

β-Lactam antibiotics that inhibit penicillin-binding proteins (PBPs) have been widely used in the treatment of bacterial infections. However, the molecular basis underlying the different inhibitory potencies of β-lactams against specific PBPs is not fully understood.
KEYWORDS: penicillin-binding protein D2, crystal structure, Listeria monocytogenes, antibiotics, β-lactams, dd-carboxypeptidase
ABSTRACT
β-Lactam antibiotics that inhibit penicillin-binding proteins (PBPs) have been widely used in the treatment of bacterial infections. However, the molecular basis underlying the different inhibitory potencies of β-lactams against specific PBPs is not fully understood. Here, we present the crystal structures of penicillin-binding protein D2 (PBPD2) from Listeria monocytogenes, a Gram-positive foodborne bacterial pathogen that causes listeriosis in humans. The acylated structures in complex with four antibiotics (penicillin G, ampicillin, cefotaxime, and cefuroxime) revealed that the β-lactam core structures were recognized by a common set of residues; however, the R1 side chains of each antibiotic participate in different interactions with PBPD2. In addition, the structural complementarities between the side chains of β-lactams and the enzyme were found to be highly correlated with the relative reactivities of penam or cephem antibiotics against PBPD2. Our study provides the structural basis for the inhibition of PBPD2 by clinically important β-lactam antibiotics that are commonly used in listeriosis treatment. Our findings imply that the modification of β-lactam side chains based on structural complementarity could be useful for the development of potent inhibitors against β-lactam-resistant PBPs.
INTRODUCTION
Peptidoglycan is a major component of the bacterial cell wall and plays critical roles in maintaining cellular morphology and protecting cells against osmotic pressure. The peptidoglycan layer of the cell wall is comprised of a polymeric repeating disaccharide unit of N-acetylglucosamine (NAG) and N-acetylmuramic acid (NAM), cross-linked by short stem peptides (1, 2). Peptidoglycan biosynthesis is mediated by the following two essential enzymatic reactions: transglycosylation, which polymerizes disaccharides to form the glycan chain, and transpeptidation, which leads to peptide cross-linking between two adjacent glycan chains to generate a mesh-like structure (3). Both transglycosylation and transpeptidation are catalyzed by penicillin-binding proteins (PBPs). PBPs can be divided into two classes based on molecular weight, namely, high-molecular-weight (HMW) PBPs and low-molecular-weight (LMW) PBPs. The HMW PBPs can be further divided into class A and class B PBPs. Class A HMW PBPs are bifunctional enzymes that catalyze both transglycosylation and transpeptidation reactions via an N-terminal glycosyltransferase domain and a C-terminal transpeptidase (TP) domain, respectively. Class B HMW PBPs exhibit only TP activity. LMW PBPs exhibit both dd-carboxypeptidase and/or dd-endopeptidase activities to modulate the degree of cross-linking of peptidoglycan (4) and are dispensable for bacterial growth in culture media (2, 5, 6). PBPs are responsible for peptidoglycan synthesis, repair, and hydrolysis, and for maintenance of the stability of the cell wall while allowing bacterial growth.
PBPs are the molecular targets for the widely used β-lactam antibiotics, which resemble the d-alanyl-d-alanine moiety of stem peptide substrates of the PBPs (7). The reaction kinetics of PBPs with β-lactams or substrates is a second-order reaction that proceeds in the following three steps (7). First, the enzyme (E) and β-lactam (I) form the reversible noncovalent complex (EI), which has a dissociation constant of KD. Second, the four-membered β-lactam ring is opened by the nucleophilic attack of the catalytic serine of PBP, which proceeds at a rate constant of k2, to produce a covalent acyl-enzyme complex (EI*). Third, the acyl-enzyme complex (EI*) is hydrolyzed at the rate constant of k3 to regenerate the enzyme (E) and an inactivated product (P). In the case of β-lactam, the acyl-enzyme complex is stably maintained because k3 has a relatively low value, resulting in bacterial cell death. The molecular basis underlying the observed differences in the potencies of various β-lactams against specific PBPs is not well understood. Previous studies have reported that the efficacies of β-lactams are primarily determined by the acylation rate rather than by the formation of the preacylation complex (8). However, β-lactam efficacy can be improved by modifying the structure of the side chain(s), thereby implying the critical role of noncovalent complex formation. This hypothesis is further supported by the fact that improved efficacies of several cephalosporins against PBP2a of methicillin-resistant Staphylococcus aureus (MRSA) are highly correlated with the lower dissociation constant but not with higher acylation rate (9). Therefore, understanding the molecular mechanisms responsible for the inhibitory potencies of β-lactams is important for the design of novel antibiotics against β-lactam-resistant pathogens.
Listeria monocytogenes is a Gram-positive foodborne bacterial pathogen that causes a serious infection called listeriosis, which is associated with mortality rates that range from 20% to 30% in humans and domestic animals (10, 11). The first-line treatment for listeriosis is penicillin G or ampicillin, alone or in combination with aminoglycosides, such as gentamicin, which have been demonstrated to enhance the bactericidal effect of therapy (12). Notably, cephalosporins are not generally used to treat listeriosis because L. monocytogenes strains are naturally resistant to these compounds, which have high MIC values (13). L. monocytogenes is known to produce nine PBPs, which were identified using the fluorescent antibiotic Bocillin FL and by genomic analysis (14). Of these PBPs, penicillin-binding protein D2 (PBPD2) has been reported to be strongly induced by the presence of antibiotics that target the cell wall, such as cefuroxime and ampicillin, via two signaling pathways mediated by CesRK proteins (15, 16), which suggests the possible engagement of this protein in antibiotic resistance of L. monocytogenes. However, the mutant strains lacking the PBPD2 gene exert weak effects on sensitivity to some antibiotics such as ampicillin (14). Although PBPD2 is classified under the LMW PBPs and is dispensable for cell survival, mutant cells were observed to have longer lengths with altered morphology because of the reduced dd-carboxypeptidase activity (14). Despite the clinical importance of L. monocytogenes, among the L. monocytogenes PBPs, only PBP4 structures have been reported to date (17). Further structural and biochemical analyses of other L. monocytogenes PBPs will be necessary to provide a detailed knowledge of the therapeutic targets for the treatment of listeriosis and to elucidate the mechanisms underlying the intrinsic cephalosporin resistance of L. monocytogenes.
In the present study, we report the high-resolution crystal structures of PBPD2 from L. monocytogenes (here LmPBPD2) in the apo form and the acylated form, in complex with the following four antibiotics: penicillin G, ampicillin, cefotaxime, and cefuroxime. We performed structural analysis of the acylated structures and binding assays for six β-lactams against LmPBPD2, including the above four target antibiotics. The results revealed that differences in the inhibitory potencies of the six β-lactam antibiotics were correlated with the structural complementarity between β-lactam side chains and the active-site groove. Our data provide a framework for understanding the role of PBPD2 in L. monocytogenes and elucidating the mechanisms underlying the observed differences in the reactivities of β-lactams against PBPD2.
RESULTS AND DISCUSSION
Relative reactivities of β-lactams against LmPBPD2.
The relative reactivities of β-lactams against an N-terminally truncated form of LmPBPD2 (residues 21 to 272) were evaluated. Three penam antibiotics (penicillin G, ampicillin, and carbenicillin) and three cephem antibiotics (cefotaxime, cefuroxime, and ceftazidime) were selected for analysis. The penams differ only in the composition of the R1 side chain, while the cephems differ in the structures of both the R1 and R2 side chains (Fig. 1). The half maximal inhibitory concentration (IC50) of each β-lactam antibiotic was determined via SDS-PAGE-based competition assays using the fluorescent penicillin Bocillin FL (Fig. 1). Among the penams and cephems, penicillin G (IC50, 7.3 μM) and cefotaxime (IC50, 8.2 μM) showed the highest reactivities against LmPBPD2, respectively. Notably, carbenicillin (IC50, 723 μM) and ceftazidime (IC50, 1,075 μM) showed extremely weak reactivity relative to that of the other antibiotics. Concerning the relative reactivities of the penams, the sizes of the chemical groups in the α-carbon of the R1 side chain were found to be inversely related to the reactivity against LmPBPD2. The carboxylate group at the α-carbon of carbenicillin is relatively larger than the amine group of ampicillin and the hydrogen of penicillin G at the same position. Interestingly, similar observations were found in cephems. Ceftazidime differs from the other two cephems by the structure of the R1 side chain containing the dimethyl/carboxylate moiety, which is considerably larger than the corresponding side chains in cefotaxime or cefuroxime. In addition, the R2 side chain of cephems could also affect antibiotic reactivity. The above findings indicated that the interactions of the β-lactams side chain(s) with the enzyme could be one of the primary factors influencing the reactivity of β-lactams against LmPBPD2.
FIG 1.

Inhibition of LmPBPD2 with three penams (A) and three cephems (B). The y axis represents the relative intensity of the Bocillin FL fluorescence, while the x axis represents the logarithmic concentration of the antibiotic. The IC50 values were determined via SDS-PAGE-based Bocillin FL competition assays. Error bars indicate standard deviations based on three independent experiments. IC50 values represent the concentration of unlabeled antibiotics required to reduce the binding of Bocillin FL by 50%. The chemical structures of the β-lactam antibiotics that were used in the present study are shown above the graphs.
Overall structure.
To examine the interactions between the antibiotics and LmPBPD2, we crystallized LmPBPD2 in the apo form and four acylated forms in complex with the following different antibiotics: penicillin G, ampicillin, cefotaxime, and cefuroxime (Table 1). All of our trials using the soaking method did not generate suitable acylated structures, even with prolonged incubation in high concentrations of antibiotics. Therefore, the crystals of all the acylated forms were obtained by the cocrystallization method. The asymmetric unit of apo-LmPBPD2 contained one molecule, while the asymmetric units of acylated LmPBPD2 interacting with penams and cephems contained four and eight molecules, respectively. Analysis of crystal packing and protein interfaces does not support the formation of oligomers, which is consistent with the result of size exclusion chromatography, in which the purified enzyme was found to be monomeric in solution (data not shown). The refined LmPBPD2 structures represented the entire LmPBPD2 molecule (residues 24 to 270), and the quality of the final models was verified using MolProbity software (18).
TABLE 1.
Data collection and refinement statistics
| Parameter | LmPBPD2 (apo) |
LmPBPD2 (acylated) in complex with: |
|||
|---|---|---|---|---|---|
| Penicillin G | Ampicillin | Cefotaxime | Cefuroxime | ||
| Data collection | |||||
| Space group | P212121 | P21 | P21 | P21 | P21 |
| Cell dimensions | |||||
| a, b, c (Å) | 37.7, 74.6, 75.0 | 74.7, 98.9, 77.5 | 74.9, 98.9, 77.4 | 74.9, 98.7, 154.6 | 75.0, 99.3, 155.1 |
| α, β, γ (°) | 90, 90, 90 | 90, 104.3, 90 | 90, 104.2, 90 | 90, 103.9, 90 | 90, 103.9, 90 |
| Resolution (Å) | 40–1.6 | 30–1.9 | 30–1.7 | 30–1.9 | 30–2.0 |
| Rsymb (%) | 8.8 (33.1)a | 14.6 (47.2) | 10.3 (29.3) | 14.6 (43.9) | 12.7 (47.5) |
| I/σ (I) | 34.3 (4.3) | 19.8 (4.6) | 17.7 (2.2) | 6.6 (1.8) | 11.7 (2.7) |
| Completeness (%) | 98.2 (79) | 98.4 (97.4) | 92.8 (85.2) | 97.4 (96.4) | 99.8 (99.7) |
| Redundancy | 5.8 (3.4) | 6.5 (6.5) | 3.9 (2.5) | 3.3 (3.4) | 6.1 (5.9) |
| Structure refinement | |||||
| Resolution (Å) | 37.3–1.6 | 30.0–1.9 | 30.0–1.7 | 30–2.0 | 30.0–2.0 |
| No. of reflections | 30,732 | 84,570 | 110,752 | 170,471 | 136,600 |
| Rwork/Rfreec | 16.5/18.9 | 21.6/24.9 | 22.9/25.9 | 21.7/24.0 | 20.0/23.5 |
| RMSD (bond lengths [Å]/angles [°]) | 0.006/1.01 | 0.007/0.88 | 0.006/0.86 | 0.007/0.83 | 0.007/0.85 |
| Average B-factor (Å2) | 14.8 | 21.6 | 23.6 | 19.2 | 16.1 |
| MolProbity statistics | |||||
| Ramachandran favored/outlier (%) | 98.8/0 | 98.4/0 | 98.1/0 | 97.13/0 | 97.4/0 |
| Rotamer outlier (%) | 0 | 1.47 | 0.97 | 0.06 | 0 |
| Clashscored | 1.32 | 8.29 | 3.75 | 2.93 | 5.23 |
| Overall scored | 0.86 | 1.58 | 1.16 | 1.24 | 1.41 |
The numbers in parentheses are statistics from the highest-resolution shell.
Rsym = Σ |Iobs − Iavg|/Iobs, where Iobs is the observed intensity of individual reflection and Iavg is the average over symmetry equivalents.
Rwork = Σ ||Fo| − |Fc||/Σ |Fo|, where |Fo| and |Fc| are the observed and calculated structure factor amplitudes, respectively. Rfree was calculated with 5% of the data.
Scores are ranked according to structures of similar resolution as formulated in MolProbity software.
Homology searches in the Dali server revealed that LmPBPD2 shares the highest similarities with the TP domains of the LMW PBPs Escherichia coli PBP6 (EcPBP6) (PDB ID 3IT9) and EcPBP5 (PDB ID 1NZO), with root mean square deviation (RMSD) values of 2.0 Å and 1.9 Å for 240 residues, respectively (19, 20). LmPBPD2 folds into a compact single-domain structure comprising two subdomains, an α/β-subdomain and an α-helical subdomain (Fig. 2A). The α/β-subdomain is formed by a central core of a five-stranded antiparallel β-sheet (β3-β4-β5-β1-β2), which is sandwiched between two helices, α8 on one side and α11 on the opposite side of the sheet. The α-helical subdomain consists of a central helix, α2, surrounded by α4 to α6, α9, and two additional β-hairpins (β2a-β2b and β2c-β2d).
FIG 2.

Structure of PBPD2 from L. monocytogenes. (A) Ribbon representation of the apo-LmPBPD2 structure. The secondary structural elements are labeled according to Pares et al. (34) and Lobkovsky et al. (35). The catalytic Ser58 residue is represented as a purple sphere. (B) Stereo view of 2Fo-Fc electron density (1.0 σ) of active-site residues, including the three conserved motifs. Water molecules are shown as red spheres, and hydrogen bonds are shown as black dashed lines. (C) Multiple-sequence alignment and secondary structure assignment of LmPBPD2 and its representative structural homologs, LmPBPD2 (Uniprot accession number Q8Y3M3), EcPBP5 (accession number P0AEB3), and EcPBP6 (accession number P08506). Red characters indicate the amino acids that are conserved among the three proteins. The three conserved motifs of the active site and the flexible loops that participate in substrate access are highlighted by red rectangular boxes and a dashed line box, respectively. Residues interacting with β-lactams are designated with black triangles.
Active site.
The active site of apo-LmPBPD2 shows a canonical conformation that is observed in other LMW PBPs, suggesting that the enzyme is catalytically competent. The three conserved SXXK, SXN, and KTG motifs characterizing the active site form an extensive hydrogen-bonding network (Fig. 2B). The nucleophilic Ser58 in the SXXK motif (S58-L59-S60-K61) is positioned favorably for acylation reaction to form an ester bond with the peptide substrate or with β-lactam antibiotics. The general base Lys61Nζ forms a hydrogen bond with Ser58Oγ at a distance of 2.6 Å, and their conformations are nearly identical to those of EcPBP6 and EcPBP5 (19, 20). Lys61 also forms two additional hydrogen bonds with Asn120Oδ1 (2.9 Å) in the SXN motif (S118-A119-N120), which is located in the loop connecting α4 and α5, and with the backbone carbonyl oxygen atom of Ser158 (3.0 Å). Based on the three hydrogen-bonding interactions and the crystallization conditions at pH 8.0, the terminal amine group of Lys61 is likely to exist in a protonation state, which is necessary for deprotonation of the catalytic Ser58 residue for initiating the acylation reaction. The other conserved Lys222 residue, which is located at the third KTG motif (K222-T223-G224), also participates in three hydrogen bonds, one with Ser118Oγ (2.9 Å) in the SXN motif, one with the backbone carbonyl of Thr223 (2.9 Å), and one with a water molecule that mediates hydrogen-bonding interactions with Thr208 and the backbone carbonyl of Leu115 (2.8 Å). As demonstrated with other PBPs the hydrogen-bonding network of the conserved residues could contribute to proton transfer during catalysis by LmPBPD2 (21, 22).
Active site flexibility associated with acylation.
To determine the effects of acylation on LmPBPD2, the acylated structures were superimposed on the apo structure. The pairwise RMSD ranged from 0.35 to 0.47 Å between the Cα atoms of the apo structure and those of the acylated structures, indicating that acylation does not alter the overall conformation of the LmPBPD2 structure. However, a local conformational change was observed around the active-site groove with movement of the β2a-β2b loop region (residues 88 to 94), which was also observed to be a flexible region based on pairwise comparisons between the acylated structures. For example, the Cα atom of Ala93 in the loop moves by approximately 7.3 Å toward the outward direction in the cefotaxime-bound structure (molecule F) compared to that of the apo structure (Fig. 3). This way, the steric clash between Val94 and the bound cefotaxime could be avoided. Although the degree of movement of the β2a-β2b loop by acylation varies for each of the acylated structures, all β-lactam-bound structures showed the same direction of loop movement, which increases the volume of the active site pocket. The corresponding β2a-β2b loop in the apo-form crystal is involved in the crystallographic packing interaction. Therefore, the entrance to the active-site pocket cannot be widened to allow access to the antibiotics. The above observations explain why acylated structures were not successfully obtained via the soaking experiments, and also suggest that the flexibility of the β2a-β2b loop could play a critical role in substrate access to the active site. Indeed, the corresponding loop region of the LMW PBPs, including EcPBP5 or EcPBP6, has also been suggested to be involved in substrate recognition and product release during the catalytic process (19, 21).
FIG 3.
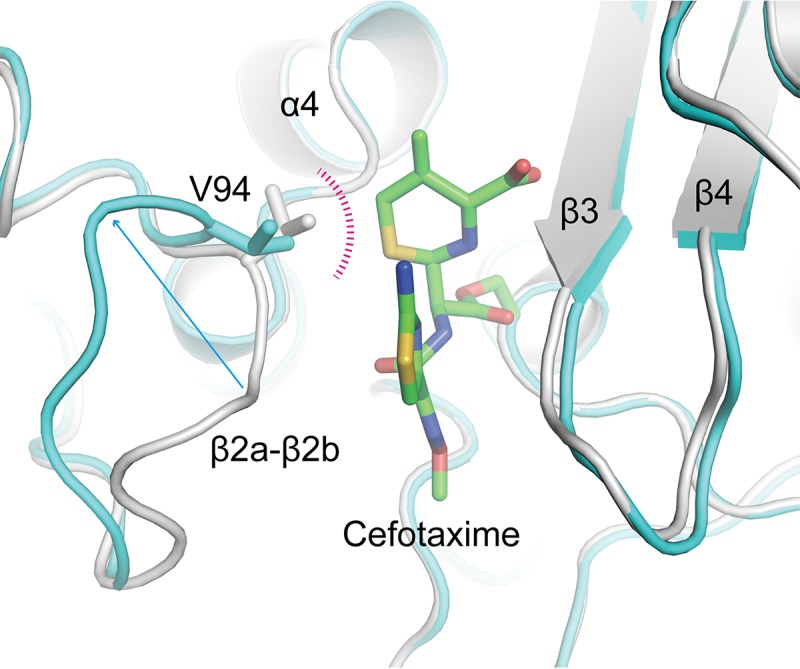
Conformational flexibility of the β2a-β2b loop. The apo form is superposed on the acylated LmPBPD2 (cefotaxime bound; molecule F). The apo form is colored in gray, and the acylated structure is colored in cyan; both structures are shown as a ribbon model. The cefotaxime molecule is shown as a stick model with green C atoms. The loop residue Val94, which causes a steric hindrance with cefotaxime, is presented as a stick model.
Molecular basis of the penams binding.
The hydrogen-bonding interactions between LmPBPD2 and the two penams are almost identical (Fig. 4A and B). In both acylated structures, the carbonyl oxygen of the β-lactam, which forms an oxyanion during the acylation reaction, is located in the oxyanion hole formed by the backbone NH groups of Ser58 and Phe225. The C3 carboxylate group of β-lactam forms a salt bridge with the side chain of Arg257 and forms a hydrogen bond with the side chain of Thr223. The amide groups of penams form two hydrogen bonds with the backbone carbonyl group of Phe225 and the side chain of Asn120, mimicking the anti-parallel β-sheet.
FIG 4.
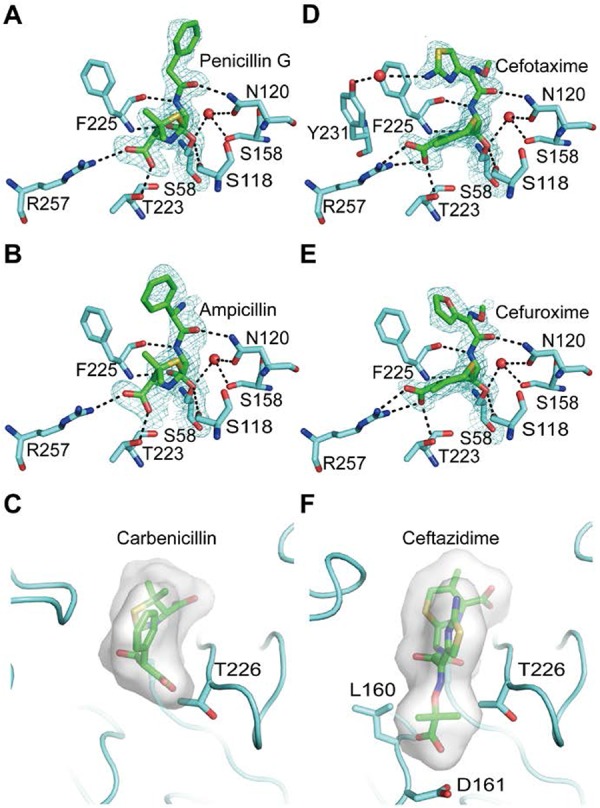
Interactions of LmPBPD2 with β-lactam antibiotics. Protein residues interacting with β-lactams are shown as a stick model in cyan. The Ser58 residues are shown in yellow, and the antibiotics are shown in green. Water molecules and hydrogen bonds are shown as red spheres and black dashed lines, respectively. Electron density maps (2Fo-Fc) covering acylated lactams are shown at a 1-σ contour level (A to D). (A) Penicillin G in the acyl-enzyme complex (molecule A). (B) Ampicillin in the acyl-enzyme complex (molecule A). (C) Steric hindrance of carbenicillin to the active site of LmPBPD2. The R1 side chain of carbenicillin is modeled based on the structure of the ampicillin-bound form and is shown as a stick model. The molecular surface corresponds to the volume of the β-lactam. The residues causing steric clashes are shown as a stick model. (D) Cefotaxime in the acyl-enzyme complex (molecule A). (E) Cefuroxime in the acyl-enzyme complex (molecule A). (F) Steric hindrance of ceftazidime to the active site of LmPBPD2. The bulky R1 side chain of ceftazidime is modeled based on the structure of the cefotaxime-acylated form. Molecules are presented as described in the legend of Fig. 4C.
The bound forms of penicillin G and ampicillin differ based on the conformation of the benzyl group in the R1 side chain (Fig. 4A and B). The benzyl group of penicillin G forms van der Waals interactions with Ser92 and Ala93 in the β2a-β2b loop region, while that of ampicillin has little contact with the same loop region. Indeed, the loop regions of the penicillin G-bound structures were found to be considerably more ordered relative to those of the ampicillin-bound structures, in which the electron density is almost absent. Therefore, we speculated that the conformation of the benzyl group could be related to the reactivity difference between penicillin G and ampicillin toward LmPBPD2. To verify our hypothesis, we conducted a modeling experiment to determine whether the benzyl group of ampicillin can adopt the same conformation as that of penicillin G in the acylated complex. In the model, the R1 amine group of ampicillin, which is the only chemical difference between penicillin G and ampicillin, causes steric hindrance with the side chain of Thr226 in the β3-β4 loop. This finding indicated that, unlike penicillin G, ampicillin cannot be accommodated in the active-site groove under the energetically favorable conformation. The above structural observations could explain why penicillin G reactivity is higher than that of ampicillin, as shown by the results of the SDS-PAGE competition assay. Although the acylated structures cannot provide relevant information on the interactions of the β-lactam side chain during the preacylation complex formation as discussed in previous studies (23), the narrow active-site groove of LmPBPD2 could also restrict the accessible conformation of antibiotic molecules prior to the acylation step.
Among the three penams tested, carbenicillin exhibited almost 100-fold lower reactivity against LmPBPD2 than did penicillin G (Fig. 1A). Although the acylated structure with carbenicillin was not obtained, carbenicillin-bound models could be built by superposing the carbenicillin onto the penicillin G-bound and ampicillin-bound structures. The two models show steric clashes between the R1 carboxylate group of carbenicillin and Thr226 or Phe225, when the R1 benzyl group of carbenicillin has the same conformation as that of ampicillin or penicillin G, respectively (Fig. 4C). Therefore, steric hindrance caused by the carboxylate group of carbenicillin could limit the accessibility of antibiotics into the narrow active-site groove of LmPBPD2, thereby weakening the inhibitory potency of carbenicillin.
Molecular basis of the cephems binding.
The electron density maps of cefotaxime and cefuroxime are well resolved for all molecules, except for the R2 side chain (Fig. 4D and E). The R2 side chain appears to be released during the catalytic opening of the β-lactam ring, by which a methylene group is formed on the end of the thiazine ring, as observed in other cephalosporin-acylated forms (24). Superposition of all of the acylated structures revealed that the conformations of the residues that participate in hydrogen-bonding interactions with penicillin G or ampicillin were unchanged in the complex structures of the cefotaxime and cefuroxime, and most of the hydrogen-bonding interactions were conserved, except for that formed by Arg257 (Fig. 4D and E). The carboxylate group of the dihydrothiazine in the cephems forms a bidentate interaction with the side chain of Arg257, while those of the penams form one hydrogen bond with Arg257. In addition, the R1 side chain of the two cephems participates in hydrophobic interactions with Phe225 in a face-to-edge manner. The only apparent difference between the two cephems is that the amine group of the thiazolidine ring of cefotaxime forms a water-mediated hydrogen bond with the side chain of Tyr231, which is not observed in cefuroxime. Therefore, we speculated that this hydrogen bond is responsible for the stronger reactivity of cefotaxime against LmPBPD2 than that of cefuroxime.
To confirm our speculation, a Y231F mutant was used to measure the relative reactivities of cefotaxime and cefuroxime under the same experimental conditions. The Y231F mutation increased the IC50 of cefotaxime by 1.5-fold, but not that of cefuroxime (Fig. 1B). These observations indicated that the water-mediated hydrogen bond partially contributes to the reactivity of cefotaxime. The IC50 of cefotaxime against the Y231F mutant was 5-fold lower than that of cefuroxime, indicating that the different inhibitory potencies of the two cephems cannot be fully explained by the acylated structures as mentioned above (23). The differences in inhibitory potencies between cefotaxime and cefuroxime could also be attributed to the structural complementarity between the R2 side chain of the cephem and the enzyme during the formation of the preacylation complex.
Ceftazidime, a third-generation cephalosporin, is clinically effective and safe for the treatment of many nosocomial bacterial infections, including Pseudomonas infection (25). However, the inhibitory activity of ceftazidime against LmPBPD2 was observed to be 130 times lower than that of cefotaxime (Fig. 1B). As mentioned above, the major structural difference between ceftazidime and cefotaxime is the presence of the bulky dimethyl/carboxylate moiety of the R1 side chain in ceftazidime, which is replaced by the small methyl group in cefotaxime. Although the acylated structure with ceftazidime was not obtained, a model for the bulky dimethyl/carboxylate moiety could be built by substituting the methyl group of the oxime of the cefotaxime-bound structures, which can be used to understand the molecule bases of the observed difference in inhibitory potencies. In our ceftazidime-bound model, the dimethyl/carboxylate moiety caused steric hindrance with the Leu160, Asp161, and Thr226 residues located in the active-site groove in any rotameric conformation, which explains the extremely low reactivity against LmPBPD2 (Fig. 4F). Therefore, ceftazidime could not access the active site without the large structural rearrangement of the active site groove, which requires energy, as shown in the case of carbenicillin. Meanwhile, the differences in inhibitory potencies between penams and cephems cannot be rationalized based on the analysis of the acylated structures because of the differences in the β-lactam core structures, which can induce distinct conformational changes in the side chain(s) during cleavage of the β-lactam ring (26).
Conclusion.
In the present study, the crystal structure of dd-carboxypeptidase PBPD2 from L. monocytogenes was determined, and it represents the second published PBP structure from this important pathogen. The relative reactivities of three penams (penicillin G, ampicillin, and carbenicillin) and three cephems (cefotaxime, cefuroxime, and ceftazidime) were measured via SDS-PAGE-based competition assays using Bocillin FL, and the binding modes of four antibiotics (penicillin G, ampicillin, cefotaxime, and cefuroxime) were determined based on the obtained acylated crystal structures. In the acylated structures, the β-lactam core structures, including the four-membered cyclic amide and the thiazolidine ring for penams or the dihydrothiazine ring for cephems, are recognized by a common set of residues in LmPBPD2. However, the R1 side chain of each antibiotic adopts different conformations in the active-site groove, leading to different interactions with the enzyme. Interestingly, the degree of structural complementarity between the R1 side chains of antibiotics and the active-site groove of LmPBPD2 is highly correlated with the relative reactivity, at least within the same β-lactam class sharing the core structure. In particular, the significantly lower inhibitory potencies of both ceftazidime and carbenicillin against LmPBPD2 can be explained by the steric hindrance caused by the bulky R1 side chains of these antibiotics. Our current findings suggested that the structural complementarity between the side chain(s) of β-lactam and the enzyme is a primary factor that contributes to the reactivity of β-lactams against LmPBPD2. Therefore, modifications of the interactions between specific side chains of β-lactams and their target enzymes can significantly improve the inhibitory potencies of the inhibitors against pathogens, including β-lactam-resistant pathogens.
MATERIALS AND METHODS
Protein expression and purification.
LmPBPD2 was expressed and purified as previously described (27). Briefly, LmPBPD2 was expressed as an N-terminal His6-tagged fusion protein in the Escherichia coli BL21(DE3)RIL strain. The protein sample was purified using a Ni-nitrilotriacetic acid (NTA) resin-based chromatography column and treated with tobacco etch virus (TEV) protease to remove the hexahistidine tag. The protein was further purified on a HiTrap Q anion exchange column followed by that on a Superdex 200 column equilibrated with 10 mM Tris-HCl (pH 8.0) and 100 mM NaCl. The fractions containing LmPBPD2 were pooled and concentrated to 20 mg · ml−1. Aliquots were flash frozen in liquid nitrogen and stored at 200 K for later use in crystallization and biochemical experiments. Site-directed mutagenesis was performed using the QuikChange kit (Stratagene) according to the manufacturer's instructions. The Y231F mutation was confirmed by sequencing, and the mutant protein was expressed and purified by the same method described above.
Crystallization and structure determination.
Apo-LmPBPD2 crystals were obtained via the sitting-drop vapor-diffusion method, as previously described (27). The crystallization conditions for the apo form were 20% (wt/vol) polyethylene glycol (PEG) 3350 and 0.2 M potassium chloride. The acylated forms were obtained by cocrystallization experiments. Crystals of ampicillin-bound or penicillin G-bound complexes were grown at 22°C via the sitting-drop vapor-diffusion method by mixing and equilibrating 2 μl of each protein solution and a precipitant solution containing 0.2 M sodium formate, 20% (wt/vol) PEG 3350, and 5 mM (each) of the target β-lactam antibiotics. Crystals of cefotaxime or cefuroxime-bound complexes were grown in solution containing 0.1 M Tris-HCl (pH 8.0), 25% (wt/vol) PEG 3350, 0.2 M sodium chloride, and 5 mM (each) the target β-lactam antibiotics. For data collection, the crystals were briefly immersed in the same precipitant containing an additional 15% glycerol and immediately placed in a 100-K nitrogen gas stream. X-ray diffraction data were collected for the apo form on beamline BL-1A of the Photon Factory (Japan), and data sets for the acylated forms were collected on beamline BL-5C of Pohang Light Source-II (Republic of Korea). All data were processed with HKL2000 (28). The apo-LmPBPD2 structure was determined by molecular replacement using Phaser (29). The TP domain structure of PBP6 from E. coli (PDB code 3IT9) was used as a search model. Model building and refinement were performed using Coot (30) and the Phenix package (31), respectively. The acylated structures were determined by molecular replacement using Phaser with the apo-LmPBPD2 structure as the starting model. The structures were rebuilt and refined in the same manner. The X-ray diffraction and structure refinement statistics are summarized in Table 1.
SDS-PAGE-based competition assays.
To assess the reactivity of β-lactams against LmPBPD2, SDS-PAGE-based competition assays using the fluorescent penicillin Bocillin FL were performed as previously described (32). Briefly, all reagents were diluted in reaction buffer (50 mM Tris-HCl [pH 8.0] and 100 mM NaCl) prior to use. The competition experiments were carried out by incubating 1 μM LmPBPD2 with various concentrations (0.05 to 10,000 μM) of unlabeled antibiotics (penicillin G, ampicillin, carbenicillin, cefotaxime, cefuroxime, and ceftazidime) in the presence of 10 μM Bocillin FL for 10 min at 22°C. Reactions were performed in aliquots and quenched by mixing with SDS-PAGE loading buffer. Samples were then separated by SDS-PAGE. The fluorescent-labeled proteins were visualized using a ChemiDoc X-ray spectrometry (XRS) imager (Bio-Rad) using a fluorescence emission filter (520 nm). Densitometry analysis was performed using ImageJ (33). The data points were normalized to the maximum value of the fluorescence intensity representing the complete saturation of LmPBPD2 by Bocillin FL. The half maximal inhibitory concentration (IC50), which is defined as the drug concentration at which 50% of the binding of Bocillin FL to LmPBPD2 is blocked, was calculated using Prism software (GraphPad Software, Inc.). At least three independent measurements were performed for each β-lactam concentration. In addition, the reactivity of cefotaxime or cefuroxime against the Y231F mutant was measured as described above.
Accession number(s).
The atomic coordinates and structure factors described here were deposited in the Protein Data Bank under PDB IDs 5ZQA (apo form), 5ZQB (penicillin G bound), 5ZQC (ampicillin bound), 5ZQD (cefotaxime bound), and 5ZQE (cefuroxime bound).
ACKNOWLEDGMENTS
This research was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (grant 2017R1D1A1B03030395).
We declare that we have no conflicts of interest.
REFERENCES
- 1.Vollmer W, Blanot D, de Pedro MA. 2008. Peptidoglycan structure and architecture. FEMS Microbiol Rev 32:149–167. doi: 10.1111/j.1574-6976.2007.00094.x. [DOI] [PubMed] [Google Scholar]
- 2.Popham DL, Young KD. 2003. Role of penicillin-binding proteins in bacterial cell morphogenesis. Curr Opin Microbiol 6:594–599. doi: 10.1016/j.mib.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 3.van Heijenoort J. 2001. Formation of the glycan chains in the synthesis of bacterial peptidoglycan. Glycobiology 11:25R–36R. doi: 10.1093/glycob/11.3.25R. [DOI] [PubMed] [Google Scholar]
- 4.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. 2008. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev 32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 5.Nelson DE, Young KD. 2001. Contributions of PBP 5 and DD-carboxypeptidase penicillin binding proteins to maintenance of cell shape in Escherichia coli. J Bacteriol 183:3055–3064. doi: 10.1128/JB.183.10.3055-3064.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nelson DE, Young KD. 2000. Penicillin binding protein 5 affects cell diameter, contour, and morphology of Escherichia coli. J Bacteriol 182:1714–1721. doi: 10.1128/JB.182.6.1714-1721.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zapun A, Contreras-Martel C, Vernet T. 2008. Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev 32:361–385. doi: 10.1111/j.1574-6976.2007.00095.x. [DOI] [PubMed] [Google Scholar]
- 8.Lu WP, Kincaid E, Sun YP, Bauer MD. 2001. Kinetics of beta-lactam interactions with penicillin-susceptible and -resistant penicillin-binding protein 2x proteins from Streptococcus pneumoniae—involvement of acylation and deacylation in beta-lactam resistance. J Biol Chem 276:31494–31501. doi: 10.1074/jbc.M102499200. [DOI] [PubMed] [Google Scholar]
- 9.Fuda C, Hesek D, Lee M, Heilmayer W, Novak R, Vakulenko SB, Mobashery S. 2006. Mechanistic basis for the action of new cephalosporin antibiotics effective against methicillin- and vancomycin-resistant Staphylococcus aureus. J Biol Chem 281:10035–10041. doi: 10.1074/jbc.M508846200. [DOI] [PubMed] [Google Scholar]
- 10.Southwick FS, Purich DL. 1996. Intracellular pathogenesis of listeriosis. N Engl J Med 334:770–776. doi: 10.1056/NEJM199603213341206. [DOI] [PubMed] [Google Scholar]
- 11.Swaminathan B, Gerner-Smidt P. 2007. The epidemiology of human listeriosis. Microbes Infect 9:1236–1243. doi: 10.1016/j.micinf.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 12.Hof H, Nichterlein T, Kretschmar M. 1997. Management of listeriosis. Clin Microbiol Rev 10:345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Espaze EP, Reynaud AE. 1988. Antibiotic susceptibilities of Listeria: in vitro studies. Infection 16(Suppl 2):S160–S164. doi: 10.1007/BF01639741. [DOI] [PubMed] [Google Scholar]
- 14.Korsak D, Markiewicz Z, Gutkind GO, Ayala JA. 2010. Identification of the full set of Listeria monocytogenes penicillin-binding proteins and characterization of PBPD2 (Lmo2812). BMC Microbiol 10:239. doi: 10.1186/1471-2180-10-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nielsen PK, Andersen AZ, Mols M, van der Veen S, Abee T, Kallipolitis BH. 2012. Genome-wide transcriptional profiling of the cell envelope stress response and the role of LisRK and CesRK in Listeria monocytogenes. Microbiology 158:963–974. doi: 10.1099/mic.0.055467-0. [DOI] [PubMed] [Google Scholar]
- 16.Gottschalk S, Bygebjerg-Hove I, Bonde M, Nielsen PK, Nguyen TH, Gravesen A, Kallipolitis BH. 2008. The two-component system CesRK controls the transcriptional induction of cell envelope-related genes in Listeria monocytogenes in response to cell wall-acting antibiotics. J Bacteriol 190:4772–4776. doi: 10.1128/JB.00015-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeong JH, Kim YS, Rojviriya C, Ha SC, Kang BS, Kim YG. 2013. Crystal structures of bifunctional penicillin-binding protein 4 from Listeria monocytogenes. Antimicrob Agents Chemother 57:3507–3512. doi: 10.1128/AAC.00144-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. 2010. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Y, Zhang W, Shi Q, Hesek D, Lee M, Mobashery S, Shoichet BK. 2009. Crystal structures of penicillin-binding protein 6 from Escherichia coli. J Am Chem Soc 131:14345–14354. doi: 10.1021/ja903773f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nicholas RA, Krings S, Tomberg J, Nicola G, Davies C. 2003. Crystal structure of wild-type penicillin-binding protein 5 from Escherichia coli: implications for deacylation of the acyl-enzyme complex. J Biol Chem 278:52826–52833. doi: 10.1074/jbc.M310177200. [DOI] [PubMed] [Google Scholar]
- 21.Nicola G, Peddi S, Stefanova M, Nicholas RA, Gutheil WG, Davies C. 2005. Crystal structure of penicillin-binding protein 5 bound to a tripeptide boronic acid inhibitor: a role for Escherichia coli Ser-110 in deacylation. Biochemistry 44:8207–8217. doi: 10.1021/bi0473004. [DOI] [PubMed] [Google Scholar]
- 22.Davies C, White SW, Nicholas RA. 2001. Crystal structure of a deacylation-defective mutant of penicillin-binding protein 5 at 2.3-Å resolution. J Biol Chem 276:616–623. doi: 10.1074/jbc.M004471200. [DOI] [PubMed] [Google Scholar]
- 23.Nicola G, Tomberg J, Pratt RF, Nicholas RA, Davies C. 2010. Crystal structures of covalent complexes of beta-lactam antibiotics with Escherichia coli penicillin-binding protein 5: toward an understanding of antibiotic specificity. Biochemistry 49:8094–8104. doi: 10.1021/bi100879m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoshida H, Kawai F, Obayashi E, Akashi S, Roper DI, Tame JR, Park SY. 2012. Crystal structures of penicillin-binding protein 3 (PBP3) from methicillin-resistant Staphylococcus aureus in the apo and cefotaxime-bound forms. J Mol Biol 423:351–364. doi: 10.1016/j.jmb.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 25.Sharma R, Park TE, Moy S. 2016. Ceftazidime-avibactam: a novel cephalosporin/beta-lactamase inhibitor combination for the treatment of resistant Gram-negative organisms. Clin Ther 38:431–444. doi: 10.1016/j.clinthera.2016.01.018. [DOI] [PubMed] [Google Scholar]
- 26.Kelly JA, Knox JR, Zhao H, Frere JM, Ghaysen JM. 1989. Crystallographic mapping of beta-lactams bound to a d-alanyl-d-alanine peptidase target enzyme. J Mol Biol 209:281–295. doi: 10.1016/0022-2836(89)90277-5. [DOI] [PubMed] [Google Scholar]
- 27.Cha HJ, Jeong JH, Kim YG. 2014. Crystallization and preliminary X-ray crystallographic analysis of PBPD2 from Listeria monocytogenes. Acta Crystallogr F Struct Biol Commun 70:535–537. doi: 10.1107/S2053230X14005470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 29.McCoy AJ. 2007. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr 63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 31.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fedarovich A, Nicholas RA, Davies C. 2012. The role of the beta5-alpha11 loop in the active-site dynamics of acylated penicillin-binding protein A from Mycobacterium tuberculosis. J Mol Biol 418:316–330. doi: 10.1016/j.jmb.2012.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pares S, Mouz N, Petillot Y, Hakenbeck R, Dideberg O. 1996. X-ray structure of Streptococcus pneumoniae PBP2x, a primary penicillin target enzyme. Nat Struct Biol 3:284–289. doi: 10.1038/nsb0396-284. [DOI] [PubMed] [Google Scholar]
- 35.Lobkovsky E, Moews PC, Liu H, Zhao H, Frere JM, Knox JR. 1993. Evolution of an enzyme activity: crystallographic structure at 2-Å resolution of cephalosporinase from the ampC gene of Enterobacter cloacae P99 and comparison with a class A penicillinase. Proc Natl Acad Sci U S A 90:11257–11261. [DOI] [PMC free article] [PubMed] [Google Scholar]