancreas, liver, and intestine, reflecting the existence of epigenetic memory in somatic cells. Surprisingly, these mice develop irreversible, OSKM‐independent cancers consisting of undifferentiated dysplastic cells in multiple organs after 1‐week induction followed by the withdrawal of OSKM.74 Notably, cancers in the kidney resemble Wilms’ tumor, which is one of the most common kidney cancers in children. The mouse cancer cells show a partial loss of original cell identity and acquisition of pluripotency‐associated gene signatures, but lack detectable genetic aberrations. Furthermore, these cancer cells often harbor aberrant DNA methylation at differentially methylated regions (DMRs) of imprinting genes, which includes hypermethylation at H19 DMR. Previous studies have suggested that the increased methylation at H19 DMR, which is one of the most common abnormalities in Wilms’ tumors, is causative of the tumor development, possibly through the increased expression of Igf2. These findings suggest that reprogramming‐associated epigenetic alteration might drive particular types of cancer. Given that the reprogramming process does not require changes in genetic information, these results could provide an in vivo proof of concept for epigenetics‐driven cancer, which occurs independent of genetic transformation but arises mainly as a result of epigenetic disruption triggered by dedifferentiation. Collectively, partial reprogramming in vivo has revealed an unappreciated impact of epigenetic regulation in cancer development (Figure 4).
14. UNVEILING NONCELL AUTONOMOUS EFFECTS OF SENESCENT CELLS ON ENHANCED CELLULAR PLASTICITY
Recent studies using in vivo reprogrammable mice highlight the intriguing role of noncell autonomous effects of senescent cells on cell fate conversion in vivo.79, 80 Those studies demonstrated that the forced expression of OSKM in vivo widely induces various types of stresses that are associated with unrestrained cell proliferation, DNA damage, inflammation, and cellular senescence in tissues. The effects indicate that OSKM expression provokes two contrary cellular outcomes, cellular reprogramming and damage‐induced cellular senescence. Interestingly, reprogrammed Nanog‐positive cells generally appear in neighboring clusters of senescent cells in OSKM‐induced tissues. Mechanistically, these senescent cells promoted in vivo reprogramming through a SASP, particularly interleukin‐6, suggesting that senescent cells enhance the cellular plasticity of adjacent cells.79 The increased interleukin‐6 in tissue could eventually facilitate trans‐ or dedifferentiation for damage‐induced tissue regeneration (Figure 4). Moreover, it is possible that the SASP‐mediated enhancement of cellular plasticity might facilitate an adaptation of neoplastic cells to their microenvironment, which eventually might contribute to cancer progression through some selection process. Therefore, these results could have implications not only in tissue regeneration but also in the SASP‐mediated progression of cancer development.
15. CONCLUSION: FUTURE PERSPECTIVES OF IN VIVO REPROGRAMMING
Although the molecular mechanisms of OSKM‐induced reprogramming in vitro have been well characterized, little is known about the mechanisms behind in vivo reprogramming. Nevertheless, emerging evidence using in vivo reprogramming suggests that OSKM‐induced senescence increases cellular plasticity of surrounding cells, which is associated with enhanced tissue regeneration in multicellular organisms through noncell autonomous effects from the senescent cells. Although OSKM induces senescence in vivo, another study using the same reprogrammable mice revealed that short‐term OSKM expression loses the senescence signature. Furthermore, the fact that the premature termination of in vivo reprogramming results in cancer development further highlights the complexity of in vivo reprogramming. Given that reprogramming is governed by epigenetic reorganization, in vivo reprogramming technology serves as an attractive tool for unveiling the epigenetic regulation underlying various pathophysiological phenomena in the complex tissue environments of multicellular organisms. Overall, further studies are needed to answer a number of interesting questions about the actions of in vivo reprogramming on cancer development, senescence‐induced cellular plasticity, tissue regeneration, and organismal rejuvenation.81
CONFLICT OF INTEREST
The authors have no conflict of interest to disclose.
Supporting information
ACKNOWLEDGMENTS
This work was supported in part by a cancer research grant from P‐CREATE, Japan Agency for Medical Research and Development (AMED) (JP18cm0106203 h0003 to Y.Y.), the Japan Society for the Promotion of Science (KAKENHI, 18H04026), the Takeda Science Foundation, the Naito Foundation, AMED‐CREST (JP18gm1110004 to Y.Y. and T.Y. and JP18gm0610017 to T.Y.), the Core Center for iPS Cell Research, Research Center Network for Realization of Regenerative Medicine, AMED, and the iPS Cell Research Fund (T.Y.).
Sogabe Y, Seno H, Yamamoto T, Yamada Y. Unveiling epigenetic regulation in cancer, aging, and rejuvenation with in vivo reprogramming technology. Cancer Sci. 2018;109:2641–2650. 10.1111/cas.13731
REFERENCES
- 1. Gurdon JB. The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles. J Embryol Exp Morphol. 1962;10:622‐640. [PubMed] [Google Scholar]
- 2. Wilmut I, Schnieke AE, McWhir J, Kind AJ, Campbell KH. Viable offspring derived from fetal and adult mammalian cells. Nature. 1997;385:810‐813. [DOI] [PubMed] [Google Scholar]
- 3. Tada M, Takahama Y, Abe K, Nakatsuji N, Tada T. Nuclear reprogramming of somatic cells by in vitro hybridization with ES cells. Curr Biol. 2001;11:1553‐1558. [DOI] [PubMed] [Google Scholar]
- 4. Cowan CA, Atienza J, Melton DA, Eggan K. Nuclear reprogramming of somatic cells after fusion with human embryonic stem cells. Science. 2005;309:1369‐1373. [DOI] [PubMed] [Google Scholar]
- 5. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663‐676. [DOI] [PubMed] [Google Scholar]
- 6. Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861‐872. [DOI] [PubMed] [Google Scholar]
- 7. Davis RL, Weintraub H, Lassar AB. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. 1987;51:987‐1000. [DOI] [PubMed] [Google Scholar]
- 8. Shen CN, Slack JM, Tosh D. Molecular basis of transdifferentiation of pancreas to liver. Nat Cell Biol. 2000;2:879‐887. [DOI] [PubMed] [Google Scholar]
- 9. Heins N, Malatesta P, Cecconi F, et al. Glial cells generate neurons: the role of the transcription factor Pax6. Nat Neurosci. 2002;5:308‐315. [DOI] [PubMed] [Google Scholar]
- 10. Wu C, Morris JR. Genes, genetics, and epigenetics: a correspondence. Science. 2001;293:1103‐1105. [DOI] [PubMed] [Google Scholar]
- 11. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415‐428. [DOI] [PubMed] [Google Scholar]
- 12. Ocampo A, Reddy P, Belmonte JCI. Anti‐aging strategies based on cellular reprogramming. Trends Mol Med. 2016;22:725‐738. [DOI] [PubMed] [Google Scholar]
- 13. Lopez‐Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194‐1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sen P, Shah PP, Nativio R, Berger SL. Epigenetic mechanisms of longevity and aging. Cell. 2016;166:822‐839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu Z, Chen K, Xia Z, et al. Nucleosome loss leads to global transcriptional up‐regulation and genomic instability during yeast aging. Genes Dev. 2014;28:396‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Han S, Brunet A. Histone methylation makes its mark on longevity. Trends Cell Biol. 2012;22:42‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McColl G, Killilea DW, Hubbard AE, Vantipalli MC, Melov S, Lithgow GJ. Pharmacogenetic analysis of lithium‐induced delayed aging in Caenorhabditis elegans. J Biol Chem. 2008;283:350‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jin C, Li J, Green CD, et al. Histone demethylase UTX‐1 regulates C. elegans life span by targeting the insulin/IGF‐1 signaling pathway. Cell Metab. 2011;14:161‐172. [DOI] [PubMed] [Google Scholar]
- 19. Maures TJ, Greer EL, Hauswirth AG, Brunet A. The H3K27 demethylase UTX‐1 regulates C. elegans lifespan in a germline‐independent, insulin‐dependent manner. Aging Cell. 2011;10:980‐990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729‐740. [DOI] [PubMed] [Google Scholar]
- 21. Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223‐233. [DOI] [PubMed] [Google Scholar]
- 22. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585‐621. [DOI] [PubMed] [Google Scholar]
- 23. Versteege I, Sevenet N, Lange J, et al. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203‐206. [DOI] [PubMed] [Google Scholar]
- 24. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593‐602. [DOI] [PubMed] [Google Scholar]
- 26. Scaffidi P, Misteli T. Lamin A‐dependent nuclear defects in human aging. Science. 2006;312:1059‐1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sarg B, Koutzamani E, Helliger W, Rundquist I, Lindner HH. Postsynthetic trimethylation of histone H4 at lysine 20 in mammalian tissues is associated with aging. J Biol Chem. 2002;277:39195‐39201. [DOI] [PubMed] [Google Scholar]
- 28. Rai TS, Cole JJ, Nelson DM, et al. HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Genes Dev. 2014;28:2712‐2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dang W, Steffen KK, Perry R, et al. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature. 2009;459:802‐807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shah PP, Donahue G, Otte GL, et al. Lamin B1 depletion in senescent cells triggers large‐scale changes in gene expression and the chromatin landscape. Genes Dev. 2013;27:1787‐1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bracken AP, Kleine‐Kohlbrecher D, Dietrich N, et al. The Polycomb group proteins bind throughout the INK4A‐ARF locus and are disassociated in senescent cells. Genes Dev. 2007;21:525‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Acosta JC, O'Loghlen A, Banito A, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006‐1018. [DOI] [PubMed] [Google Scholar]
- 33. Krizhanovsky V, Yon M, Dickins RA, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657‐667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Acosta JC, Banito A, Wuestefeld T, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978‐990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA. 2001;98:12072‐12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Demaria M, Ohtani N, Youssef SA, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF‐AA. Dev Cell. 2014;31:722‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xue W, Zender L, Miething C, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kang TW, Yevsa T, Woller N, et al. Senescence surveillance of pre‐malignant hepatocytes limits liver cancer development. Nature. 2011;479:547‐551. [DOI] [PubMed] [Google Scholar]
- 39. Munoz‐Espin D, Canamero M, Maraver A, et al. Programmed cell senescence during mammalian embryonic development. Cell. 2013;155:1104‐1118. [DOI] [PubMed] [Google Scholar]
- 40. Storer M, Mas A, Robert‐Moreno A, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155:1119‐1130. [DOI] [PubMed] [Google Scholar]
- 41. Capell BC, Drake AM, Zhu J, et al. MLL1 is essential for the senescence‐associated secretory phenotype. Genes Dev. 2016;30:321‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schellenberg A, Lin Q, Schuler H, et al. Replicative senescence of mesenchymal stem cells causes DNA‐methylation changes which correlate with repressive histone marks. Aging. 2011;3:873‐888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Koch CM, Reck K, Shao K, et al. Pluripotent stem cells escape from senescence‐associated DNA methylation changes. Genome Res. 2013;23:248‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bork S, Pfister S, Witt H, et al. DNA methylation pattern changes upon long‐term culture and aging of human mesenchymal stromal cells. Aging Cell. 2010;9:54‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Booth LN, Brunet A. The aging epigenome. Mol Cell. 2016;62:728‐744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Heyn H, Li N, Ferreira HJ, et al. Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci USA. 2012;109:10522‐10527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson‐Gilford progeria syndrome. Nat Med. 2005;11:440‐445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. De Cecco M, Criscione SW, Peterson AL, Neretti N, Sedivy JM, Kreiling JA. Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging. 2013;5:867‐883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23‐28. [DOI] [PubMed] [Google Scholar]
- 50. Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89‐92. [DOI] [PubMed] [Google Scholar]
- 51. Chen RZ, Pettersson U, Beard C, Jackson‐Grusby L, Jaenisch R. DNA hypomethylation leads to elevated mutation rates. Nature. 1998;395:89‐93. [DOI] [PubMed] [Google Scholar]
- 52. Herman JG, Latif F, Weng Y, et al. Silencing of the VHL tumor‐suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci USA. 1994;91:9700‐9704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Laird PW, Jackson‐Grusby L, Fazeli A, et al. Suppression of intestinal neoplasia by DNA hypomethylation. Cell. 1995;81:197‐205. [DOI] [PubMed] [Google Scholar]
- 54. Yamada Y, Jackson‐Grusby L, Linhart H, et al. Opposing effects of DNA hypomethylation on intestinal and liver carcinogenesis. Proc Natl Acad Sci USA. 2005;102:13580‐13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lin H, Yamada Y, Nguyen S, et al. Suppression of intestinal neoplasia by deletion of Dnmt3b. Mol Cell Biol. 2006;26:2976‐2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Linhart HG, Lin H, Yamada Y, et al. Dnmt3b promotes tumorigenesis in vivo by gene‐specific de novo methylation and transcriptional silencing. Genes Dev. 2007;21:3110‐3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lawrence MS, Stojanov P, Mermel CH, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jones S, Wang TL, Shih Ie M, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Feinberg AP, Koldobskiy MA, Gondor A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17:284‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ito Y, Hoare M, Narita M. Spatial and temporal control of senescence. Trends Cell Biol. 2017;27:820‐832. [DOI] [PubMed] [Google Scholar]
- 61. Niwa T, Tsukamoto T, Toyoda T, et al. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Can Res. 2010;70:1430‐1440. [DOI] [PubMed] [Google Scholar]
- 62. Banito A, Rashid ST, Acosta JC, et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009;23:2134‐2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Banito A, Gil J. Induced pluripotent stem cells and senescence: learning the biology to improve the technology. EMBO Rep. 2010;11:353‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hong H, Takahashi K, Ichisaka T, et al. Suppression of induced pluripotent stem cell generation by the p53‐p21 pathway. Nature. 2009;460:1132‐1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Utikal J, Polo JM, Stadtfeld M, et al. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature. 2009;460:1145‐1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li H, Collado M, Villasante A, et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009;460:1136‐1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cruickshanks HA, McBryan T, Nelson DM, et al. Senescent cells harbour features of the cancer epigenome. Nat Cell Biol. 2013;15:1495‐1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lapasset L, Milhavet O, Prieur A, et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011;25:2248‐2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Liu GH, Barkho BZ, Ruiz S, et al. Recapitulation of premature ageing with iPSCs from Hutchinson‐Gilford progeria syndrome. Nature. 2011;472:221‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhang J, Lian Q, Zhu G, et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell. 2011;8:31‐45. [DOI] [PubMed] [Google Scholar]
- 71. Marion RM, Strati K, Li H, et al. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell. 2009;4:141‐154. [DOI] [PubMed] [Google Scholar]
- 72. Mertens J, Paquola ACM, Ku M, et al. Directly reprogrammed human neurons retain aging‐associated transcriptomic signatures and reveal age‐related nucleocytoplasmic defects. Cell Stem Cell. 2015;17:705‐718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hashimoto K, Yamada Y, Semi K, et al. Cellular context‐dependent consequences of Apc mutations on gene regulation and cellular behavior. Proc Natl Acad Sci USA. 2017;114:758‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ohnishi K, Semi K, Yamamoto T, et al. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell. 2014;156:663‐677. [DOI] [PubMed] [Google Scholar]
- 75. Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760‐764. [DOI] [PubMed] [Google Scholar]
- 76. Baker DJ, Wijshake T, Tchkonia T, et al. Clearance of p16Ink4a‐positive senescent cells delays ageing‐associated disorders. Nature. 2011;479:232‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Baker DJ, Childs BG, Durik M, et al. Naturally occurring p16(Ink4a)‐positive cells shorten healthy lifespan. Nature. 2016;530:184‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Abad M, Mosteiro L, Pantoja C, et al. Reprogramming in vivo produces teratomas and iPS cells with totipotency features. Nature. 2013;502:340‐345. [DOI] [PubMed] [Google Scholar]
- 79. Mosteiro L, Pantoja C, Alcazar N, et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science. 2016;354:aaf4445. [DOI] [PubMed] [Google Scholar]
- 80. Chiche A, Le Roux I, von Joest M, et al. Injury‐induced senescence enables in vivo reprogramming in skeletal muscle. Cell Stem Cell. 2017;20:407‐414. e4 [DOI] [PubMed] [Google Scholar]
- 81. Taguchi J, Yamada Y. Unveiling the role of senescence‐induced cellular plasticity. Cell Stem Cell. 2017;20:293‐294. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials