

Abstract
Hereditary leiomyomatosis and renal cell cancer (HLRCC) is a hereditary cancer syndrome characterized by inactivation of the Krebs cycle enzyme fumarate hydratase (FH). HLRCC patients are at high risk of developing kidney cancer of type 2 papillary morphology that is refractory to current radiotherapy, immunotherapy and chemotherapy. Hence, an effective therapy for this deadly form of cancer is urgently needed. Here, we show that FH inactivation (FH −/−) proves synthetic lethal with inducers of ferroptosis, an iron‐dependent and nonapoptotic form of cell death. Specifically, we identified gene signatures for compound sensitivities based on drug responses for 9 different drug classes against the NCI‐60 cell lines. These signatures predicted that ferroptosis inducers would be selectively toxic to FH −/− cell line UOK262. Preferential cell death against UOK262‐FH −/− was confirmed with 4 different ferroptosis inducers. Mechanistically, the FH −/− sensitivity to ferroptosis is attributed to dysfunctional GPX4, the primary cellular defender against ferroptosis. We identified that C93 of GPX4 is readily post‐translationally modified by fumarates that accumulate in conditions of FH −/−, and that C93 modification represses GPX4 activity. Induction of ferroptosis in FH‐inactivated tumors represents an opportunity for synthetic lethality in cancer.
Keywords: ferroptosis, fumarate hydratase, GPX4, hereditary leiomyomatosis and renal cell cancer, synthetic lethal
1. INTRODUCTION
Hereditary leiomyomatosis and renal cell cancer (HLRCC) is a hereditary cancer syndrome characterized by the variable development of uterine fibroids, cutaneous leiomyomas and kidney cancer of type 2 papillary morphology.1 HLRCC‐associated kidney cancers are notably aggressive, have poor prognosis, and are resistant to current radiotherapy, chemotherapy and immunotherapy, prompting an urgent need to identify new treatment avenues.2
Hereditary leiomyomatosis and renal cell cancer is caused by loss‐of‐function mutations to the gene encoding the TCA cycle enzyme, fumarate hydratase (FH). HLRCC patients harbor a germline, inactivating mutation to one of the FH alleles.1 Loss of heterozygosity at the FH locus, which results in the complete loss of FH enzymatic function, is invariably found in the diseased tissues, solidifying FH inactivation as the tumor‐initiating event in HLRCC.
The loss of FH enzymatic function imparts unique molecular changes to the cells. Upon FH inactivation, its substrate, fumarate, accumulates to a high level in the cells. This accumulated fumarate can form adducts on cysteine residues of proteins in a process known as succination.3 Several proteins, including Kelch‐like ECH‐associated protein 1 (KEAP1),4 aconitase 2 (ACO2)5 and iron regulatory protein 2 (IRP2),6 have been reported to be succinated in FH‐inactivated cells, and the succination events have been implicated in altered cellular signaling. For example, succination of KEAP1 allows for the accumulation and activation of the nuclear factor (erythroid‐2)‐like 2 (NRF2) transcription factor, which orchestrates the dominant cellular transcriptional programming observed in HLRCC cells.4, 7 More recently, we showed that NRF2 activation and IRP2 succination increase cellular ferritin level in a concerted manner, and the ferritin drives HLRCC cells’ proliferation.6 Despite the unique and distinguishing biology driven by the FH loss, a strategy to specifically target HLRCC cells has yet to come to fruition. We reasoned that the unique transcriptional changes induced by the FH loss would enable us to identify targetable vulnerabilities through bioinformatics approaches. We used the k‐Top Scoring Pair (k‐TSP) algorithm on previously published NCI‐60 mechanism of action‐based drug screening data to develop gene expression identifiers that could predict sensitivity against 9 classes of drugs. Using these identifiers, we identified and validated that drugs capable of inducing ferroptosis, an iron‐dependent and nonapoptotic form of cell death, are synthetic lethal with FH inactivation. We went on to elucidate the mechanism behind the synthetic lethality.
2. MATERIALS AND METHODS
2.1. Chemicals and reagents
Erastin (Selleck, Houston, TX, USA), RSL3 (Cayman Chemicals, Ann Arbor, MI, USA), ML162 (Cayman Chemicals), dimethyl fumarate (DMF; Santa Cruz Biotechnology, Dallas, TX, USA), Tert‐butyl Hydroperoxide (TBHP; Santa Cruz), 5,5′‐dithiobis‐(2‐nitrobenzoic acid) (DTNB; Santa Cruz), crystal violet (Santa Cruz), monosodium glutamate (TCI, Portland, OR, USA), reduced glutathione (GSH; Millipore‐Sigma, St. Louis, MO, USA) and deferoxamine (DFO, Millipore‐Sigma) were purchased from the indicated companies.
2.2. Cell culture
UOK262 cells were a generous gift of Dr Marston Linehan (National Cancer Institute, NIH, Bethesda, MD, USA). All cells were cultured in RPMI1640 supplemented with 10% heat‐inactivated FBS at 37°C in atmospheric air supplemented with 5% CO2. Lentivirus packaging was conducted in DMEM media with 1 mmol/L sodium pyruvate to improve virus yield. Transfections were performed using Attractene (Qiagen, Hildebrand, Germany). UOK262‐FH res and UOK262‐EV were generated previously.6
2.3. Generation of HK2‐FH−/+
HK2‐FH −/+ cells were created using the CRISPR/Cas9 system with single guide RNA (sgRNA) (5′‐CACCGGGAGGCACTGCTGTTGGTAC‐3′ and 5′‐CACCGGAGCTCATAGATTCTTGGCA‐3′).8 Cells were transfected without a homology‐directed repair arm. Edited cells were screened by Sanger sequencing to identify cells harboring indel mutations in one of the FH alleles.
2.4. Generation of HK2 fumarate hydratase KO cell lines
CRISPR/Cas9 technology was used to knock out FH in HK2 cells. Two nontargeting sgRNA (Control‐1: 5′‐GTAGCACATGGCGACTCTTA‐3′ and Control‐2: 5′‐GGCTCAACGGACTGTCACGG‐3′) and 2 sgRNA targeting FH (sgFH‐3: 5′‐AATTCTCCCAGACCTGACCG‐3′ and sgFH‐5:5′‐CCAGTCTGCCATACCACGAG‐3′) were cloned into the pL‐CRISPR.EFS.GFP, a generous gift from Benjamin Ebert (Addgene #57818).9 The resulting virus particles were used to transduce HK2‐FH −/+ to generate control and FH −/− HK2 cells.
2.5. Generation of HT1080‐FH −/−
CRISPR/Cas9 technology was used to knock out FH in HT1080 cells with sgRNA 5′‐CACCGGGTATCATATTCTATCCGGA‐3′. A homology‐directed repair (HDR) arm was generated to allow for the insertion of a puromycin selection cassette into the editing locus. Puromycin‐resistant clones were screened for FH knockout by immunoblot.
2.6. Dose‐response viability assays
Cell viability following treatment was measured using the CellTiter 96 AQueous One Solution assay (Promega, Madison, WI, USA) at 72 hours post‐treatment. Dose‐response analyses were performed using the nonlinear regression model implemented in the DRC package in the R statistical environment.10, 11, 12 Statistical significance difference between testing groups was assessed by ANOVA test. All curve comparisons were considered significant unless otherwise noted in the figure legend.
2.7. Crystal violet staining
For crystal violet staining, cells were fixed in 4% paraformaldehyde at 72 hours post‐treatment and then stained with crystal violet solution (0.5% w/v crystal violet in 20% v/v methanol).
2.8. Immunoblotting
Primary antibodies used for immunoblotting were as follows: ACTB (Millipore‐Sigma A1978), FH (Cell Signaling 4567, Danvers, MA, USA), Flag (Cell Signaling 8146), GPX4 (Abcam 41787, Cambridge, MA, USA), 2‐succinylcysteine (Discovery Antibodies crb2005017, Billingham, UK) and NRF2 (Santa Cruz sc‐13032). ACTB was used as a loading control. Band densitometry was quantified by Image Lab software (Bio‐Rad, Hercules, CA, USA).
2.9. mRNA qPCR analysis
RNA was isolated and prepared for qPCR analyses as described previously. The following TaqMan probes were purchased (Thermo Fisher Scientific, Waltham, MA, USA): ACTB (4352935), FTL (Hs00830226_gH), AKR1B10 (Hs00252524_m1), GCLM (Hs00157694_m1), NQO1 (Hs01045994_m1), SLC7A11 (Hs00921938_m1), TXNRD1 (Hs00917067_m1) and FH (Hs00895618_m1). Results were analyzed using the 2−ΔΔCT method.13
2.10. Isolation and mass spectrometry analysis of GPX4
The open‐reading frame of GPX4 with an in‐frame c‐terminal Flag sequence together with the GPX4 3′UTR of human GPX4 mRNA (NM_002085.4) was cloned into pCDNA3.1(+) (Thermo) to make pCDNA‐GPX4cFlag. This plasmid is used to ectopically express GPX4 in HEK293 cells, which was purified by immunoprecipitation using anti‐FLAG beads (Sigma M8823). The immunoprecipitated GPX4‐FLAG protein was visualized by Coomassie Blue Staining and excised for mass spectrometry analysis.
2.11. Glutathione peroxidase enzyme assay
Lysates or enzymes were incubated with 500 μmol/L GSH and TBHP (purified GPX4: 16 μmol/L, whole cell lysate: 62.5 μmol/L) in 300 mmol/L Tris‐HCl buffer pH 8.0 for 45 minutes. Remaining GSH was measured by quenching the enzymatic reaction with 3 volumes of 2 mmol/L DTNB and measuring absorbance at 412 nm. The average amount of GSH used over time was calculated from 3 biological replicates.
2.12. Fumarate measurements
Fumarate concentrations were measured using a fumarate assay kit (ab102516, Abcam).
2.13. Reduced glutathione measurements
Reduced GSH levels were measured using the GSH‐Glo kit (V6911, Promega).
2.14. Determining sensitive and resistant cell lines by mechanisms of action
Analyses were performed using R statistical programming language. Antitumor compounds with known mechanisms of action were previously tested against the NCI‐60 cancer cell line for relative sensitivity or resistance.14 Raw data were retrieved from Shimada et al14 Only compounds with known mechanisms of action were used in downstream analyses. Only mechanisms of action with more than 1 compound in the class were evaluated. Within each mechanism of action, Euclidean distances between each drug were determined. Mean distances were calculated for each drug. An optimal cutoff was used to remove drugs that elicited atypical responses compared with others in their mechanism of action. Ward clustering of drugs was performed according to a clustering algorithm proposed by Murtagh and Legendre.15 Drugs with the same mechanism of action that clustered together were defined as a drug class.
In the available dataset, median‐subtracted −log10(GI50) values indicated that a cell line was sensitive to a drug, while positive values indicated resistance. Within each drug class, a cell line was determined to be sensitive if it was sensitive to at least 95% of the drugs in the class. A cell line was determined to be resistant if it was not sensitive to any drugs within the class. Drug classes that did not have any cell lines determined to be either sensitive or resistant were removed from the analysis.
2.15. Identification of drug sensitivity signatures
Affymetrix microarray analyses (platform Human Genome U133 Plus 2.0) of the NCI‐60 cancer cell lines were retrieved from the National Cancer Institute's CellMiner database.16, 17 Gene expression profile for the UOK262 cell line was performed on the same platform, and the data normalization and transformation were done together with the NCI‐60 data in a single batch. Briefly, raw CEL files were processed using a robust multichip‐averaging algorithm implemented in the Affy package. A custom CDF for Affymetrix Human Genome U133 Plus 2.0 (hgu133plus2hsentrezgcdf) was used. This CDF implement selected 20 056 high‐confident probes. Quality control was implemented by removing 1 outlier replicate for each cell line, with outliers defined as replicates with the maximum standard deviation from mean of that particular cell line.
Following the quality control filtering, gene expression classifiers to identify sensitivity or resistance to each drug class were predicted using the k‐Top Scoring Pairs algorithm.18, 19 Following identification of the gene pair signature, UOK262 microarray data were compared against NCI‐60 microarray data for the defined sensitive and resistance cell lines.
3. RESULTS
3.1. Ferroptosis‐inducers may target hereditary leiomyomatosis and renal cell cancer
To find therapeutic vulnerabilities associated with HLRCC, we utilized a legacy dataset consisting of median‐subtracted −log10(GI50) data of 156 chemotherapeutic compounds tested against the NCI‐60 cell lines.14 The mechanisms of action (MOA) of these 156 compounds are known. Through cluster refinement, we grouped them into 9 different MOA (Figure 1A, Supplementary Table S1), and then identified the NCI‐60 member cell lines that were sensitive or resistant to each MOA (Supplementary Table S2). Guided by the identified sensitive and resistant cell lines for each MOA, we performed machine‐learning using the k‐tsp algorithm on the gene expression microarray data for the corresponding NCI‐60 cell lines to identify classifiers to predict sensitivity for each of the 9 MOA. Using the identified classifiers, we predicted that HLRCC cells would be sensitive only to ferroptosis inducers (FIN) (Figure 1B). HLRCC cells were predicted to be minimally responsive (eg, Ds), nonresponsive (eg, YK) or resistant (eg, HDAC) to all other mechanisms of action tested (Figure 1B), which is consistent with previous drug screening studies.20
Figure 1.
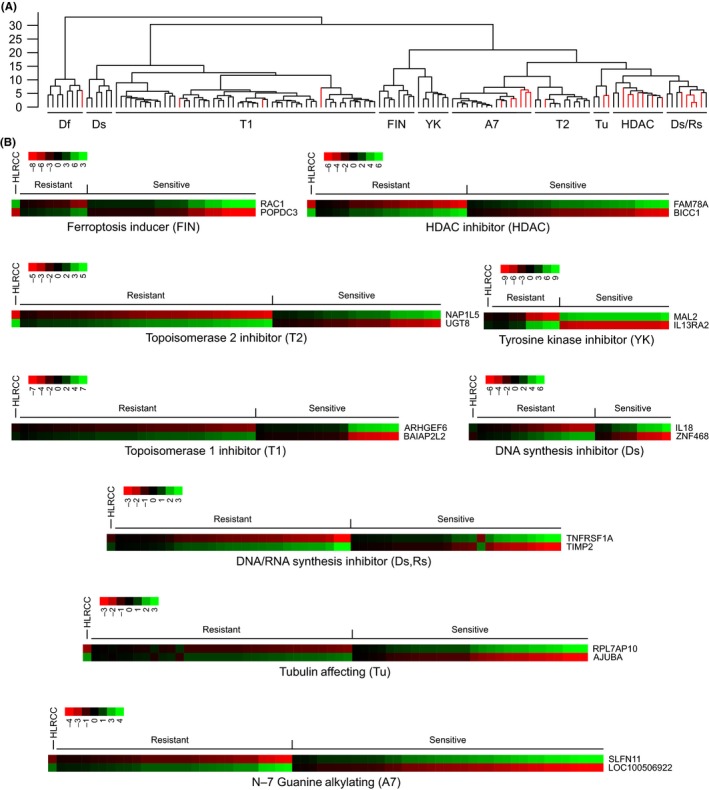
Hereditary leiomyomatosis and renal cell cancer (HLRCC) cells are predicted to be sensitive to ferroptosis inducers. A, Ward clustering analysis of different drugs reveals that most drugs within a cluster have the same known mechanism of action. Clustering was based on relative sensitivity or resistance of the NCI‐60 cancer cell lines to various compounds with known mechanisms of action. Drugs within a cluster not conforming to the dominant mechanism of action (red) were removed from later analyses. B, Gene expression signature of NCI‐60 cancer cell lines that are sensitive or resistant to various drug classes. Cell lines that were sensitive or resistant to a drug class were utilized in a k‐TSP prediction algorithm to identify a 2‐gene classifier that predicts sensitivity. Each box represents a gene expression of a tested cell line determined to be sensitive or resistant to a particular drug class. Red (green) indicates low (high) gene expression for the k‐TSP predicted gene. Gene expression levels of UOK262, an HLRCC cell line, of the k‐TSP predictor genes for each drug class are also presented. The predictions indicate that HLRCC‐derived cell line UOK262 will only be sensitive to ferroptosis inducers (FIN)
3.2. Ferroptosis induction can selectively kill hereditary leiomyomatosis and renal cell cancer cells
Ferroptosis is a nonapoptotic, iron‐dependent cell death originally identified as the mechanism by which the RAS synthetic lethal compound, erastin, selectively killed the RAS‐activated, fibrosarcoma HT1080 cell line.21 To validate the predicted sensitivity of HLRCC cells to ferroptosis, we compared the erastin LC50 value of the HLRCC cell line UOK262 to that of the well‐established erastin‐sensitive HT1080 cell line and found UOK262 to be more sensitive (Figure 2A, Table 1). Next, we determined whether erastin was inducing iron‐dependent, ferroptotic cell death in UOK262 by cotreating the cells with erastin and 100 μmol/L deferoxamine (DFO), an iron chelator. As expected, DFO abrogated the erastin‐induced cell death, indicating that erastin engenders ferroptosis in UOK262 cells (Figure 2B).
Figure 2.
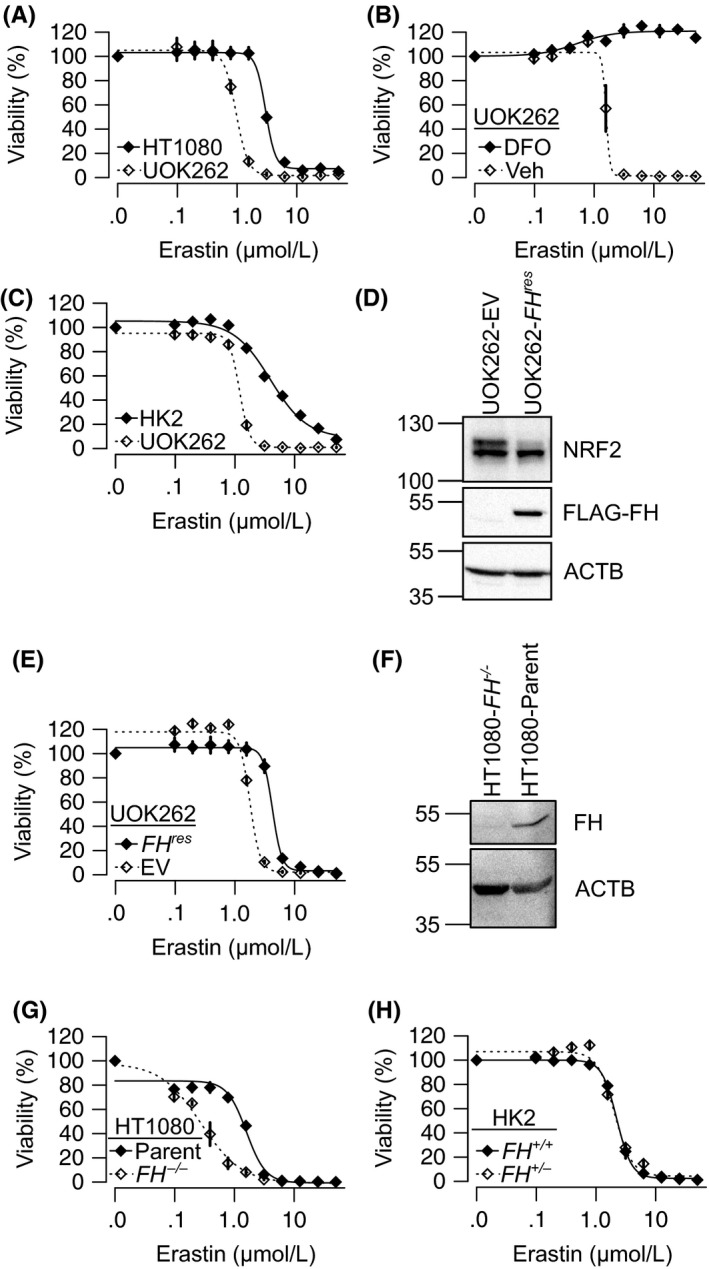
FH inactivation sensitizes cells to erastin‐induced cell death. Erastin dose‐response curves of: (A) canonically ferroptosis‐sensitive HT1080 cell line and HLRCC cell line, UOK262, (B) UOK262 and immortalized nonmalignant kidney epithelial cells HK2, (C) UOK262 with 100 μmol/L deferoxamine (DFO) or vehicle (Veh). D, Immunoblot of UOK262 stably transduced with either empty vector (UOK262‐EV) or flag‐tagged fumarate hydratase (FH) (UOK262‐FH res). E, Ectopic expression of FH (FH res) in UOK262 significantly reduced sensitivity to erastin. F, Immunoblots showing successful FH knockout in HT1080‐FH −/− compared to parental HT1080 (HT1080‐parent). G, Erastin dose‐response curves of HT1080‐FH −/− and HT1080‐parent reveal loss of FH significantly sensitizes HT1080 to erastin. H, Erastin dose‐response curves of HK2‐FH +/+ and HK2‐FH +/− show no difference between FH wild type and heterozygous mutant. ANOVA P = .81. All points on dose‐response curves are presented as mean ± standard deviations of a representative experiment
Table 1.
LC50 values of cell lines to various ferroptosis‐inducing treatments (FIN)
| Cell line/Treatment | FIN | LC50 ± SD (μmol/L) |
|---|---|---|
| UOK262parental | Erastin | 1.19 ± 0.04 |
| UOK262parental + 40 μmol/L DMF | Erastin | 0.42 ± 0.02 |
| HK2 | Erastin | 3.92 ± 0.57 |
| HK2 + 40 DMF | Erastin | 2.60 ± 0.11 |
| HT1080 | Erastin | 3.00 ± 0.06 |
| HT1080‐FH −/− | Erastin | 0.27 ± 0.03 |
| A498 | Erastin | 2.48 ± 0.17 |
| A498 + 40 DMF | Erastin | 1.12 ± 0.24 |
| UOK262‐FH −/− | Erastin | 1.78 ± 0.15 |
| UOK262‐FH res | Erastin | 4.25 ± 0.15 |
| HK2‐FH +/+ | Erastin (DMEM media) | 2.23 ± 0.03 |
| HK2‐FH +/− | Erastin (DMEM media) | 2.06 ± 0.15 |
| UOK262‐FH −/− | RSL3 | 28.68 ± 1.43 nmol/L |
| UOK262‐FH res | RSL3 | 50.36 ± 6.14 nmol/L |
| UOK262‐FH −/− | ML162 | 35.20 ± 1.00 nmol/L |
| UOK262‐FH res | ML162 | 59.62 ± 4.29 nmol/L |
| UOK262‐FH −/− | Glutamate | 1.98 ± 0.70 mmol/L |
| UOK262‐FH res | Glutamate | 80.03 ± 3.03 mmol/L |
| HT1080 | Glutamate | 43.90 ± 3.15 mmol/L |
| HT1080‐FH −/− | Glutamate | 16.25 ± 2.07 mmol/L |
To evaluate the potential cancer cell selectivity of the erastin‐induced cell death, we compared the effectiveness of erastin in killing UOK262 cells to that of an immortalized, nonmalignant kidney epithelia cell line, HK2. Again, we found that UOK262 was more sensitive, indicating that HLRCC cells could be preferentially more sensitive to ferroptosis than normal kidney cells (Figure 2C, Table 1).
3.3. Fumarate hydratase inactivation sensitizes hereditary leiomyomatosis and renal cell cancer cells to ferroptosis
Hereditary leiomyomatosis and renal cell cancer is caused by FH inactivation, which results in intracellular fumarate accumulation and increased protein succination. Thus, we hypothesized that FH inactivation sensitizes HLRCC cells to ferroptosis. To test this hypothesis, we generated isogenic FH‐rescued (UOK262‐FH res) and empty vector control (UOK262‐EV) derivative UOK262 lines. UOK262‐FH res stably expresses a fully functional, flag‐tagged version of FH. As expected, the ectopic expression of FH reduces NRF2 protein levels, indicating that it lowered intracellular fumarate levels and reduced protein succination (Figure 2D). Indeed, qPCR analysis showed mRNA levels of NRF2 target genes decreased following FH reintroduction (Supplementary Figure S1A). We found that UOK262‐EV is more sensitive to erastin than its FH‐rescued counterpart UOK262‐FH res (Figure 2E, Table 1), supporting our hypothesis that FH inactivation is responsible for ferroptosis sensitivity in HLRCC. To further test our hypothesis, we generated an FH knockout derivative cell line of HT1080 to see if we could further sensitize the cell line to ferroptosis (Figure 2F). As expected, HT1080‐FH −/− cells were more sensitive than HT1080‐parental cells to erastin treatment (Figure 2G).
Hereditary leiomyomatosis and renal cell cancer patients harbor a germline inactivating mutation to an FH allele in all healthy cells in their bodies. Therefore, for a treatment modality to be truly selective in HLRCC patients, it must distinguish between homozygous and heterozygous FH mutant cells. To test this, we generated a heterozygous FH mutant HK2 cell line (HK2‐FH +/−). This cell line harbors an inactivating frameshift mutation in exon 6 of FH (Supplementary Figure S1B). Dose‐response studies showed that the HK2‐FH +/− was no more sensitive to erastin than HK2‐FH +/+, indicating that erastin is selectively killing the FH −/− HLRCC cells (Figure 2H). Furthermore, CRISPR/Cas9‐mediated complete ablation of FH (using 2 independent FH targeting sgRNAs) in HK2‐FH +/− sensitized it to ferroptosis induction (Supplementary Figure S1C, D).
3.4. Fumarate hydratase−/− sensitizes cells to multiple ferroptosis inducers
Mechanistic studies on ferroptosis‐inducing compounds have identified GPX4 inhibition as the converging mechanism of action for those compounds.22 GPX4 is an essential selenocysteine‐containing enzyme responsible for clearing cellular lipid peroxides generated from the iron‐catalyzed fenton reaction.23 GPX4 utilizes glutathione as a coenzyme; thus, ferroptosis inducers can inhibiting GPX4 either by limiting intracellular glutathione availability or by directly binding to and inhibiting GPX4. Erastin induces ferroptosis through inhibition of SLC7A11, the antiporter responsible for import of cystine in exchange for intracellular glutamate.21 Cystine is necessary for glutathione synthesis. While erastin appears to be toxic to HLRCC cells due to glutathione depletion, ferroptosis can be induced by mechanisms beyond SLC7A11 inhibition. RSL3 and ML162 induce ferroptosis through direct GPX4 inhibition.22 Consistently, UOK262‐EV was more sensitive to both RSL3 and ML162 than UOK262‐FH res (Figure 3A‐B).
Figure 3.
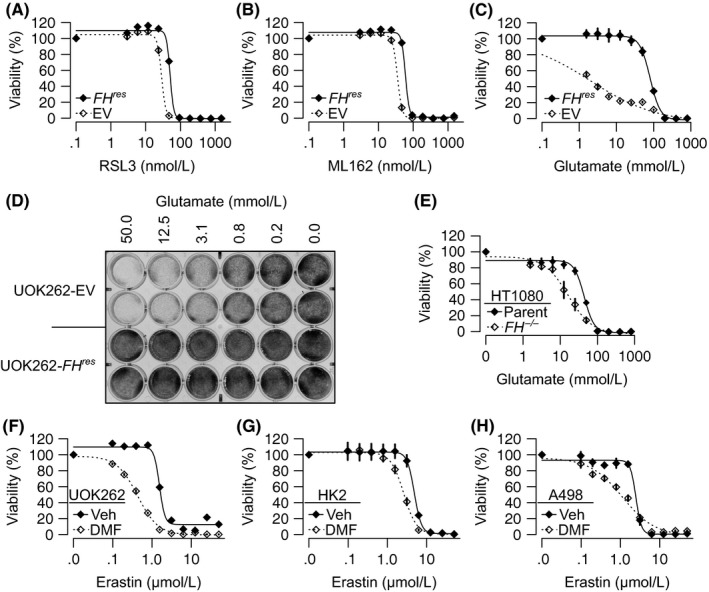
Cells with fumarate accumulation are sensitive to multiple ferroptosis inducers. A‐C, Dose‐response curves of UOK262‐EV and UOK262‐FH res treated with (A) RSL3 (B) ML162 or (C) glutamate showing that UOK262‐EV is significantly more sensitive to all 3 compounds. D, Crystal violet staining of UOK262‐EV and UOK262‐FH res cells following treatment with monosodium glutamate. E, Dose‐response analysis showing HT1080‐FH −/− is significantly more sensitive to glutamate than HT1080‐parent. F‐H, 40 μmol/L dimethyl fumarate (DMF) significantly enhances erastin activity in UOK262 (F), HK2 (G) and A498 cells (H). All points on dose‐response curves are presented as mean ± SD of a representative experiment
One further mechanism to induce ferroptosis exploits the glutamate‐cystine concentration gradients necessary for SLC7A11 activity. Extracellular glutamate can induce ferroptosis by blockading SLC7A11 antiporter activity.21 UOK262‐EV cells were much more sensitive to treatment with monosodium glutamate than UOK262‐FH res (Figure 3C‐D). DFO treatment abrogated glutamate sensitivity in both UOK262‐EV and UOK262‐FH res (Supplementary Figure S2A). HT1080‐FH −/− cells were also more sensitive to glutamate than HT1080‐parental cells (Figure 3E). As glutamate can enter the Krebs cycle, we sought to evaluate whether the glutamate treatment alters fumarate levels. Consistent with previous reports,24 FH inactivation increased fumarate levels in UOK262‐EV relative to UOK262‐FH res and in HT1080‐FH −/− relative to HT1080‐parental. However, glutamate treatment did not significantly alter fumarate levels except in UOK262‐FHres, whereby fumarate levels increased slightly (Supplementary Figure S2B). To validate that glutamate was inducing ferroptosis by reducing GSH levels, we measured GSH following glutamate treatment. Consistent with previous reports on Fh −/− mice,7 reduced glutathione (GSH) levels in FH‐inactivated cells UOK262‐EV and HT1080‐FH −/− were higher than their counterpart cell lines with functional FH (UOK262‐FH res and HT1080‐parental). As expected, glutamate decreased free glutathione levels across all tested cells (Supplementary Figure S2C).
3.5. Fumarate hydratase inactivation sensitizes cells to ferroptosis through intracellular fumarate accumulation
The main biochemical consequence of FH inactivation is fumarate accumulation. To evaluate whether fumarate can sensitize cells to ferroptosis, we treated UOK262, HK2 and a clear cell kidney cancer cell line, A498, with 40 μmol/L of the membrane‐permeable form of fumarate, dimethyl fumarate (DMF). Across all cell lines, DMF sensitizes cells to erastin‐induced cell death, indicating that FH inactivation sensitizes cells to ferroptosis through intracellular fumarate accumulation (Figures 3F‐H). It is worth noting that while DMF enhances fumarate levels,25 DMF treatment also depletes glutathione, which could be contributing to the enhanced ferroptosis induction in this model system (Supplementary Figure S2D).
3.6. Fumarates covalently modifying C93 of GPX4 and inhibiting its activity
NRF2 could confer protection against ferroptotic cell death through several mechanisms. Importantly, GPX4 is a transcription target of NRF2; thus, sustained NRF2 activation may lead to increased GPX4 expression and, therefore, more resistance to ferroptotic cell death. Paradoxically, NRF2 is constitutively active in HLRCC cells, and UOK262‐EV cells do show higher GPX4 protein levels than UOK262‐FH res cells (Figure 4A). This contradiction led us to hypothesize that GPX4 activity is inhibited in FH‐inactivated cells, and the inhibition is due to succination of GPX4 by fumarate. To assess whether GPX4 is succinated in the presence of fumarate, HEK293 cells were transfected with a vector that ectopically expresses flag‐tagged GPX4. Transfected cells were treated with DMF to mimic fumarate accumulation, and the flag‐tagged GPX4 was isolated for mass spectrometry analysis. Monomethyl‐succinyl and dimethyl‐succinyl adducts were identified on GPX4‐C93, indicating that the C93 of GPX4 is amenable to covalent modification by fumarate (Figure 4B‐C).
Figure 4.
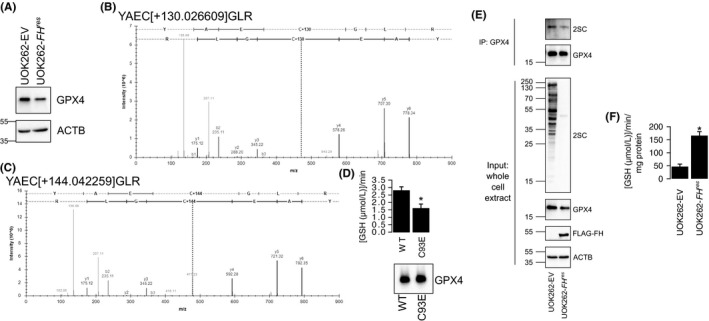
C93 of GPX4 is amenable to succination. A, GPX4 protein levels were higher in UOK262‐EV than in UOK262‐FH res. B‐C, Monomethyl‐succinyl adducts (+130 Da) (B) and dimethyl‐succinyl adducts (+144 Da) (C) were found on C93 of flag‐tagged GPX4 isolated from HEK293 cells treated with 40 μmol/L dimethyl fumarate. D, GPX4‐C93E showed reduced enzymatic activity. Immunoblot of GPX4 indicates equal loading. E, Immunoblot showing GPX4 immunoprecipitated from UOK262‐EV lysates has higher 2‐succinylcysteine (2SC) modification than that from UOK262‐FH res. F, Whole cell lysates from UOK262‐EV showed reduced GPX4 activity than UOK262‐FH res. Bars represent the mean of 3 biological replicates, and error bars represent standard deviation. *Indicates Student's t test P < .05
To assess the impact of fumarate‐mediated modifications to C93 of GPX4, we generated a C93E mutant version of GPX4 that mimics 2‐succinylcysteine. GPX4‐WT and GPX4‐C93E proteins were isolated and utilized in an enzyme assay that measured the ability of GPX4 proteins to reduce tert‐butyl hydroperoxide (TBHP) in the presence of glutathione (Supplementary Figure S3). GPX4‐C93E depleted glutathione slower than GPX4‐WT, suggesting that the succinated form of GPX4 has reduced activity (Figure 4D). To validate succination of native GPX4 in FH −/− HLRCC cells, we immunoprecipitated GPX4 from UOK262‐EV and UOK262‐FH res and immunoblotted for 2‐succinylcysteine. As expected, UOK262‐EV showed increased succinated GPX4 compared with UOK262‐FH res. Consistent with previous works,26 whole cell lysates from UOK262‐EV showed marked increase in pan‐protein succination as compared to UOK262‐FH res (Figure 4E). Enzyme assay (GSH‐dependent reduction of TBHP) also showed that UOK262‐EV whole cell lysates had significantly lower activity than UOK262‐FH res, indicating repressed GPX4 activity (Figure 4F).
4. DISCUSSION
Targeting cancer‐specific vulnerabilities offers the ability to kill cancer cells while sparing normal cells. Such targeted strategies can be seen in compounds that selectively inhibit products produced from gain‐of‐function oncogene mutants that are essential to cancer cell survival. For example, vemurafenib selectively inhibits BRAF‐V600E and offers robust and dramatic responses that prolong patients’ lives.27 In contrast to oncogene gain‐of‐function‐driven tumors, it is much more difficult to selectively target cancers that arise from loss‐of‐function mutations to tumor suppressor genes. One strategy is to target pathways activated by the tumor suppressor loss. However, such pathways are typically essential to other cellular systems, resulting in unwanted collateral damage. HLRCC is unique in the sense that the tumor suppressor (FH) loss drives an expansion of the catalogue of succinated proteins. Thus, targeting the succinated proteins that are otherwise unsuccinated in normal tissues may offer the specificity seen in strategies that target cancer‐specific gain‐of‐function mutations. In this study, we demonstrated that GPX4 succination, a feature specific to FH‐inactivated cancer cells, is targetable using ferroptosis‐inducing compounds. As such, the finding serves as a proof of concept for future drug development.
From a cell signaling standpoint, it becomes apparent that HLRCC cells are primed to die by ferroptosis. HLRCC cells require FH inactivation to be tumorigenic, and the FH inactivation increases cellular protein succination. Consequently, essential proteins, such as GPX4, become succinated and display reduced enzymatic activity. However, the cells do not spontaneously die from ferroptosis because FH inactivation also activates NRF2. NRF2 coordinately reduces the intracellular labile iron pool by increasing expression of ferroportin and ferritin, the proteins which export and store iron, respectively.28 NRF2 also drives the increased expression of GPX4 and glutathione biosynthesis. Thus, NRF2 activation may be critical in countering the effects of the reduced GPX4 activity. This delicate balance between maintaining FH inactivation while keeping the iron‐driven oxidative stress in check is a vulnerability unique to FH‐inactivated cells. Future efforts to develop treatments for this deadly disease may focus on targeting this particular vulnerability. For example, we have previously shown that ferritin knockdown retards cell proliferation.6 Prolonged ferritin knockdown is actually lethal to HLRCC cells (result not shown), potentially through ferroptotic cell death. While ferritin knockdown is harder to achieve in vivo, ferritin could still be modulated through iron chelation. For this purpose, it is important to emphasize that the type of iron chelators useful for reducing ferritin while still sensitizing cells to ferroptotic cell death would be the redox active iron chelators, such as Triapine and Dp44mT.29
The role of glutathione in the interplay between HLRCC and ferroptosis sensitivity remains enigmatic. Our work and others7 have shown that fumarate hydratase inactivation increases free GSH levels, while additional groups have shown that GSH levels remain relatively constant30 or decreased31 following fumarate hydratase inactivation. While we and others have shown that DMF consistently reduces GSH levels,30, 31 different fumarate esters have been shown to differentially modulate GSH levels.32 More in‐depth investigations on the reactions between fumarate species and glutathione, and their roles in ferroptosis induction, are necessary.
It is worth noting that the succinated GPX4 remains active, but with reduced activity. We reason that this incomplete loss of enzymatic activity could also partially explain the modest LC50 difference that we saw with current ferroptosis‐inducing compounds. On the bright side, this apparent‐altered kinetic suggests a binding or catalytic difference between the succinated form and the native form of GPX4, allowing for the development of compounds that specifically target succinated GPX4 and further reduce potential unwanted side effects. Other possibilities to further optimize ferroptosis induction as a means to treat HLRCC include combination therapies with fumarate esters such as dimethyl fumarate; HLRCC cells are unable to metabolize fumarate, and fumarate was shown to enhance sensitivity to ferroptosis.
While our data show that FH inactivation sensitizes cells to ferroptosis for multiple FIN, there remains a therapeutic gap in the relative selectivity of different agents: glutamate engendered a much larger therapeutic window than any of the other compounds. More investigations must focus on identifying ferroptosis inducers to improve potency and selectivity of current agents. Moreover, most of the identified and characterized ferroptosis inducers have poor pharmacokinetic properties that preclude their use in vivo. As others have recently highlighted,33 the development of potent, bioavailable FIN is an urgent priority in cancer development. Ferroptosis induction has shown promise in drug‐resistant cancers beyond HLRCC, such as head and neck cancers and tumor persister cells,33, 34 magnifying the necessity for further development of FIN.
Aside from FIN selectively killing FH‐inactivated cells, other synthetic lethal combinations have been identified for HLRCC. SLC7A11 inhibition by sulfasalazine was synthetic lethal in cells from Fh1 null mice.31 Heme oxygenase inhibition was shown to be synthetic lethal with Fh1 deficiency in mouse cells.35 Intriguingly, heme oxygenase is critically involved in iron homeostasis and has been shown to affect ferroptosis sensitivity.28 Toxicities of reactive‐oxygen species (ROS) inducers, notably the proteasome inhibitor bortezomib, were also shown to inversely correlate with fumarate hydratase activity.36 Ferroptosis induction is also a ROS‐dependent process. Commonalities between these different mechanisms of cell death further indicate that exploration of HLRCC treatment modalities should focus on tipping the precariously balanced ROS and iron homeostasis found in FH‐inactivated tumors toward cell death.
HLRCC is an aggressive and deadly disease that afflicts younger adults. While the incidence of HLRCC is low, its unique biology warrants the development of a curative treatment strategy that may remove it from the list of deadly diseases altogether. This understudied and underappreciated malignancy not only manifests in the form of quickly progressing and fatal renal cell carcinomas but also presents as skin lesions that are disfiguring, painful and unnecessarily detrimental to patients’ quality the potential to develop into of life. A treatment strategy that targets FH‐inactivated cells will enable the development of topical treatments that can eliminate these debilitating leiomyomas. We have shown that FH inactivation and fumarate accumulation sensitize cells to ferroptotic cell death; manipulation of this cell death pathway could yield a synthetic lethal agent that kills HLRCC‐associated renal tumors while sparing nearby healthy tissues. As we continue to unravel this novel cell death modality and identify new FIN to move it into in vivo studies, the unmet clinical needs of HLRCC patients could serve as a valuable ferroptosis proving ground.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
Supporting information
ACKNOWLEDGMENTS
This work was supported in part by DGE‐1143953 from the National Science Foundation (M.J.K.) and R21ES027920 from NIEHS (A.O.).
Kerins MJ, Milligan J, Wohlschlegel JA, Ooi A. Fumarate hydratase inactivation in hereditary leiomyomatosis and renal cell cancer is synthetic lethal with ferroptosis induction. Cancer Sci. 2018;109:2757–2766. 10.1111/cas.13701
Funding information
This work was supported in part by DGE‐1143953 from the National Science Foundation (M.J.K.) and R21ES027920 from NIEHS (A.O.).
REFERENCES
- 1. Tomlinson IP, Alam NA, Rowan AJ, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406‐410. [DOI] [PubMed] [Google Scholar]
- 2. Menko FH, Maher E, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC). Renal cancer risk, surveillance and treatment. Fam Cancer. 2014;13:637‐644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pollard P, Briere J, Alam N, et al. Accumulation of Krebs cycle intermediates and over‐expression of HIF1α in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14:2231‐2239. [DOI] [PubMed] [Google Scholar]
- 4. Ooi A, Wong J‐C, Petillo D, et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell. 2011;20:511‐523. [DOI] [PubMed] [Google Scholar]
- 5. Ternette N, Yang M, Laroyia M, et al. Inhibition of mitochondrial aconitase by succination in fumarate hydratase deficiency. Cell Rep. 2013;3:689‐700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kerins MJ, Vashisht AA, Liang BX‐T, et al. fumarate mediates a chronic proliferative signal in fumarate hydratase‐inactivated cancer cells by increasing transcription and translation of ferritin genes. Mol Cell Biol. 2017;37:e00079‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Adam J, Hatipoglu E, O'Flaherty L, et al. Renal cyst formation in Fh1‐deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011;20:524‐537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR‐Cas9 system. Nat Protoc. 2013;8:2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heckl D, Kowalczyk MS, Yudovich D, et al. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR‐Cas9 genome editing. Nat Biotechnol. 2014;32:941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ritz C, Baty F, Streibig JC, Gerhard D. Dose‐response analysis using R. PLoS One. 2015;10:e0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Team RC. R: A language and environment for statistical computing. In: R Foundation for Statistical Computing V, ed. Vienna, Austria; 2016.
- 12. Ritz C, Streibig JC. Bioassay analysis using R. J Stat Softw. 2005;12:1‐22. [Google Scholar]
- 13. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods (San Diego, Calif). 2001;25:402‐408. [DOI] [PubMed] [Google Scholar]
- 14. Shimada K, Hayano M, Pagano NC, Stockwell BR. Cell‐line selectivity improves the predictive power of pharmacogenomic analyses and helps identify NADPH as biomarker for ferroptosis sensitivity. Cell Chem Biol. 2016;23:225‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Murtagh F, Legendre P. Ward's hierarchical agglomerative clustering method: which algorithms implement Ward's criterion? J Classif. 2014;31:274‐295. [Google Scholar]
- 16. Shankavaram UT, Varma S, Kane D, et al. Cell Miner: a relational database and query tool for the NCI‐60 cancer cell lines. BMC Genom. 2009;10:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reinhold WC, Sunshine M, Liu H, et al. Cell Miner: a web‐based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI‐60 cell line set. Can Res. 2012;72:3499‐3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Damond J. ktspair: k‐Top scoring pairs for microarray classification. R package version 1; 2011.
- 19. Tan AC, Naiman DQ, Xu L, Winslow RL, Geman D. Simple decision rules for classifying human cancers from gene expression profiles. Bioinformatics. 2005;21:3896‐3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Perrier‐Trudova V, Huimin BW, Kongpetch S, et al. Fumarate hydratase‐deficient cell line NCCFH1 as a new in vitro model of hereditary papillary renal cell carcinoma Type 2. Anticancer Res. 2015;35:6639‐6653. [PubMed] [Google Scholar]
- 21. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell. 2012;149:1060‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Imai H, Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic Biol Med. 2003;34:145‐169. [DOI] [PubMed] [Google Scholar]
- 24. Ashrafian H, O'Flaherty L, Adam J, et al. Expression profiling in progressive stages of fumarate‐hydratase deficiency: the contribution of metabolic changes to tumorigenesis. Cancer Res. 2010;70:9153‐9165. [DOI] [PubMed] [Google Scholar]
- 25. Huang H, Taraboletti A, Shriver LP. Dimethyl fumarate modulates antioxidant and lipid metabolism in oligodendrocytes. Redox Biol. 2015;5:169‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bardella C, El‐Bahrawy M, Frizzell N, et al. Aberrant succination of proteins in fumarate hydratase‐deficient mice and HLRCC patients is a robust biomarker of mutation status. J Pathol. 2011;225:4‐11. [DOI] [PubMed] [Google Scholar]
- 27. Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507‐2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kerins MJ, Ooi A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxid Redox Signal. 2017. https://www.liebertpub.com/doi/abs/10.1089/ars.2017.7176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yu Y, Wong J, Lovejoy DB, Kalinowski DS, Richardson DR. Chelators at the cancer coalface: desferrioxamine to Triapine and beyond. Clin Cancer Res. 2006;12:6876‐6883. [DOI] [PubMed] [Google Scholar]
- 30. Sullivan LB, Martinez‐Garcia E, Nguyen H, et al. The proto‐oncometabolite fumarate binds glutathione to amplify ROS‐dependent signaling. Mol Cell. 2013;51:236‐248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zheng L, Cardaci S, Jerby L, et al. Fumarate induces redox‐dependent senescence by modifying glutathione metabolism. Nat Commun. 2015;6:6001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brennan MS, Matos MF, Li B, et al. Dimethyl fumarate and monoethyl fumarate exhibit differential effects on KEAP1, NRF2 activation, and glutathione depletion in vitro . PLoS One. 2015;10:e0120254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hangauer MJ, Viswanathan VS, Ryan MJ, et al. Drug‐tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551:247‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sun X, Ou Z, Chen R, et al. Activation of the p62‐Keap1‐NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Frezza C, Zheng L, Folger O, et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature. 2011;477:225‐228. [DOI] [PubMed] [Google Scholar]
- 36. Sourbier C, Valera‐Romero V, Giubellino A, et al. Increasing reactive oxygen species as a therapeutic approach to treat hereditary leiomyomatosis and renal cell carcinoma. Cell Cycle. 2010;9:4183‐4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials