

Abstract
Imatinib has revolutionized the treatment of gastrointestinal stromal tumors (GIST); however, primary and secondary resistance to imatinib is still a major cause of treatment failure. Multiple mechanisms are involved in this progression. In the present study, we reported a novel mechanism for the acquired resistance to imatinib, which was induced by enhanced Ca2+ influx via stromal‐interacting molecule 1 (STIM1)‐mediated store‐operated Ca2+ entry (SOCE). We found that the STIM1 expression level was related to the acquired resistance to imatinib in our studied cohort. The function of STIM1 in imatinib‐resistant GIST cells was also confirmed both in vivo and in vitro. The results showed that STIM1 overexpression contributed to SOCE and drug response in imatinib‐sensitive GIST cells. Blockage of SOCE by STIM1 knockdown suppressed the proliferation of imatinib‐resistant GIST cell lines and xenografts. In addition, STIM1‐mediated SOCE exerted an antiapoptotic effect via the MEK/ERK pathway. The results from this study provide a basis for further research into potential novel therapeutic strategies in acquired imatinib‐resistant GIST.
Keywords: gastrointestinal stromal tumors, imatinib, resistance, STIM1, store‐operated Ca2+ entry
1. INTRODUCTION
Gastrointestinal stromal tumors (GIST) are the most common mesenchymal tumor of the digestive tract. The incidence of GIST and so‐called subclinical GIST is nearly 10%‐30% in adults.1, 2, 3 KIT and PDGFRA mutations are recognized as the driving factors of GIST,4, 5 with the decisive therapy being selective receptor tyrosine kinase inhibitors. Imatinib has been approved by the Food and Drug Administration for the treatment of GIST since 2002 and is regarded as the first‐line drug worldwide, including in the National Comprehensive Cancer Network, European Society for Medical Oncology, and Asian consensus.6, 7, 8 Although more than 80% of patients benefit from imatinib monotherapy, half still develop acquired resistance within 2 years of treatment, leading to recurrence or metastasis, and increasing mortality and morbidity.9 Interstitial cells of Cajal (ICC) are the peacemaker cells in the gastrointestinal tract and are where GIST originates from.10 Intracellular Ca2+ plays a key role in the function of ICC;11 therefore, it is important to study the influence of intracellular Ca2+ in GIST tumorigenesis, proliferation and drug response. Store‐operated Ca2+ entry (SOCE) is mediated via store‐operated channels and is the principal approach for Ca2+ entry.12 Stromal‐interacting molecule 1 (STIM1) is a critical component of SOCE. STIM1 is an endoplasmic reticulum (ER) Ca2+ sensor. Once the ER Ca2+ concentration is depleted, STIM1 is activated and aggregates to the pore subunit of the Ca2+ channel (such as Orai1) in the plasma membrane, forming a pore channel for Ca2+ influx. From the opening of the Ca2+ channel, ER stores can be refilled again.13, 14 Recently, a growing body of literature has reported that SOCE is involved in the proliferation and migration of various cancers.15, 16, 17 A study of ovary carcinoma showed that SOCE participates in resistance to cisplatin.18 However, the mechanism by which SOCE promotes the malignancy of tumor cells remains inconclusive and the role of STIM1‐mediated SOCE in GIST is unclear.
In this study, we investigated the correlation between the STIM1 expression level and imatinib resistance in GIST patients. We found that STIM1 was upregulated in imatinib‐resistant GIST cells compared to sensitive parental cells, and knockdown or overexpression of STIM1 has significant effects on SOCE, proliferation and drug response in imatinib‐resistant GIST cells. We further revealed that STIM1‐mediated SOCE could induce apoptosis via the MEK/ERK pathway.
2. MATERIALS AND METHODS
2.1. Patients and specimens
A total of 35 specimens were collected from pathologically‐confirmed GIST patients between 2012 and 2017 from the Department of General Surgery, Xinhua Hospital, School of Medicine, Shanghai Jiao Tong University, China. Fresh GIST tissues and paired nontumor tissues used for quantitative RT‐PCR (qRT‐PCR) and western blotting were frozen and stored in liquid nitrogen within 15 minutes after removal. No patients in this study received neoadjuvant therapy before their operations. Imatinib treatment after radical surgery was performed as described in the guidelines, with a proportion of the patients developing imatinib resistance after 6 months, defined as acquired resistance. All patients received follow‐up. This study was approved by the Ethics Committee of the Xinhua Hospital and all patients provided informed consent.
2.2. Cell culture
Human GIST cell lines GIST‐T1 and GIST‐882 were obtained from the Shanghai Cancer Institute. Both cell lines were grown in DMEM (Gibco, Gaithersburg, MD, USA) that contained 10% FBS (Gibco). Cells were cultured at 37°C in 5% CO2 and at 95% relative humidity. Imatinib‐resistant sublines emerged from cell cultures with gradually increasing doses of imatinib. Imatinib‐sensitive cell lines GIST‐882 and GIST‐T1 were cultured in medium with gradually increasing doses of imatinib (.2, .4 and 1 μmol/L) and were obtained after 1, 2 and 4 months, respectively. Relative resistance assessment was applied to detect the stability of the resistant phenotype originating from each culture continuously in medium with increasing concentrations of imatinib for up to 6 months. Imatinib (STI571) was purchased from Selleck (Shanghai, China).
2.3. Quantitative RT‐PCR
Total RNA was isolated from tissue samples or cultured cells using TRIzol Reagent (Takara, Shiga, Japan). cDNA was synthesized with PrimeScript Reverse Transcriptase (Takara, Osaka, Japan) following the manufacturer's instructions. Real‐time PCR was performed with the StepOne Real‐Time PCR System (Applied Biosystems, Foster City, USA) with SYBR Green (Takara, Dalian, China). GAPDH was used as the internal standard. Primer sequences used for amplification were listed in supporting information (Supplementary Table S1).
2.4. Western blotting
Protein was extracted using radioimmunoprecipitation assay buffer (Cell Signaling, Danvers, MA, USA). A total of 25 μg of protein was loaded onto a 12% sodium dodecyl sulfate polyacrylamide electrophoresis gel and then transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5% skim milk at room temperature for 1 h and then incubated with primary antibodies at 4°C overnight. The anti‐STIM1 antibody was purchased from Proteintech (Wuhan, China). Primary antibodies against MEK, p‐MEK, ERK, p‐ERK and GAPDH were obtained from Cell Signaling Technology. Subsequently, the membranes were washed with Tris‐buffered saline containing Tween 20 (TBST) and reacted with the appropriate HRP‐conjugated secondary antibody. Blots were visualized by Gel Doc 2000 (Bio‐Rad, Hercules, CA, USA) or Amersham Imager 600 (GE).
2.5. siRNA, plasmid and Lv‐shRNA transfection
Stromal‐interacting molecule 1 were designed and synthesized by Biotend (Shanghai, China). The siRNA targeting human STIM1 are listed below: siRNA1: 5′‐UCAAUUCGGCAAAACUCUGdTdT‐3′, siRNA2: 5′‐AAGGUCUCCUCAUACUGAGdTdT‐3′, siRNA3: 5′‐AAUCGGAAUGGGUCAAAUCdTdT‐3′. siRNA was transfected into the cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. STIM1 plasmids (Longqian Biotech, Shanghai, China) or control plasmids were transfected into cells using the ViaFect Transfection Reagent (Promega, Madison, WI, USA). Purified lentiviruses encoding small hairpin RNA targeting STIM1 (5′‐GGAGGATAATGGCTCTATT‐3′) were constructed by Genechem (Shanghai, China). GIST‐882‐R cells were infected at a multiplicity of infection of 40 for 24 hours. STIM1 expression levels were detected by qRT‐PCR and western blotting.
2.6. Cell viability assays
The Cell Counting Kit‐8 (CCK‐8; Dojindo, Kumamoto, Japan) assay was used for the assessment of cell viability. Cells were plated at 2 × 103 cells per well in 96‐well plates. For proliferation assays, absorbance at 450 nm was measured for 5 days. To determine the half maximal inhibitory concentration (IC50), cells were rinsed the next day and cultured with various doses of imatinib for 48 hours. Colony formation assays were performed to assess anchorage‐independent growth. A total of 500 cells were seeded in 6‐well plates and cultured for 10 days. Then, the cells were fixed, stained and photographed.
2.7. Calcium imaging
Cells were stained with 4 μM Fluo‐4‐AM (Invitrogen) before imaging and then resuspended with Hanks’ balanced salt solution (with Ca2+, Mg2+, without phenol red, pH 7.4; Yeasen, Shanghai, China) to remove extracellular Fluo‐4‐AM. A Zeiss LSM 710 (Zeiss, Jena, Germany) measured fluo‐4‐Ca2+ fluorescence at 488‐nm emission. SOCE in Fluo‐4‐AM‐loaded cells was detected using a calcium imaging system 200 s after the intracellular Ca2+ responses were stimulated with 2‐μM thapsigargin (TG).
2.8. Flow cytometry analysis of cell apoptosis
Apoptosis was inspected with an Annexin V‐FITC Apoptosis Detection kit (BD Biosciences) according to the manufacturer's instructions. Cells were collected and twice washed with PBS, gently resuspended in 100 μL Annexin V binding buffer (1×) containing 2.5 μL FITC (BD Pharmingen) and 5 μL of 50 μg/mL PI, and then incubated at room temperature in the dark for 15 minutes. The stained cells were analyzed by flow cytometry (BD Biosciences) and data were analyzed with FlowJo (Flowjo Studio, Carrboro, NC, USA).
2.9. Xenograft nude mouse model
Animal studies were approved by the Ethics Committee of Xinhua Hospital. Nude nu/nu mice, 4‐6 weeks old, were purchased from the Shanghai Laboratory Animal Center of the Chinese Academy of Sciences (Shanghai, China). GIST‐882‐R cells were stably infected with Lv‐shNC (negative control)/Lv‐shSTIM1. A total of 5 × 106 viable cells were injected into the right axilla of nude mice. Tumor sizes were measured weekly using a vernier caliper. After 4 weeks, the mice were killed and the tumors were dissected out and weighed.
2.10. Statistical analysis
The SPSS 22.0 software program for Windows was used for statistical analysis. The difference between the STIM1 expression level and clinicopathologic parameters was found using Pearson's χ2 test or Fisher's exact test. The independent Student's t test was used when the data were normally distributed. Each experimental value was expressed as the mean ± standard deviation. A P‐value of less than .05 was considered statistically significant. All data points represent the mean of triplicate experiments.
3. RESULTS
3.1. Stromal‐interacting molecule 1 overexpression was related to acquired imatinib resistance in gastrointestinal stromal tumor patients
To investigate the role of STIM1 in GIST, we first compared the expression levels of STIM1 mRNA in 35 pairs of GIST tissue by qRT‐PCR. The relative STIMI expression levels were significantly higher in tumor tissue samples than those in corresponding nontumor tissue samples (P < .01) (Figure 1A,B). Based on the fold change, we divided patients into a high‐level group (fold change ≥ 2) and a low‐level group (fold change <2). Further clinicopathological association examination of the 35 GIST patients showed that STIM1 was significantly associated with acquired imatinib resistance (P = .022) (Table 1). STIM1 expression levels in GIST patients who developed imatinib resistance were significantly higher than in those who did not develop imatinib resistance (P < .01) (Figure 1C). Furthermore, western blotting confirmed that STIM1 protein expression levels in GIST tissues were higher than those in the corresponding non‐GIST tissues (Figure 1D).
Figure 1.
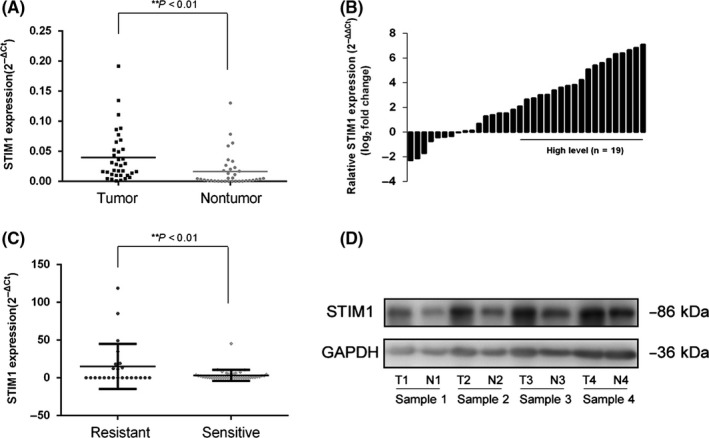
Stromal‐interacting molecule 1 (STIM1) overexpression is related to acquired imatinib resistance in gastrointestinal stromal tumors (GIST) patients. A, Scatterplots of relative STIM1 expression in GIST tissues and their matched nontumor counterparts. STIM1 expressions were calculated and are expressed as the STIM1/GADPH expression ratio (2−ΔCT). B, Comparison of STIM1 expression levels between GIST tissues and corresponding nontumor tissues. C, Scatterplots of relative STIM1 mRNA expression levels in imatinib‐resistant and imatinib‐sensitive groups. D, Relative STIM1 protein expression levels in GIST tissues and corresponding non‐GIST tissues. **P < .01
Table 1.
Association of STIM1 expression with the clinicopathological characteristics of GIST
| Variable | Category | Number of cases | STIM1 | ||
|---|---|---|---|---|---|
| Number of high‐level cases | χ2 | P | |||
| Age | <60 | 19 | 10 | .274 | .600 |
| ≥60 | 16 | 9 | |||
| Sex | Male | 20 | 11 | .077 | .782 |
| Female | 15 | 8 | |||
| Tumor location | Stomach | 18 | 10 | .274 | .600 |
| Nonstomach | 17 | 9 | |||
| Tumor size | ≤2 cm | 7 | 4 | .674 | .879 |
| >2 cm ≤ 5 cm | 11 | 5 | |||
| >5 cm ≤ 10 cm | 13 | 8 | |||
| >10 cm | 4 | 2 | |||
| Mitotic rate | ≤5 | 23 | 14 | 1.172 | .311* |
| >5 | 12 | 5 | |||
| Rupture | Yes | 6 | 1 | 4.130 | .073* |
| No | 29 | 18 | |||
| Risk classification (NIH) | Very low | 5 | 4 | 1.154 | .670 |
| Low | 4 | 2 | |||
| Moderate | 12 | 6 | |||
| High | 14 | 7 | |||
| Imatinib resistance | Yes | 9 | 8 | 5.846 | .022 * |
| No | 26 | 11 | |||
| Total | 35 | 19 | |||
Bold values indicate statistical significance; P < .05. *Fisher's exact test. GIST, gastrointestinal stromal tumors; STIM1, stromal‐interacting molecule 1.
3.2. Overexpressing of stromal‐interacting molecule 1 and enhanced store‐operated Ca2+ entry were detected in imatinib‐resistant gastrointestinal stromal tumor cells
To reveal the function of STIM1, we established 2 cell line models of acquired resistance following continuous in vitro exposure to imatinib using GIST‐T1 and GIST‐882 cells. We first investigated the peak of the Ca2+ elevation and found that SOCE was higher in imatinib‐resistant cells than that in imatinib‐sensitive cells (Figure 2A,B). STIM1, Orai1 and TRPC channel expression in imatinib‐resistant cells and their parental counterparts were compared using qRT‐PCR (Supplementary Figure S1); only the STIM1 expression level had significant change. Among the 4 cell lines, STIM1 expression decreased in imatinib‐sensitive GIST‐882 and GIST‐T1 cells, whereas it was overexpressed in the homologous imatinib‐resistant cells (Figure 2C). Consistent protein levels were observed in western blotting (Figure 2D).
Figure 2.
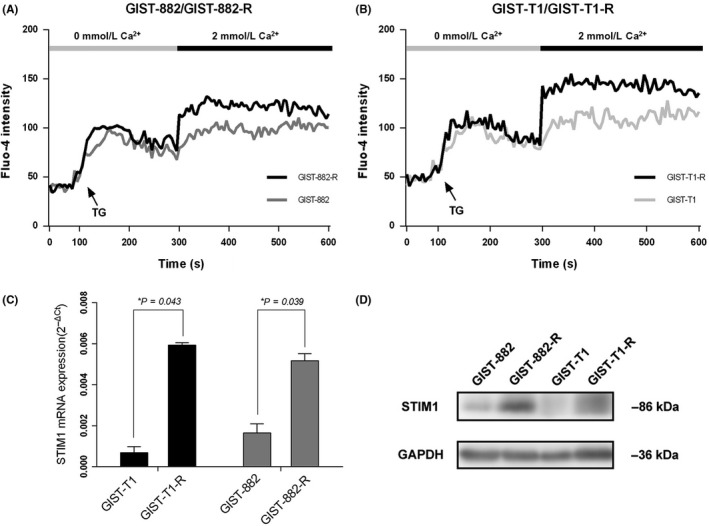
Overexpressing of stromal‐interacting molecule 1 (STIM1) and enhanced store‐operated Ca2+ entry (SOCE) are detected in imatinib‐resistant GIST cells. A and B, Compared to their parental cell lines, SOCE was increased in imatinib‐resistant gastrointestinal stromal tumors (GIST) cells. C and D, STIM1 mRNA and protein expression levels in GIST‐T1, GIST‐882 and their parental imatinib‐resistant cells. *P < .05
3.3. Knockdown of stromal‐interacting molecule 1‐suppressed proliferation of imatinib‐resistant gastrointestinal stromal tumor cells in vitro
We transfected GIST‐882‐R and GIST‐T1‐R cell lines with 3 different siRNA against STIM1. The efficiency of each siRNA was assessed by qRT‐PCR and, from this, the third siRNA was employed (Figure 3A). Western blot analysis confirmed the knockdown efficiency (Figure 3B). We used CCK‐8 and colony formation assays to explore the influence of STIM1 knockdown on GIST cell proliferation. Figure 3C shows that the viability of GIST‐882‐R and GIST‐T1‐R cells were markedly inhibited by STIM1 depletion (P < .05). In addition, compared with the si‐NC (negative control) groups, the downregulation of STIM1 reduced the capacity of colony formation in GIST‐882‐R and GIST‐T1‐R cells (Figure 3D).
Figure 3.
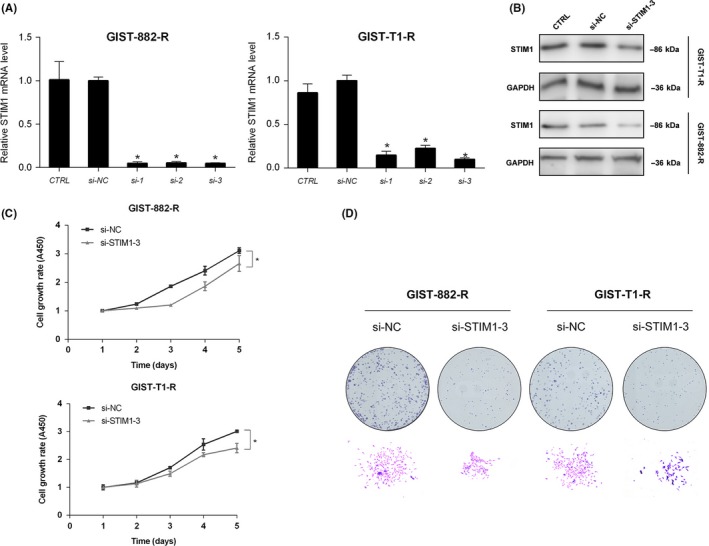
Knockdown of stromal‐interacting molecule 1 (STIM1) suppresses proliferation in imatinib‐resistant gastrointestinal stromal tumors (GIST) cells. A and B, Knockdown efficiency of STIM1 in GIST‐T1‐R and GIST‐882‐R cells tested by quantitative RT‐PCR and western blotting, respectively. GADPH was used as the loading control. C, Cell growth curves detected by Cell Counting Kit‐8 proliferation assays at various time points. D, Macroscopic images of colonies formed by treated GIST cells. *P < .05
3.4. Overexpression of stromal‐interacting molecule 1 enhanced store‐operated Ca2+ and imatinib sensitivity in gastrointestinal stromal tumor cells
We transfected STIM1 overexpression vectors into GIST‐882 and GIST‐T1 cells to determine the functions of STIM1. qRT‐PCR and western blotting verified that the expression level of STIM1 was upregulated after transfection (Figure 4A,B). We then detected that the IC50 value of imatinib was higher in the STIM1‐transfected cells than that in the empty vector‐transfected cells. The IC50 value of GIST‐882 increased from .2001 ± .0012 μmol/L to .9868 ± .0107 μmol/L, and the IC50 value of GIST‐T1 increased from .4257 ± .0053 μmol/L to 1.7610 ± .1054 μmol/L after transfection (Figure 4C). STIM1 is one of the essential components of SOCE; therefore, we also investigated the effect of STIM1 overexpression on SOCE and found that STIM1 overexpression significantly increased the peak of the Ca2+ elevation resulting from Ca2+ influx (Figure 4D).
Figure 4.
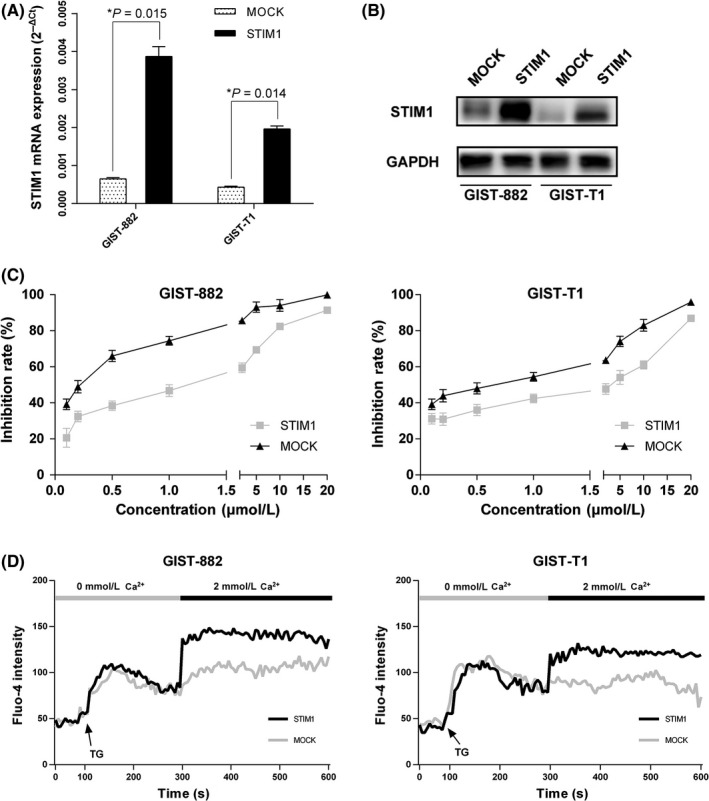
Overexpression of stromal‐interacting molecule 1 (STIM1) enhances store‐operated Ca2+ entry (SOCE) and imatinib sensitivity in gastrointestinal stromal tumors (GIST) cells. A and B, Overexpression efficiency of STIM1 in GIST‐882 and GIST‐T1 cells verified by quantitative RT‐PCR and western blotting. C, STIM1 overexpression reduced drug‐sensitivity of imatinib in GIST‐882 and GIST‐T1 cells. D, STIM1 overexpression increased SOCE in GIST‐882 and GIST‐T1 cells. *P < .05
3.5. Blockage of store‐operated Ca2+ inhibits growth of imatinib‐resistant gastrointestinal stromal tumors in vivo
To evaluate the effect of STIM1 on imatinib‐resistant GIST growth in vivo, we chose GIST‐882‐R cell lines for stable transfection with shRNA lentivirus vectors to STIM1. qRT‐PCR and western blotting verified the knockdown efficiency of the system (Figure 5A,B, respectively). In addition, we confirmed that STIM1 downregulation decreased the functional SOCE in GIST‐882‐R cells (Figure 5C). With the above verification, GIST‐882‐R cell lines stably expressing shRNA‐STIM1 or the negative control were injected into the axilla of nude mice, and the tumor volume was regularly monitored for the following 4 weeks (Figure 5D). Our results showed that the growth of STIM1‐knockdown xenografts was significantly inhibited compared to the tumors formed by control cells (Figure 5E,F). These results showed that blockage of SOCE is essential for growth inhibition of imatinib‐resistant GIST.
Figure 5.
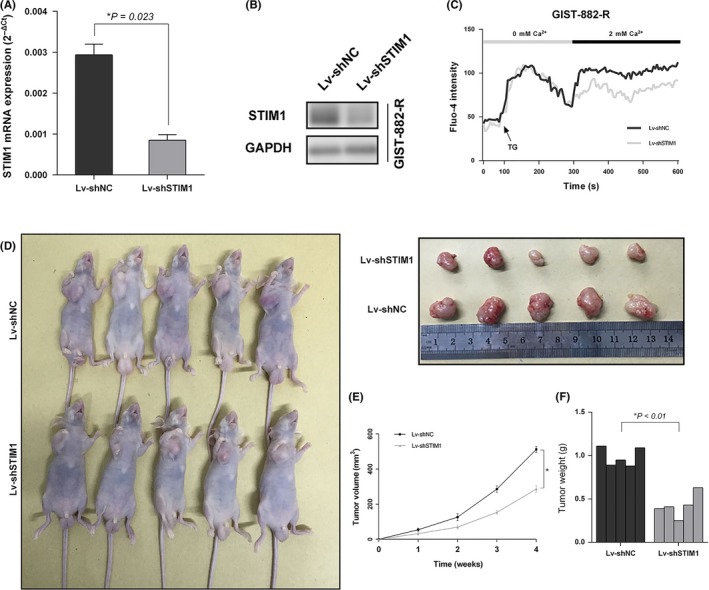
Blockage of store‐operated Ca2+ entry (SOCE) inhibits growth of imatinib‐resistant GIST in vivo. A and B, The lentiviral knockdown efficiency was examined by quantitative RT‐PCR and western blotting in gastrointestinal stromal tumors (GIST)‐882‐R cells. C, Stromal‐interacting molecule 1 (STIM1) knockdown via transfection with Lv‐shSTIM1 decreased SOCE in GIST‐882‐R cells. D, Representative photographs of tumor formation in nude mice injected with Lv‐shSTIM1 and Lv‐shNC GIST‐882‐R cells. E and F, The tumor volume and weight were measured in Lv‐shSTIM1 and Lv‐shNC groups
3.6. Stromal‐interacting molecule 1‐mediated store‐operated Ca2+ exerted an antiapoptosis effect through the MEK/ERK pathway
To explore the molecular mechanism of the proliferate inhibition induced by STIM1, flow cytometry was used to investigate the effect of STIM1 on apoptosis by measuring the apoptotic index, which is defined as the percentage of apoptotic cells. The results showed that apoptotic and dead cells increased significantly in the cells with STIM1 siRNA transfection. The apoptosis indexes of the si‐NC group and the si‐STIM1 group in GIST‐882‐R cells were 1.44% and 9.42%, respectively; and the apoptosis indexes of the si‐NC group and the si‐STIM1 group in GIST‐T1‐R cells were 2.48% and 13.42%, respectively (P < .05; Figure 6A). Considering that the deregulation of MEK/ERK phosphorylation represents an important antiapoptotic mechanism in various tumors,19 we studied the phosphorylation status of proteins involved in the above signaling pathway using western blotting. We found that p‐MEK and p‐ERK expression levels decreased significantly when STIM1 was silenced (Figure 6B). These results indicated that the MEK/ERK pathway might be regulated by the STIM1‐mediated SOCE in imatinib‐resistant GIST cells.
Figure 6.
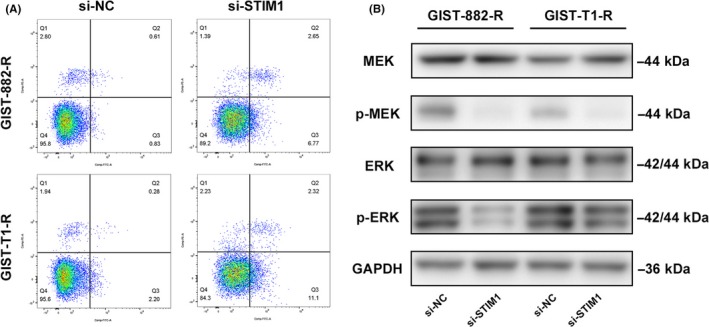
Stromal‐interacting molecule 1 (STIM1)‐mediated store‐operated Ca2+ entry (SOCE) exerts an antiapoptosis effect through the MEK/ERK pathway. A, STIM1 knockdown induced apoptosis in gastrointestinal stromal tumors (GIST)‐882‐R and GIST‐T1‐R cells. Cells stained with FITC were considered apoptotic. The percentages of apoptotic cells are shown. B, Phosphorylation of MEK/ERK pathway‐associated proteins was detected in the imatinib‐resistant GIST cells by western blotting. GAPDH was used as the loading control
4. DISCUSSION
Imatinib has revolutionized the treatment of GIST; however, primary and secondary resistance to imatinib is still a major cause of treatment failure. Several mechanisms have been found to be responsible for imatinib resistance, including secondary point mutations, gene amplification, autophagy and apoptosis, and other mechanisms. These mechanisms are complex, often heterogeneous, and not fully understood.20 Among all possible reasons, second point mutations in the kinase or loop domain of KIT or PDGFRA are considered the main cause.21 However, more than a few imatinib‐resistant GIST do not detect such mutations,22, 23 indicating the existence of additional mechanisms and the existence of additional mechanisms. Here, we report on a novel mechanism of acquired resistance to imatinib, which was induced by enhanced Ca2+ entry via STIM1‐mediated SOCE. This hypothesis was verified both in vivo and in vitro by regulating the STIM1 expression level. The results showed that STIM1 was related to acquired imatinib resistance in GIST. Inhibition of STIM1‐mediated SOCE suppressed the proliferation of imatinib‐resistant GIST cell lines and xenografts. Further study indicated that STIM1 plays a critical role in apoptosis of imatinib‐resistant GIST cell lines, which was confirmed by detecting the expression of the MEK/ERK pathway.
In fact, Ca2+ entry participates in a variety of fundamental cellular mechanisms and ensures that cells acclimatize to external environmental change.24 Recent research has suggested that deregulation of SOCE contributes to tumor proliferation, invasion and migration. SOCE channels have been proven to be potential targets for several malignant tumors.16, 25, 26 STIM1, which is the essential component of SOCE and the focus of the present study, is located on human chromosome region 11p15.5, and the loss of 11p13‐11p15.5 has been associated with several malignancies. Therefore, further research on why overexpressed STIM1 exerts an antidrug effect in GIST is required. According to previous research in prostate cancer,27, 28 although different studies have contrary results for the expression level of STIM1, STIM1 plays an oncogene role. Difference in sample size and detecting methods may explain these discrepancies. The present study examined STIM1 expression in both translational and post‐translational levels and matched it to the clinicopathological characteristics of GIST patients. We failed to find correlation between STIM1 and risk stratification (Table 1); however, we found that STIM1 was related to acquired imatinib resistance in the studied cohort. Therefore, we investigated the function of STIM1‐mediated SOCE in imatinib‐resistant GIST.
Based on our data, we suggest that the antiapoptotic effect induced by SOCE is one of the mechanisms leading to imatinib resistance. Apoptosis has been widely investigated in GIST and is closely related to tumor progression. Wang et al29 found a significant negative correlation between apoptosis and the degree of GIST differentiation. Moreover, apoptosis is also involved in imatinib response. Liu et al30 report that GIST cells survived imatinib treatment by escaping from apoptosis. Another study reports that apoptosis induced by miR‐518a‐5p affected the cellular response to the drug, causing imatinib resistance in GIST.31 We have also found previously that STIM1‐mediated SOCE contributes to apoptosis via phosphorylation of the MEK/ERK pathway, which is a classical signaling pathway that participates in cell growth, EMT and apotosis.32 STIM1‐knockdown cells in the present study had lower levels of phosphorylated MEK and ERK than that of control cells, whereas total protein levels were unaffected. Thus, the results suggest that the MEK/ERK pathway might be involved in the apoptosis of acquired imatinib‐resistant GIST.
In summary, to determine a strategy to fight imatinib‐resistant GIST, many aspects still need to be explicated and understood despite the achievements researchers have already made. Our research improves the understanding of how STIM1‐mediated SOCE is involved in acquired imatinib‐resistant GIST and expounds the biological functions of STIM1. Future studies on the effects of SOCE inhibitors or STIM1‐targeted drugs will be of further assistance for the development of clinical therapies.
CONFLICT OF INTEREST
The authors declare no conflict of interest for this article.
Supporting information
{kind=link}
ACKNOWLEDGMENTS
This work was funded by the Shanghai Sailing Program (16YF1407100), The Key Project of Science and Technology of Shanghai (No. 13JC1401202) and The Shanghai Key Laboratory of Biliary Tract Disease Research Foundation (17DZ2260200).
Yang Z, Pan L, Liu S, et al. Inhibition of stromal‐interacting molecule 1‐mediated store‐operated Ca2+ entry as a novel strategy for the treatment of acquired imatinib‐resistant gastrointestinal stromal tumors. Cancer Sci. 2018;109:2792–2800. 10.1111/cas.13718
Yang and Pan contributed equally to this work.
Contributor Information
Yijun Shu, Email: shuyijun@xinhuamed.com.cn.
Ping Dong, Email: dongping@xinhuamed.com.cn.
REFERENCES
- 1. Nishida T, Goto O, Raut CP, Yahagi N. Diagnostic and treatment strategy for small gastrointestinal stromal tumors. Cancer. 2016;122:3110‐3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abraham SC, Krasinskas AM, Hofstetter WL, Swisher SG, Wu TT. “Seedling” mesenchymal tumors (gastrointestinal stromal tumors and leiomyomas) are common incidental tumors of the esophagogastric junction. Am J Surg Pathol. 2007;31:1629‐1635. [DOI] [PubMed] [Google Scholar]
- 3. Agaimy A, Wunsch PH, Hofstaedter F, et al. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c‐KIT mutations. Am J Surg Pathol. 2007;31:113‐120. [DOI] [PubMed] [Google Scholar]
- 4. Hirota S, Isozaki K, Moriyama Y, et al. Gain‐of‐function mutations of c‐kit in human gastrointestinal stromal tumors. Science. 1998;279:577‐580. [DOI] [PubMed] [Google Scholar]
- 5. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708‐710. [DOI] [PubMed] [Google Scholar]
- 6. Koo DH, Ryu MH, Kim KM, et al. Asian consensus guidelines for the diagnosis and management of gastrointestinal stromal tumor. Cancer Res Treat. 2016;48:1155‐1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. von Mehren M, Randall RL, Benjamin RS, et al. Soft tissue sarcoma, Version 2.2016, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2016;14:758‐786. [DOI] [PubMed] [Google Scholar]
- 8. ESMO . Gastrointestinal stromal tumours: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol. 2014;25(Suppl 3):i21‐i26. [DOI] [PubMed] [Google Scholar]
- 9. Kee D, Zalcberg JR. Current and emerging strategies for the management of imatinib‐refractory advanced gastrointestinal stromal tumors. Ther Adv Med Oncol. 2012;4:255‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Min KW, Leabu M. Interstitial cells of Cajal (ICC) and gastrointestinal stromal tumor (GIST): facts, speculations, and myths. J Cell Mol Med. 2006;10:995‐1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhu MH, Sung TS, O'Driscoll K, Koh SD, Sanders KM. Intracellular Ca(2 + ) release from endoplasmic reticulum regulates slow wave currents and pacemaker activity of interstitial cells of Cajal. Am J Physiol Cell Physiol. 2015;308:C608‐C620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Prevarskaya N, Skryma R, Shuba Y. Calcium in tumour metastasis: new roles for known actors. Nat Rev Cancer. 2011;11:609‐618. [DOI] [PubMed] [Google Scholar]
- 13. Liou J, Kim ML, Heo WD, et al. STIM is a Ca2 + sensor essential for Ca2 + ‐store‐depletion‐triggered Ca2 + influx. Curr Biol. 2005;15:1235‐1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Feng M, Grice DM, Faddy HM, et al. Store‐independent activation of Orai1 by SPCA2 in mammary tumors. Cell. 2010;143:84‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang S, Zhang JJ, Huang XY. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009;15:124‐134. [DOI] [PubMed] [Google Scholar]
- 16. Yang N, Tang Y, Wang F, et al. Blockade of store‐operated Ca(2 + ) entry inhibits hepatocarcinoma cell migration and invasion by regulating focal adhesion turnover. Cancer Lett. 2013;330:163‐169. [DOI] [PubMed] [Google Scholar]
- 17. Wei J, Zhang J, Si Y, et al. Blockage of LMP1‐modulated store‐operated Ca(2 + ) entry reduces metastatic potential in nasopharyngeal carcinoma cell. Cancer Lett. 2015;360:234‐244. [DOI] [PubMed] [Google Scholar]
- 18. Schmidt S, Liu G, Liu G, et al. Enhanced Orai1 and STIM1 expression as well as store operated Ca2 + entry in therapy resistant ovary carcinoma cells. Oncotarget. 2014;5:4799‐4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shu YJ, Bao RF, Jiang L, et al. MicroRNA‐29c‐5p suppresses gallbladder carcinoma progression by directly targeting CPEB4 and inhibiting the MAPK pathway. Cell Death Differ. 2017;24:445‐457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tamborini E. Mechanism of resistance in gastrointestinal stromal tumors. Handbook of experimental pharmacology. Berlin, Germany: Springer; 2017;3. [DOI] [PubMed] [Google Scholar]
- 21. Gramza AW, Corless CL, Heinrich MC. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin Cancer Res. 2009;15:7510‐7518. [DOI] [PubMed] [Google Scholar]
- 22. Antonescu CR, Besmer P, Guo T, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11:4182‐4190. [DOI] [PubMed] [Google Scholar]
- 23. Wardelmann E, Merkelbach‐Bruse S, Pauls K, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res. 2006;12:1743‐1749. [DOI] [PubMed] [Google Scholar]
- 24. Clapham DE. Calcium signaling. Cell. 2007;131:1047‐1058. [DOI] [PubMed] [Google Scholar]
- 25. Hou MF, Kuo HC, Li JH, et al. Orai1/CRACM1 overexpression suppresses cell proliferation via attenuation of the store‐operated calcium influx‐mediated signalling pathway in A549 lung cancer cells. Biochim Biophys Acta. 2011;1810:1278‐1284. [DOI] [PubMed] [Google Scholar]
- 26. Chen YF, Chiu WT, Chen YT, et al. Calcium store sensor stromal‐interaction molecule 1‐dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc Natl Acad Sci USA. 2011;108:15225‐15230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu Y, Zhang S, Niu H, et al. STIM1 accelerates cell senescence in a remodeled microenvironment but enhances the epithelial‐to‐mesenchymal transition in prostate cancer. Sci Rep. 2015;5:11754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhou Y, Gu P, Li J, et al. Suppression of STIM1 inhibits the migration and invasion of human prostate cancer cells and is associated with PI3K/Akt signaling inactivation. Oncol Rep. 2017;38:2629‐2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang Q, Kou YW. Study of the expressions of p53 and bcl‐2 genes, the telomerase activity and apoptosis in GIST patients. World J Gastroenterol. 2007;13:2626‐2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu Y, Perdreau SA, Chatterjee P, Wang L, Kuan SF, Duensing A. Imatinib mesylate induces quiescence in gastrointestinal stromal tumor cells through the CDH1‐SKP2‐p27Kip1 signaling axis. Cancer Res. 2008;68:9015‐9023. [DOI] [PubMed] [Google Scholar]
- 31. Shi Y, Gao X, Hu Q, et al. PIK3C2A is a gene‐specific target of microRNA‐518a‐5p in imatinib mesylate‐resistant gastrointestinal stromal tumor. Lab Invest. 2016;96:652‐660. [DOI] [PubMed] [Google Scholar]
- 32. Wang ZC, Gao Q, Shi JY, et al. Protein tyrosine phosphatase receptor S acts as a metastatic suppressor in hepatocellular carcinoma by control of epithermal growth factor receptor‐induced epithelial‐mesenchymal transition. Hepatology. 2015;62:1201‐1214. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials