

Abstract
Epidermal growth factor receptor (EGFR)‐activating mutations confer sensitivity to tyrosine kinase inhibitor (TKI) treatment for non‐small‐cell lung cancer (NSCLC). ASP8273 is a highly specific, irreversible, once‐daily, oral, EGFR TKI that inhibits both activating and resistance mutations. This ASP8273 dose‐escalation/dose‐expansion study (NCT02192697) was undertaken in two phases. In phase I, Japanese patients (aged ≥20 years) with NSCLC previously treated with ≥1 EGFR TKI received escalating ASP8273 doses (25‐600 mg) to assess safety/tolerability and to determine the maximum tolerated dose (MTD) and/or the recommended phase II dose (RP2D) by the Bayesian Continual Reassessment Method. In phase II, adult patients with T790M‐positive NSCLC in Japan, Korea, and Taiwan received ASP8273 at RP2D to further assess safety/tolerability and determine antitumor activity, which was evaluated according to Simon's two‐stage design (threshold response = 30%, expected response = 50%, α = 0.05, β = 0.1). Overall, 121 (n = 45 [33W/12M] phase I, n = 76 [48W/28M]) phase 2) patients received ≥1 dose of ASP8273. In phase I, RP2D and MTD were established as 300 and 400 mg, respectively. As 27 of the 63 patients treated with ASP8273 300 mg achieved a clinical response, ASP8273 was determined to have antitumor activity. The overall response rate at week 24 in all patients was 42% (n = 32/76; 95% confidence interval, 30.9‐54.0). Median duration of progression‐free survival was 8.1 months (95% confidence interval, 5.6, upper bound not reached). The most commonly reported treatment‐related adverse event in phase II was diarrhea (57%, n = 43/76). ASP8273 300 mg was generally well tolerated and showed antitumor activity in Asian patients with both EGFR‐activating and T790M mutations.
Keywords: clinical trial, epidermal growth factor receptor, non‐small‐cell carcinoma, signal transduction inhibitors/kinase inhibitor, tyrosine kinase inhibitor
Abbreviations
- AE
adverse event
- ALT
alanine transaminase
- AST
aspartate aminotransferase
- AUC
area under the plasma concentration‐time curve
- CI
confidence interval
- CR
complete response
- CTCAE
Common Terminology Criteria for Adverse Events
- DCR
disease control rate
- DLT
dose‐limiting toxicity
- EGFR
epidermal growth factor receptor
- ex19del
exon 19 deletion
- MTD
maximum tolerated dose
- NCSLC
non‐small‐cell lung cancer
- ORR
overall response rate
- PD
progressive disease
- PFS
progression‐free survival
- PR
partial response
- RP2D
recommended phase II dose
- SD
stable disease
- TEAE
treatment‐emergent adverse event
- TKI
tyrosine kinase inhibitor
- TRAE
treatment‐related adverse event
1. INTRODUCTION
The presence of EGFR‐activating mutations in patients with NSCLC can result in increased malignant cell survival, proliferation, invasion, metastatic spread, and tumor angiogenesis.1, 2 These mutations are estimated to be present in approximately 50% of patients with NSCLC in East Asian countries.3 Exon 19 deletions and exon 21 L858R substitutions are the most common EGFR mutations.1, 4 These mutations confer sensitivity to TKIs and account for approximately 90% of EGFR mutations in patients with NSCLC.5
Patients with NSCLC with EGFR‐activating mutations have experienced antitumor activity and prolonged PFS following treatment with the reversible EGFR TKIs such as gefitinib and erlotinib.6, 7 However, this clinical efficacy is often limited by an acquired drug resistance, most commonly caused by a point mutation (T790M) in the gene encoding EGFR. Approximately 50%‐60% of patients treated with TKIs develop T790M‐mediated resistance, suggesting that, along with activating mutations, the T790M mutation is an important factor in determining the appropriate treatment strategy in these patients.8, 9
ASP8273 is an oral, irreversible EGFR TKI that inhibits the kinase activity of EGFR containing the ex19del‐ or L858R‐activating mutation and the T790M resistance mutation with higher potency than WT EGFR. Based on preclinical activity, ASP8273 was evaluated in a phase I/II study in patients with EGFR‐mutant lung cancer in Japan.
The primary objectives for phase I of this study were to assess safety/tolerability of ASP8273 as well as to determine the MTD and/or the RP2D based on the DLT profile. Secondary objectives were to determine the pharmacokinetics and antitumor activity of ASP8273. In phase II, the primary objective was to determine the antitumor activity of ASP8273; secondary objectives were to determine the safety and pharmacokinetics of ASP8273. Here, we report the results from study initiation date, January 2014, until the cut‐off date, 15 January 2016.
2. MATERIALS AND METHODS
2.1. Study design and treatment
This dose‐escalation/dose‐expansion study (NCT02192697) was undertaken in two phases. Phase I, consisting of a dose‐escalation cohort, an additional T790M cohort, and a re‐enrollment cohort, was undertaken in four centers in Japan and phase II was held in 15 centers across Japan, Taiwan, and Korea (Figure 1). Eligible patients with NSCLC were aged ≥20 years, had given written informed consent, had an ECOG performance status ≤1, had a histologically or cytologically confirmed diagnosis of NSCLC, were confirmed to have the ex19del, L858R, G719X, or L861Q mutation among the EGFR‐activating mutations, and had a life expectancy ≥12 weeks based on investigator's judgment. Eligible patients also met all of the following requirements for laboratory tests within 7 days before enrollment: neutrophil count ≥1500/mm3, platelet count ≥75 000/mm3, hemoglobin ≥9 g/dL, serum creatinine <1.5 mg/dL, total bilirubin <1.5× the upper limit of normal (this did not apply to patients with Gilbert's syndrome), and AST and ALT <2.5× the upper limit of normal.
Figure 1.
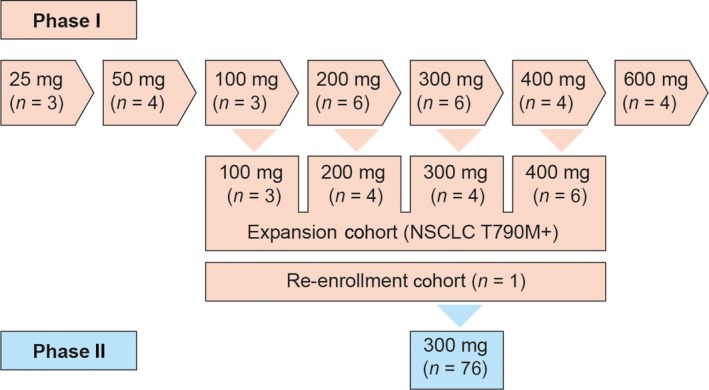
Study design involving patients with non‐small‐cell lung cancer (NSCLC) with epidermal growth factor receptor‐activating and T790M mutations treated with ASP8273. Phase I, dose escalation; phase II, dose expansion
For enrollment in phase I, patients were not expected by the investigator to show a therapeutic response to existing treatments. Patients were enrolled irrespective of T790M mutation status. For phase II, patients had confirmed PD after previous treatment with EGFR TKIs, and had expression of the EGFR T790M mutation centrally confirmed by a tumor biopsy of the primary or metastatic lesions or by a tumor tissue sample that had been collected and archived after confirmation of PD. Patients were excluded from participation if any of the following applied at the time of enrollment: (i) persistent clinical evidence of previous antitumor treatment‐related toxicity grade ≥2 (using the NCI‐CTCAE version 4.0), except alopecia and skin toxicities considered irrelevant by the investigator during study enrollment; (ii) history of or concurrent interstitial lung disease; (iii) received treatment with gefitinib within 8 days or erlotinib within 5 days before the start of study treatment; (iv) received previous treatment (except reversible EGFR TKIs) intended to have antitumor effects or treatment with another investigational drug or an investigational device within 14 days prior to the start of study treatment; and (v) received treatment with EGFR TKIs (eg, rociletinib, osimertinib) that can inhibit EGFR with the T790M mutation (this did not apply to the re‐enrollment cohort). The dose‐escalation and additional EGFR‐T790M parts consisted of two periods: a single‐dose period (cycle 0 lasting 2 days) and a multiple‐dose period (from cycle 1 onwards, each cycle lasting 21 days).
During phase I, patients received escalating ASP8273 doses (25‐600 mg). At least three patients were enrolled at each dose level, depending on the occurrence of DLTs in cycles 0 and 1. Patients enrolled at one dose level were not enrolled at other dose levels. ASP8273 was given at an initial dose of 25 mg, and then escalated to higher dose levels set at 50, 100, 200, 400, and 600 mg. Among these dose levels, the next recommended dose level was selected according to the posterior mean of DLT rate estimated by the Bayesian continual reassessment method. Dose‐limiting toxicities were defined as the following study drug‐related AEs: (i) grade 4 neutropenia; (ii) grade ≥3 febrile neutropenia; (iii) grade 4 thrombocytopenia or grade 3 thrombocytopenia with bleeding that requires platelet transfusion; (iv) grade ≥3 nonhematologic toxicity; (v) transient electrolyte imbalance that is not accompanied by any clinical signs or symptoms and that does not require continuous therapeutic intervention; (vi) diarrhea, nausea, and vomiting that can be improved to grade ≤2 with appropriate treatment; (vii) grade 3 AST and ALT levels that improve to grade ≤2 within 7 days after onset; and (viii) any toxicity resulting in a delay (≥11 days) of study treatment with ASP8273. The final judgment on DLTs was made by the investigator and the sponsor, and the severity of each of the AEs was assessed according to NCI‐CTCAE version 4.0. The next dose level was determined after the end of cycle 1 assessments in all patients enrolled in the last dose‐escalation cohort. The decision to escalate to the next dose was based on safety and efficacy data and was determined by the investigators and study sponsor during a dose‐escalation meeting. In the re‐enrollment portion of the study, a patient who discontinued the study due to PD in the 25 mg cohort during phase I (dose‐escalation) could be re‐enrolled and treated with ASP8273 as multiple oral doses, once‐daily in each 21‐day cycle, at the RP2D of 300 mg until discontinuation. In this study, DLTs were assessed only during cycles 0 and 1 of the dose‐escalation and additional EGFR T790M parts of phase I. The MTD was defined as the highest dose of ASP8273 at which the incidence of DLTs was estimated to be the closest to 33%.
The study was designed by the study sponsor in collaboration with the investigators and was carried out in accordance with the protocol, the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use guidelines, applicable regulations and guidelines governing clinical study conduct, and the ethical principles of the Declaration of Helsinki.
2.2. Study end‐points and assessments
A primary end‐point of phase I was to determine the safety and tolerability of ASP8273 based on DLTs (excluding the re‐enrollment part), AEs, clinical laboratory tests, bone turnover markers, vital signs, percutaneous oxygen saturation, body weight, 12‐lead electrocardiograms, ophthalmologic examination, chest X‐ray examination, chest computed tomography examination, and ECOG performance status. Adverse events were graded using the NCI‐CTCAE version 4.0. Laboratory safety assessments included monitoring hematology and blood chemistry. The other primary end‐point for phase I was to determine the MTD and/or RP2D of ASP8273 based on the DLT profiles.
Secondary objectives of phase I during dose‐escalation and additional T790M cohorts were to determine the antitumor activity of ASP8273. Tumor lesions were assessed with an imaging technique such as radiography, chest computed tomography, or MRI on days 1 (predose) and 21 for each treatment cycle. Antitumor activity was determined according to RECIST version 1.1 and measured by best overall response, ORR (CR + PR), DCR (CR + PR + SD), PFS, and duration of response.
The primary objective in phase II was to determine the antitumor activity of ASP8273 as measured by best overall response at week 24 and cut‐off date, ORR (based on central review at week 24 and cut‐off date), as well as DCR at week 24 and cut‐off date; safety and tolerability of ASP8273 was a secondary end‐point of phase II. ASP8273 single‐ and repeat‐dose pharmacokinetics profiles, including dose proportionality (phase I) and ethnic differences (phase II) were secondary end‐points in both phases.
Pharmacokinetic parameters of ASP8273 were evaluated by plasma sampling in both phase I and phase II. During phase I, blood was drawn on cycle 0 day 1 (predose) and 0.5, 1, 2, 4, 6, 8, 10, 24, 34, and 48 hours postdose before dosing on cycle 1 day 1 and days 8, 15, 18, and 21. Phase II blood sampling was carried out cycle 1 day 1 (predose) and 0.5, 1, 2, 4, 6, 8, and 24 hours postdose; also before dosing on cycle 1 day 2, and days 8, 15, and 21. An EGFR‐mutation test using histological or cytological samples, optional in phase I but required in phase II, were analyzed at a central EGFR gene testing laboratory using the therascreen EGFR RGQ PCR kit (Qiagen, Hilden, Germany). For this purpose, either tumor biopsy samples of the primary or metastatic lesions or an archived tumor tissue sample were used. Samples for EGFR‐activating mutations (ex19del, L858R, L861Q, and G719X) and for the EGFR T790M mutation were also analyzed.
2.3. Statistical analysis
All patients who received at least one dose of ASP8273, who had acceptable images for baseline tumor assessment, and who were evaluated for at least one efficacy end‐point after start of treatment were included in the full analysis set. Patients who received at least one dose of ASP8273 were included in the safety analysis set.
For phase I, the ORR, defined as the proportion of patients with best overall response over the entire exposure period was rated as CR or PR, was calculated by dose in each part and its 95% CI (Clopper‐Pearson) is presented. For phase II, the ORR at week 24 (by central and local review) and for the entire treatment period (by central review only) were calculated. The antitumor activity of ASP8273 based on RECIST version 1.1 in phase II was evaluated according to Simon's two‐stage design (uninteresting response = 30%, desired response = 50%, α = 0.05, β = 0.1). If 9 or more of the 24 ASP8273‐treated patients achieved response in the first stage, then 39 additional patients would be enrolled. If ≥25 of 63 total patients achieved response, ASP8273 would be considered to have antitumor effects based on the observed response rate equal to or more than the threshold response rate of 30%. Adverse events were coded according to the Medical Dictionary for Regulatory Activities version 16.1. It was used to summarize AEs by System Organ Class and preferred term for both study phases. The number and percentage of patients experiencing DLTs were summarized by study drug dose.
3. RESULTS
3.1. Disposition, demographics, and disease characteristics of the overall study
In phase I of this study, 49 patients provided informed consent to participate in the clinical study (Figure 2). A total of 47 patients were enrolled and received the study drug; 45 patients were included in safety and efficacy analyses. The majority of patients were women (n = 33; 73%), all were clinical stage IV, and the majority of patients never smoked (n = 33; 73%). The EGFR ex19del (n = 30; 67%) and L858R (n = 14; 31%) activating mutations were common in this population; 23 patients (51%) harbored a T790M resistance mutation, with the T790M mutation status unknown for 16 patients (Table 1). The median duration of exposure was 170 days overall and the mean compliance rate was 92%.
Figure 2.
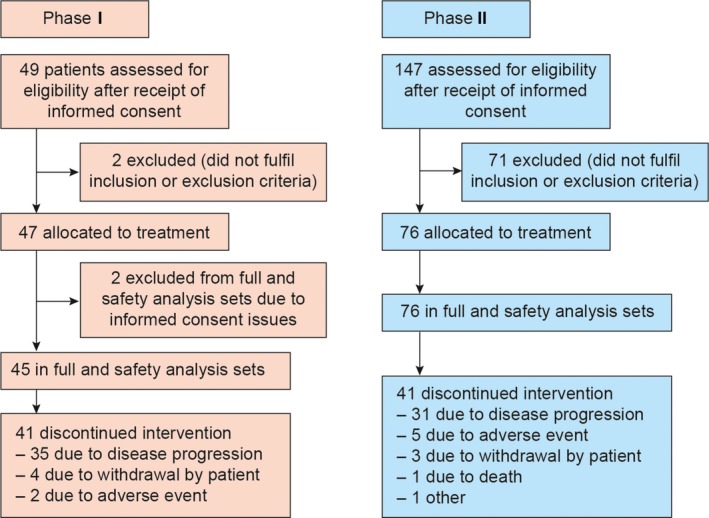
Study disposition involving patients with non‐small‐cell lung cancer (NSCLC) with epidermal growth factor receptor‐activating and T790M mutations treated with ASP8273. Phase I, dose escalation; phase II, dose expansion
Table 1.
Demographic and baseline disease characteristics of patients with non‐small‐cell lung cancer with epidermal growth factor receptor (EGFR)‐activating and T790M mutations treated with ASP8273
| Phase I | Phase II | ||||||
|---|---|---|---|---|---|---|---|
| 25‐100 mg (n = 11) | 200 mg (n = 10) | 300 mg (n = 10) | 400 mg (n = 10) | 600 mg (n = 4) | Total (N = 45) | Total (N = 76) | |
| Median age, years (range) | 69 (43‐74) | 65 (47‐74) | 60 (28‐74) | 67 (42‐78) | 66 (60‐78) | 65 (28‐78) | 63 (39‐83) |
| Sex, n (%) | |||||||
| Male | 5 (45) | 1 (10) | 3 (30) | 2 (20) | 1 (25) | 12 (27) | 28 (37) |
| Female | 6 (55) | 9 (90) | 7 (70) | 8 (80) | 3 (75) | 33 (73) | 48 (63) |
| ECOG performance status, n (%) | |||||||
| 0 | 2 (18) | 5 (50) | 3 (30) | 3 (30) | 3 (75) | 16 (36) | 20 (26) |
| 1 | 9 (82) | 5 (50) | 7 (70) | 7 (70) | 1 (25) | 29 (64) | 56 (74) |
| Number of prior EGFR TKI treatments, n (%) | |||||||
| 1 | 4 (36) | 6 (60) | 6 (60) | 6 (60) | 3 (75) | 25 (56) | 45 (59) |
| 2 | 7 (64) | 4 (40) | 1 (10) | 4 (40) | 1 (25) | 17 (38) | 21 (28) |
| 3 | 0 (0) | 0 (0) | 3 (30) | 0 (0) | 0 (0) | 3 (7) | 10 (13) |
| Patients with EGFR‐activating mutation, n (%) | |||||||
| Exon 19 Del | 6 (55) | 6 (60) | 8 (80) | 7 (70) | 3 (75) | 30 (67) | 52 (68) |
| L858R | 5 (45) | 3 (30) | 2 (20) | 3 (30) | 1 (25) | 14 (31) | 22 (29) |
| T790M mutation status, n (%) | |||||||
| Positive | 3 (27) | 8 (80) | 5 (50) | 7 (70) | 0 (0) | 23 (51) | 76 (100) |
| Negative | 0 (0) | 2 (20) | 3 (30) | 0 (0) | 1 (25) | 6 (13) | 0 (0) |
| Unknown | 8 (73) | 0 (0) | 2 (20) | 3 (30) | 3 (75) | 16 (36) | 0 (0) |
| Minor mutations, n (%) | |||||||
| L861Q | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (3) |
| G719X | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (1) |
| Other | 0 (0) | 1 (10) | 0 (0) | 1 (10) | 1 (25) | 3 (7) | 6 (8) |
| Tobacco history, n (%) | |||||||
| Never smoked | 6 (55) | 8 (80) | 8 (80) | 8 (80) | 3 (75) | 33 (73) | 51 (67) |
| Current smoker | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Former smoker | 5 (45) | 2 (20) | 2 (20) | 2 (20) | 1 (25) | 12 (27) | 25 (33) |
| Clinical stage, n (%) | |||||||
| 0 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| IA | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| IB | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| IIA | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 3 (4) |
| IIB | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (1) |
| IIIA | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (3) |
| IIIB | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (3) |
| IV | 11 (100) | 10 (100) | 10 (100) | 10 (100) | 4 (100) | 45 (100) | 68 (90) |
TKI, tyrosine kinase inhibitor.
Of the 147 patients who provided informed consent to participate in phase II of this study (carried out in Japan, Korea, and Taiwan), 76 were enrolled and received the study drug; all 76 patients were included in the safety analysis set and full analysis set. Similar to phase I, more women (n = 48; 63%) than men (n = 28; 37%) were enrolled, the majority of patients never smoked (n = 51; 67%), and were clinical stage IV (n = 68; 90%). All patients (n = 76) had an EGFR T790M resistance mutation, 52 (68%) had an ex19del‐activating mutation, and 22 (29%) had an L858R EGFR‐activating mutation. For this cohort, the median duration of study drug exposure was 196 days and the mean compliance rate was 94%.
3.2. Phase I
3.2.1. Safety and tolerability of ASP8273 (25‐600 mg)
Across the wide ASP8273 dose‐escalation range (25‐600 mg), TEAEs occurring in ≥20% of all patients were diarrhea (76%; n = 34), nausea (53%; n = 24), vomiting (49%; n = 22), increased ALT (47%; n = 21), decreased appetite and peripheral sensory neuropathy (38%; n = 17 each), increased AST (36%; n = 16), increased blood creatinine and constipation (33%; n = 15 each), hyponatremia and platelet count decreased (31%; n = 14 each), hypoalbuminemia (22%; n = 10), and anemia, dry skin, dysgeusia, and malaise (20%; n = 9 each). No deaths were reported during phase I. Serious TEAEs reported in phase I were dehydration, hyponatremia, and diarrhea (4%; n = 2 each).
As detailed in Table 2, diarrhea (76%; n = 34), vomiting and increased ALT (44%; n = 20), nausea (42%; n = 19), peripheral sensory neuropathy (36%; n = 16), increased AST, increased blood creatinine, and decreased platelet count (31%; n = 14 each) were the most commonly reported TRAEs in phase I. Most of the TRAEs were grade ≤2 in severity; the grade ≥3 TRAEs occurring in ≥2 patients were hyponatremia (20%; n = 9), diarrhea (18%; n = 8), increased AST and anemia (7%; n = 3 each), and colitis, hypoalbuminemia, nausea, and peripheral sensory neuropathy (4%; n = 2 each).
Table 2.
Incidence of overall treatment‐related adverse events (TRAEs) and grade ≥3 TRAEs occurring in ≥20% of patients with non‐small‐cell lung cancer treated with ASP8273 (phase I)
| TRAE, n (%) | 25‐100 mg (n = 11) | 200 mg (n = 10) | 300 mg (n = 10) | 400 mg (n = 10) | 600 mg (n = 4) | Total (N = 45) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Overall | Grade ≥3 | Overall | Grade ≥3 | Overall | Grade ≥3 | Overall | Grade ≥3 | Overall | Grade ≥3 | Overall | Grade ≥3 | |
| Diarrhea | 5 (45) | 0 (0) | 8 (80) | 0 (0) | 7 (70) | 1 (10) | 10 (100) | 5 (50) | 4 (100) | 2 (50) | 34 (76) | 8 (18) |
| Nausea | 3 (27) | 0 (0) | 3 (30) | 0 (0) | 4 (40) | 0 (0) | 7 (70) | 1 (10) | 2 (50) | 1 (25) | 19 (42) | 2 (4) |
| Vomiting | 2 (18) | 0 (0) | 3 (30) | 0 (0) | 6 (60) | 0 (0) | 6 (60) | 0 (0) | 3 (75) | 0 (0) | 20 (44) | 0 (0) |
| ALT increased | 1 (9) | 0 (0) | 5 (50) | 0 (0) | 5 (50) | 2 (20) | 7 (70) | 1 (10) | 2 (50) | 0 (0) | 20 (44) | 3 (7) |
| Decreased appetite | 2 (18) | 0 (0) | 4 (40) | 1 (10) | 2 (20) | 0 (0) | 2 (20) | 0 (0) | 2 (50) | 0 (0) | 12 (27) | 1 (2) |
| Peripheral sensory neuropathy | 4 (36) | 1 (9) | 3 (30) | 0 (0) | 2 (20) | 0 (0) | 4 (40) | 1 (10) | 3 (75) | 0 (0) | 16 (36) | 2 (4) |
| AST increased | 0 (0) | 0 (0) | 4 (40) | 0 (0) | 4 (40) | 0 (0) | 4 (40) | 0 (0) | 2 (50) | 0 (0) | 14 (31) | 0 (0) |
| Blood creatinine increased | 2 (18) | 0 (0) | 2 (20) | 0 (0) | 2 (20) | 0 (0) | 5 (50) | 0 (0) | 3 (75) | 0 (0) | 14 (31) | 0 (0) |
| Constipation | 3 (27) | 0 (0) | 3 (30) | 0 (0) | 0 (0) | 0 (0) | 3 (30) | 0 (0) | 1 (25) | 0 (0) | 10 (22) | 0 (0) |
| Hyponatremia | 0 | 0 (0) | 4 (40) | 3 (30) | 0 (0) | 0 (0) | 5 (50) | 5 (50) | 2 (50) | 1 (25) | 11 (24) | 9 (20) |
| Platelet count decreased | 3 (27) | 0 (0) | 2 (20) | 0 (0) | 3 (30) | 0 (0) | 3 (30) | 0 (0) | 3 (75) | 0 (0) | 14 (31) | 0 (0) |
| Dysgeusia | 1 (9) | 0 (0) | 3 (30) | 0 (0) | 1 (10) | 0 (0) | 1 (10) | 0 (0) | 3 (75) | 0 (0) | 9 (20) | 0 (0) |
| Malaise | 2 (18) | 0 (0) | 1 (10) | 0 (0) | 0 (0) | 0 (0) | 5 (50) | 1 (10) | 1 (25) | 0 (0) | 9 (20) | 1 (2) |
| TRAEs of special interest | ||||||||||||
| Rash | 1 (9) | 0 (0) | 0 (0) | 0 (0) | 2 (20) | 0 (0) | 1 (10) | 0 (0) | 1 (25) | 0 (0) | 5 (11) | 0 (0) |
| QTc prolongation | 1 (9) | 0 (0) | 0 (0) | 0 (0) | 2 (20) | 0 (0) | 1 (10) | 0 (0) | 0 (0) | 0 (0) | 4 (9) | 0 (0) |
| Interstitial lung disease | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (25) | 0 (0) | 1 (2) | 0 (0) |
ALT, alanine transaminase; AST, aspartate aminotransferase.
Dose‐limiting toxicities were experienced by 9 patients during the study; specifically, 5 patients in the 400 mg dose group and 4 patients in the 600 mg dose group. All DLTs were of grade ≥3 nonhematologic toxicity, and the most common DLTs were diarrhea (7%; n = 3), followed by colitis, nausea (4%; n = 2 each), and malaise, biliary tract infection, and hyponatremia (2%; n = 1 each). Based on the Bayesian continual reassessment method, the 400 mg dose was recommended as the MTD.
3.2.2. Pharmacokinetics of ASP8273
ASP8273 showed linear pharmacokinetics and dose proportionality over the dose range of 100 to 400 mg in phase I (Figure 3; Tables S1‐S5). Steady‐state ASP8273 was achieved by cycle 1 day 8 after once‐daily dosing. After 21 days of daily dosing, approximately 30%‐60% accumulation AUC was observed. During phase II, approximately 20% accumulation AUC was observed after 21 days of daily dosing. The exposures of ASP8273 were comparable among the group (Korean, Taiwanese, and Japanese).
Figure 3.
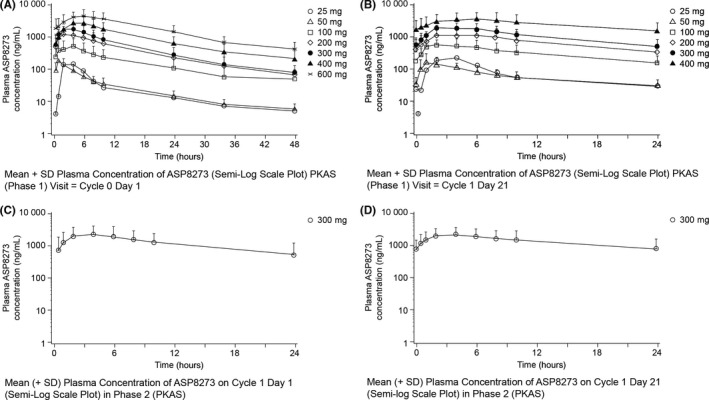
Pharmacokinetic profile of ASP8273 in Phase I (dose escalation) and Phase II (dose expansion). A, Single‐dose ASP8273 plasma concentration (phase I). B, Repeat‐dose ASP8273 plasma concentration (phase I). C, Single‐dose ASP8273 plasma concentration (phase II). D, Repeat‐dose ASP8273 plasma concentration (phase II)
3.2.3. Phase I antitumor activity
Across the dose range (25‐600 mg), the ORR was 49% (95% CI, 33.7, 64.2). Although no patient achieved a CR, 22 patients achieved PR; additionally, 18 patients achieved SD and five had PD (Table 3). Disease control rate, defined as CR + PR + SD, was high (89%; n = 40) and the median duration of PFS was 5.6 months (95% CI: 3.6, 11.1). The duration of response, as assessed by local review, was 8.4 months (95% CI, 4.2, 11.0) in the total population. The proportion of patients with maximum shrinkage from baseline in the target lesion ≥30% was 55% (n = 24/44) (Figure 4). Most patients in all dose groups had shrinkage in the target lesion with no clear correlation between degree of percent change from baseline in the target lesion and the dose level of ASP8273.
Table 3.
Best overall response rate (ORR) across all doses (local review) among patients with non‐small‐cell lung cancer treated with ASP8273
| 25‐100 mg (n = 11) | 200 mg (n = 10) | 300 mg (n = 10) | 400 mg (n = 10) | 600 mg (n = 4) | Total (N = 45) | |
|---|---|---|---|---|---|---|
| CR, n (%) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| PR, n (%) | 2 (18) | 5 (50) | 5 (50) | 7 (70) | 3 (75) | 22 (49) |
| SD, n (%) | 8 (73) | 4 (40) | 3 (30) | 3 (30) | 0 (0) | 18 (40) |
| ORR, % (95% CIa) | 18 (2.28, 51.8) | 50 (18.7, 81.3) | 50 (18.7, 81.3) | 70 (34.8, 93.3) | 75 (19.4, 99.4) | 49 (33.7, 64.2) |
| DCR, % (95% CIa) | 90 (58.7, 99.8) | 90 (55.5, 99.7) | 80 (44.4, 97.5) | 100 (69.2, 100) | 75 (19.4, 99.4) | 89 (75.9, 96.3) |
Based on exact binomial confidence interval (CI; Clopper–Pearson).
CR, complete response; DCR, disease control rate (CR + PR + SD); PR, partial response; SD, stable disease.
Figure 4.
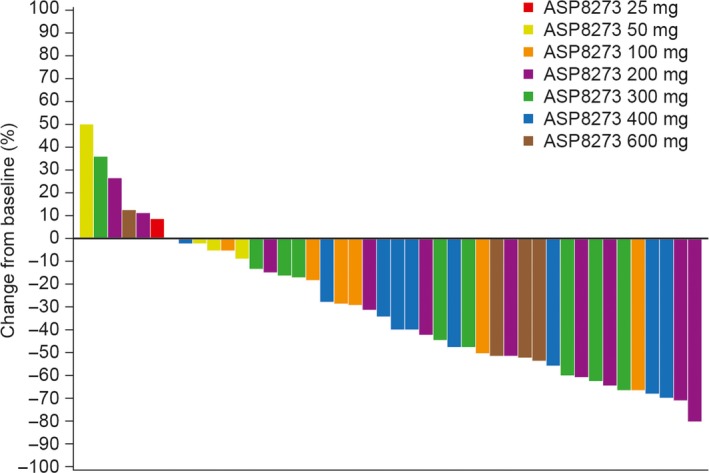
Maximum percent change from baseline in tumor size in patients with non‐small‐cell lung cancer treated with ASP8273 (phase I, local review)
When antitumor activity was assessed by T790M status, ORRs and DCRs with ASP8273 (25‐600 mg) were higher in patients with a known T790M mutation than patients with no T790M mutation (Table 4). Patients with unknown T790M status had response rates that were similar to patients with known T790M. However, due to the small number of patients in these subgroups, these data should be interpreted with caution.
Table 4.
Best overall response rate (ORR) to ASP8273 among patients with non‐small‐cell lung cancer by epidermal growth factor receptor T790M mutation status, phase I
| T790M+ (n = 23) | T790M− (n = 6) | T790M unknown (n = 16) | |
|---|---|---|---|
| CR, n (%) | 0 (0) | 0 (0) | 0 (0) |
| PR, n (%) | 12 (52) | 2 (33) | 8 (50) |
| SD, n (%) | 10 (44) | 1 (17) | 7 (44) |
| ORR, % (95% CI)a | 52 (30.6, 73.2) | 33 (4.3, 77.7) | 50 (24.7, 75.3) |
| DCR, % (95% CI)a | 96 (78.1, 99.9) | 50 (11.8, 88.2) | 94 (69.8, 99.8) |
Data presented as n (%).
Based on exact binomial confidence interval (CI; Clopper‐Pearson).
CR, complete response; DCR, disease control rate; PR, partial response; SD, stable disease.
Based on these efficacy data and a comprehensive evaluation of safety, including the DLT profile and pharmacokinetic data, the RP2D was determined to be 300 mg.
3.3. Phase II
3.3.1. Antitumor activity
As 27 of the 63 patients treated with ASP8273 300 mg, in the first and second stages of phase II combined, achieved a clinical response (based on independent central review), ASP8273 was determined to have shown antitumor activity (ORR 43%; 95% CI, 30.5, 56.0).
The ORR at week 24, which was assessed at an independent central site, for the phase II population (N = 76) treated with ASP8273 300 mg, was 42% (n = 32; 95% CI, 30.9, 54.0). A total of 32 patients achieved PR, 29 achieved SD, and 8 had PD (Table 5). Disease control rate was 80% and the median duration of PFS by central review was 8.1 months (95% CI, 5.6, upper bound not reached). The proportion of evaluable patients with maximum shrinkage from baseline in the target lesion ≥30%, which was centrally assessed, was 67% (n = 46). As seen in Figure 5, almost all patients had shrinkage in target lesions. Based on central‐tissue testing, the ORR and median duration of PFS were also estimated by the presence or absence of EGFR subtypes ex19del and L8585R, and small differences were observed (Table 6).
Table 5.
Best overall response rate (ORR) at week 24 (central review) among patients with non‐small‐cell lung cancer treated with ASP8273, phase II (full analysis set)
| 300 mg (N = 76) | |
|---|---|
| CR, n (%) | 0 |
| PR, n (%) | 32 (42) |
| SD, n (%) | 29 (38) |
| ORR, % (95% CI)a | 42 (30.9, 54.0) |
| DCR, % (95% CI)a | 80 (69.5, 88.5) |
Data presented as n (%).
Based on exact binomial confidence interval (CI, Clopper‐Pearson).
CR, complete response; DCR, disease control rate; PR, partial response; SD, stable disease.
Figure 5.
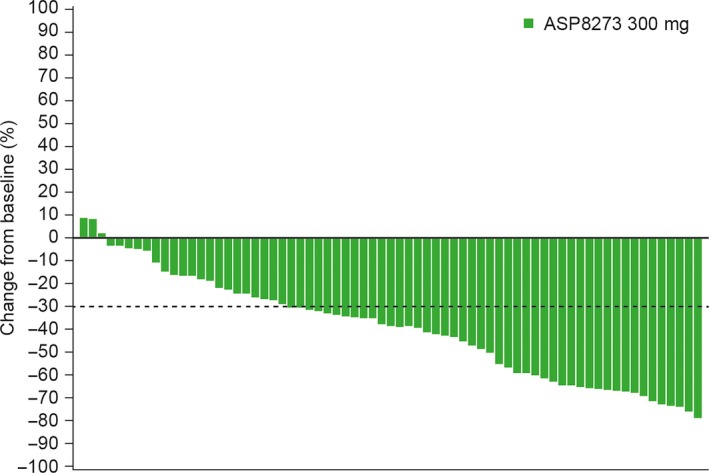
Maximum percent change from baseline in tumor size in patients with epidermal growth factor receptor T790M mutation‐positive non‐small‐cell lung cancer (phase II, central assessment)
Table 6.
Overall response rate (ORR) and median duration of progression‐free survival (PFS) at week 24 (phase II) among patients with non‐small‐cell lung cancer treated with ASP8273, by exon 19 deletion (ex19del) and L858R
| Ex19del+ (n = 49) | Ex19del− (n = 27) | L858R+ (n = 23) | L858R− (n = 53) | |
|---|---|---|---|---|
| PFS months (95% CI) | 8.1 (5.6, upper bound not reached) | 5.6 (3.0, upper bound not reached) | 5.6 (2.6, upper bound not reached) | 8.1 (5.7, upper bound not reached) |
| ORR (95% CI) | 47% (32.5, 61.7) | 33% (16.5, 54.0) | 35% (16.4, 57.3) | 45% (31.6, 59.6) |
CI, confidence interval; –, upper bound not reached.
3.3.2. Safety and tolerability of ASP8273 300 mg
At the 300 mg dose, TEAEs occurring in ≥20% of patients were diarrhea (66%; n = 50), nausea (41%; n = 31), increased ALT (36%; n = 27), vomiting and decreased appetite (34%; n = 26 each), hyponatremia (33%; n = 25), constipation (26%; n = 20), peripheral sensory neuropathy (25%; n = 19), and increased AST (24%; n = 18). Fatal TEAEs occurred in two patients: one patient experienced grade 5 TEAEs, pneumonia and dyspnea, 88 days after initiating treatment with 300 mg ASP8273; the other patient experienced grade 5 TEAEs leptomeningeal carcinomatosis, cough, and dyspnea, 108 days after initiating treatment. None of these fatal TEAEs was considered related to ASP8273. Serious drug‐related TEAEs were reported in 21% of patients (16/76); the most common (≥2 patients) were hyponatremia (7%; n = 5/76), increased ALT, and decreased appetite (3%; n = 2/76 each). Treatment‐emergent AEs leading to permanent discontinuation were reported by 6/76 patients; these were infectious pleural effusion, pneumonia, pyelonephritis, sepsis, hyponatremia, metastases to meninges, and organizing pneumonia.
In phase II, TRAEs were reported by 93% of patients (n = 71); the most frequently reported were diarrhea (57%; n = 43), hyponatremia and increased ALT (29%; n = 22 each), and vomiting (28%; n = 21). Most TRAEs were mild in severity, however, 15 reports of hyponatremia were grade ≥3 (Table 7).
Table 7.
Incidence of overall treatment‐related adverse events (TRAEs) and grade ≥3 TRAEs occurring in ≥10% of patients with non‐small‐cell lung cancer treated with ASP8273 (phase II) at initial dose level (safety analysis set)
| 300 mg (N = 76) | ||
|---|---|---|
| All grades, n (%) | Grade ≥3, n (%) | |
| Overall | 71 (93) | 29 (38) |
| Constipation | 9 (12) | 0 (0) |
| Diarrhea | 43 (57) | 0 (0) |
| Nausea | 20 (26) | 0 (0) |
| Vomiting | 21 (28) | 0 (0) |
| Malaise | 10 (13) | 0 (0) |
| Alanine aminotransferase increased | 22 (29) | 3 (4) |
| Aspartate aminotransferase increased | 13 (17) | 2 (3) |
| Platelet count decreased | 13 (17) | 0 (0) |
| Decreased appetite | 12 (16) | 3 (4) |
| Hyponatremia | 22 (29) | 15 (20) |
| Dysgeusia | 9 (12) | 0 (0) |
| Hypoesthesia | 8 (11) | 0 (0) |
| Peripheral sensory neuropathy | 18 (24) | 0 (0) |
| TRAEs of special interest | ||
| Rash | 5 (7) | 0 (0) |
| QTc prolongation | 5 (7) | 0 (0) |
| Interstitial lung disease | 0 (0) | 0 (0) |
4. DISCUSSION
Treatment of patients with NSCLC remains a challenge because many of these patients acquire drug resistance with a secondary mutation (T790M) following treatment with EGFR TKIs. In this phase I/II study of patients with EGFR‐mutant lung cancer in Japan, Korea, and Taiwan, AEs during both phases were similar to those observed with other drugs in this class,10 and 63 patients achieved a clinical response (based on independent central review) after treatment with ASP8273 for both stages of phase II combined (ORR 42.9%; 95% CI, 30.5, 56.0). These findings provide further insight into achieving the optimal antitumor effects of third‐generation EGFR TKIs for NSCLC.
ASP8273 given orally daily was generally well tolerated and AEs were manageable in patients with NSCLC. During phase I of the study (dose‐escalation), DLTs were experienced by 20% of patients (9/45), and ASP8273 300 mg was identified as the RP2D and 400 mg as the MTD. During phase II (dose‐expansion), 93.4% of patients reported drug‐related AEs, the most common of which was diarrhea (57%; n = 43/76). These events were anticipated from other EGFR TKIs in patients with NSCLC.10 During phase I, one patient who was treated with 600 mg ASP8273 experienced interstitial lung disease of grade 2 or lower. Hyponatremia and peripheral sensory neuropathy occurred more frequently with ASP8273 compared with other drugs in the class, which was consistent with findings from a phase I study of patients with EGFR‐mutation‐positive NSCLC undertaken in the USA (NCT02113813). Hyponatremia is a serious adverse event that is common among patients with metastatic lung cancer. In a clinical trial with EGFR TKI gefitinib in patients with advanced NSCLC, a total of 3 of 40 patients (8%) experienced grade 3 hyponatremia. During phase II of this study, hyponatremia grade ≥3 occurred in 20% of patients treated with ASP8273. Although this rate is higher than targeted lung cancer agents, in this study, treatment‐emergent hyponatremia was not considered to be cause for discontinuation of the clinical trial. Instances of treatment‐related peripheral sensory neuropathy (occurring among 18/76, 24% of patients) were mild in severity (grade 1 or 2).
ASP8273 showed linear pharmacokinetics and dose proportionality over the dose range of 100‐400 mg. Oral absorption of ASP8273 occurred rapidly; maximum concentrations were achieved within 1 to 6 hours after a single dose and at steady state. Steady‐state ASP8723 was achieved by day 8 after once‐daily dosing. Median terminal elimination half‐life of ASP8273 ranged from approximately 11 to 14 hours across the 50 to 600 mg dose range.
Tumor imaging data suggested that responses occurred in patients in all dose groups above 100 mg; additionally, when antitumor activity was assessed by T790M status, ORRs, PFS, and DCRs with ASP8273 (25‐600 mg) were higher in patients with a known T790M mutation than patients with no T790M mutation. However, due to the small number of patients in these subgroups, these data should be interpreted with caution.
In summary, the phase I portion of this study identified the RP2D and MTD of ASP8273 as 300 and 400 mg, respectively, when given orally to patients with clinical stage IV NSCLC. Additionally, we observed during phase II that ASP8273 has a manageable toxicity profile and patients achieved a response rate (CR + PR) of 43%, suggesting ASP8273 has potential antitumor effects.
CONFLICT OF INTEREST
CT, SK, JK, KA, AI, and MF have nothing to disclose. MS, TN, KP, RA, YK, and Kentaro Takeda are Astellas employees. SM received personal fees from Astellas during the conduct of this study. HH reports grants and personal fees from Ono Pharmaceutical, personal fees from Bristol‐Myers Squibb, Chugai Pharmaceutical, MSD, Taiho Pharmaceutical, and personal fees and other from AstraZeneka, Eli Lilly Japan, Boehringer Ingelheim Japan, outside the submitted work. KN reports grants and personal fees from Astellas Pharma, AstraZeneca, Chugai Pharmaceutical, Nippon Boehringer Ingelheim, and Taiho Pharmaceutical during the conduct of the study, personal fees from EPS Holdings, Showa Yakuhin Kako, Symbio Pharmaceutical, grants from EPS Associates, Quintiles, Japan Clinical Research Operations, Eisai, PPD‐SNBL, Takeda Pharmaceutical, GlaxoSmithKline, AbbVie, Yakult Honsha, PAREXEL International, Otsuka Pharmaceutical, AC MEDICAL, Merck Serono, Oncotherapy Science, personal fees from Kissei Pharmaceutical, AYUMI Pharmaceutical, and grants and personal fees from Ono Pharmaceutical, Eli Lilly Japan, Novartis Pharma, MSD, Bristol‐Myers Squibb, Pfizer Japan, Kyowa Hakko Kirin, and Daiichi Sankyo outside the submitted work. KK reports grants from Chugai Pharmaceutical and personal fees from Pfizer Japan and Novartis Pharma during the conduct of the study, personal fees from Taiko Pharmaceutical and Eli Lilly Japan, grants from AstraZeneca, Nippon Boehringer Ingelheim, Daiichi Sankyo Pharmaceutical, and Shionogi & Co. outside the submitted work. MN received research funding from Novartis, ONO Pharmaceutical, Chugai Pharmaceutical, Bristol‐Myers Squibb, Taiho Pharmaceutical, Eli Lilly, Pfizer, Astellas Pharma, AstraZeneca, and honoria from Pfizer, Bristol‐Myers Squibb, ONO Pharmaceutical, Chugai Pharmaceutical, Eli Lilly, Taiho Pharmaceutical, and AstraZeneca. Koji Takeda received grants from Astellas during the conduct of the study, grants and personal fees from AstraZeneca, Boehringer Ingelheim, Bristol‐Myers Squibb, Chugai Pharmaceutical, and Eli Lilly, grants from Eisai, Kyowa Hakko Kirin, Ono Pharmaceutical, and Merck Serono, and personal fees from Novartis, Taiho Pharmaceutical, Daiichi Sankyo outside the submitted work. JY reports personal fees from Boehringer Ingelheim, Eli Lilly, Bayer, Roche/Genentech, Chugai, Astellas, MSD, Merck Serono, Pfizer, Novartis, Clovis Oncology, Celgene, Merrimack, Yuhan Pharmaceuticals, BMS, Ono Pharmaceuticals, Daiichi Sankyo, AstraZeneca, and Hansoh Pharmaceuticals outside the submitted work. HM reports grants and nonfinancial support from Astellas Pharma during the conduct of the study, personal fees from AstraZeneca, Lilly Japan, Chugai Pharma, Boehringer Ingelheim, Pfizer, Taiho Pharmaceutical, Novartis, Bristol‐Myers Squibb Japan, Ono Pharmaceutical, and Teijin Pharma outside the submitted work. TS reports grants from Astellas Pharma during the conduct of the study, personal fees from Astellas Pharma, AstraZeneca, Bayer Yakuhin, Bristol‐Myers Squibb, Fuji Pharma, Hisamitsu Pharmaceutical, Kissei Pharmaceutical, Kyowa Hakko Kirin, Mochida Pharmaceutical, Nippon Kayaku, Ono Pharmaceutical, Roche Diagnostics, Sanofi, Showa Yakuhin Kako, Sumitomo Dainippon Pharma, Taiho Pharmaceutical, Takeda Pharmaceutical, and Roche Singapore, grants from Astellas Pharma, Bayer Yakuhin, Merck Serono, Novartis Pharma, and Verastem, and grants and personal fees from AstraZeneca, Chugai Pharmaceutical, Daiichi Sankyo, Eisai, Eli Lilly Japan, MSD, Nippon Boehringer Ingelheim, Pfizer Japan, and Yakult outside the submitted work. HN reports grants from Merck Serono, Pfizer Eisai, Novartis, Daiichi Sankyo, GlaxoSmithKline, Quintiles, Astellas Pharma, grants and personal fees from Taiho Pharmaceutical, Chugai Pharma, Eli Lilly, AstraZeneca, Boehringer Ingelheim, and Ono Pharmaceutical, and personal fees from Sanofi and Bristol‐Myers Squibb outside the submitted work.
Supporting information
ACKNOWLEDGEMENTS
We would like to acknowledge all investigators, coordinators, and study site personnel, as well as patients and their families for their participation in this study. This research was sponsored by Astellas Pharma, Inc. (Tokyo, Japan). Financial support for the development of this manuscript, including writing and editorial assistance under the authors’ guidance, was provided by Melissa Creasey and Regina Switzer of SuccinctChoice Medical Communications (Chicago, IL, USA) and was funded by the study sponsor.
Murakami H, Nokihara H, Hayashi H, et al. Clinical activity of ASP8273 in Asian patients with non‐small‐cell lung cancer with EGFR activating and T790M mutations. Cancer Sci. 2018;109:2852–2862. 10.1111/cas.13724
At the time of data collection, Dr. Jin‐Hyoung Kang was affiliated with the Asan Medical Center, University of Ulsan College of Medicine, Seoul Korea.
Clinical trial registration number: NCT02192697.
REFERENCES
- 1. Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367‐1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene. 2000;19:6550‐6565. [DOI] [PubMed] [Google Scholar]
- 3. Shi Y, Au JS, Thongprasert S, et al. A prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non‐small‐cell lung cancer of adenocarcinoma histology (PIONEER). J Thorac Oncol. 2014;9:154‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ladanyi M, Pao W. Lung adenocarcinoma: guiding EGFR‐targeted therapy and beyond. Mod Pathol. 2008;21(Suppl 2):S16‐22. [DOI] [PubMed] [Google Scholar]
- 5. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169‐181. [DOI] [PubMed] [Google Scholar]
- 6. Zhou Q, Zhang XC, Chen ZH, et al. Relative abundance of EGFR mutations predicts benefit from gefitinib treatment for advanced non‐small‐cell lung cancer. J Clin Oncol. 2011;29:3316‐3321. [DOI] [PubMed] [Google Scholar]
- 7. Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first‐line treatment for European patients with advanced EGFR mutation‐positive non‐small‐cell lung cancer (EURTAC): a multicentre, open‐label, randomised phase 3 trial. Lancet Oncol. 2012;13:239‐246. [DOI] [PubMed] [Google Scholar]
- 8. Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor‐resistant disease. J Clin Oncol. 2013;31:1070‐1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non‐small‐cell lung cancer to gefitinib. N Engl J Med. 2005;352:786‐792. [DOI] [PubMed] [Google Scholar]
- 10. Janne PA, Yang JC, Kim DW, et al. AZD9291 in EGFR inhibitor‐resistant non‐small‐cell lung cancer. N Engl J Med. 2015;372:1689‐1699. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials